
Abstract
The mammalian intestinal tract harbors a vast and diverse ecosystem of microbes that are separated from the sterile host milieu by a single layer of epithelial cells. While this bio-geographical configuration is critical for host biological processes, it imposes a risk for microbial penetration and life-threatening systemic invasion. Inflammasomes are cytosolic multi-protein platforms that sense both microbial and damage-associated molecular patterns and initiate a potent innate immune anti-microbial response. In this review, we will highlight the role of inflammasomes in the orchestration and regulation of the intestinal immune response, focusing on the roles of inflammasomes in maintenance of intestinal homeostasis, enteric infection, auto-inflammation, and tumorigenesis. We highlight the centrality of inflammasome signaling in the complex cross-talk between host mucosal immune arms and the environment, in particular the microflora, with emphasis on the spatial and temporal integration of inflammasome activation with signals from other innate signaling platforms.
Similar content being viewed by others

Introduction
Mucosal surfaces of complex organisms are in constant interaction with a multitude of microorganisms that include bacteria, viruses, parasites, fungi, and protozoa. In normal circumstances, these interactions optimize nutrient uptake and shape appropriate physiological immune responses. However, an alteration in the fine balance between the host and commensal microorganisms can lead to chronic inflammation, tumorigenesis, and metabolic abnormalities.
In mammals, a wide array of recognition systems have evolved to preserve normal interactions with commensal flora and to initiate immune responses towards invading pathogens. Toll-like receptors (TLR) recognize distinct conserved pathogen-associated molecular patterns (PAMPs) at the extracellular and endosomal compartments, and play a critical role in host interaction with the commensal microflora.1 In addition, a number of cytosolic receptors recognize intracellular PAMPs and factors released during perturbations in tissue homeostasis known as damage-associated molecular patterns (DAMPs). Inflammasomes are a group of protein complexes that assemble upon recognition of a diverse set of noxious stimuli and are now considered the cornerstone of the intracellular surveillance system.2 Recent evidence indicates that inflammasomes play critical roles in mucosal immune responses and their disruption leads to severe local abnormalities (e.g., propensity for dissemination of infection, intestinal auto-inflammation, and tumorigenesis) with systemic implications (e.g., metabolic syndrome).3, 4
Inflammasomes
Inflammasomes are multi-protein complexes assembled around a set of core components that include a sensor protein, an adaptor protein (apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (CARD) (ASC)), and an inflammatory caspase. Sensor proteins belong to two families of proteins: the NLR family and the PYHIN family. Inflammasome-forming NLR family members include NLRP1, NLRP2, NLRP3, NLRP6, NLRP7, NLRC4, and potentially NLRP12.5 They all contain a nucleotide-binding domain (NBD), carboxy-terminal leucine-rich repeat (LRR) domain, and can contain a PYD, a CARD or both. The LRR domain is involved in auto-inhibition that is disabled upon direct or indirect sensing of the activating signal, whereas the NBD is required for homo-oligomerization or hetero-oligomerization that leads to inflammasome assembly.5 Similarly, inflammasome-forming PYHIN family members (AIM2 and IFI16) are characterized by having, in addition to a PYD, a HIN200 domain, which is involved in direct ligand binding.6, 7
Thus, inflammasomes comprise, in essence, a multi-protein platform for the activation of inflammatory caspases, of which caspase-1 seems to have a dominant role in inflammatory reactions.8 Caspase-1 is synthesized as an inactive zymogen (pro-caspase-1) and becomes proteolytically active after controlled dimerization in the inflammasomes. Tight control of caspase-1 activation by the inflammasomes is paramount since the processing and release of IL-1β and IL-18, two quintessential inflammatory cytokines, as well as a subset of leaderless proteins that facilitate tissue repair, is directly regulated by caspase-1. Moreover, caspase-1 is required for pyroptosis, a form of cell death frequently observed during microbial infection that combines characteristics of apoptosis (DNA fragmentation) and necrosis (inflammation and cytokine release). The exact contribution of IL-1β, IL-18, and pyroptosis in intestinal mucosal processes will be discussed in further detail below.
Models of Inflammasome Activation
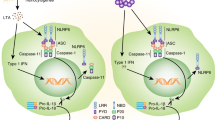
The diversity of exogenous and endogenous signals that activate the inflammasomes highlights their relevance as cytoplasmic sensors (Figure 1). Several models have been proposed to explain how a wide range of dissimilar signals are sensed; these include models based on the direct or indirect recognition of activating signals or on the recognition of general cellular stress. In a minority of cases, direct binding of specific ligands, such as muramyl dipeptide (MDP) and double-stranded DNA (dsDNA), leads to conformational changes in NLRP1 and AIM2, respectively, resulting in inflammation assembly and activation.6, 9, 10, 11 Furthermore, oxidized mitochondrial DNA has been described as a trigger of the NLRP3 inflammasome.12 Alternatively, the NLRP3 and NLRC4 inflammasomes are activated indirectly through interactions with other sensor proteins. For instance, NAIP proteins sense proteins from bacteria with type III and type IV secretion systems (T3SS and T4SS) such as Salmonella, Pseudomonas, Legionella, and Yersinia, which result in the recruitment of NLRC4 and assembly of the inflammasome.13, 14 Similarly, increased amounts of reactive oxygen species (ROS) are sensed by a complex of thioredoxin and thioredoxin-interacting protein (TXNIP), leading to the dissociation of this complex. Subsequent binding of TXNIP to NLRP3 leads to the activation of NLRP3, the recruitment of ASC and pro-caspase-1, and formation of the active inflammasome complex.15 Finally, inflammasomes sense and are activated by general perturbations of the cytoplasmic milieu. The phagocytosis of specific crystalline and particulate structures can lead to lysosome destabilization and the release of lysosomal content, including proteases, which could lead to a proteolytic inactivation of a yet uncharacterized negative regulator or to activation of a positive regulator of NLRP3, resulting in NLRP3 inflammasome assembly.16

Activators of the inflammasomes. The NLRP3, NLRC4, NLRP1, AIM2, and NLRP6 inflammasomes are activated by sterile (host- and environmentally derived) and pathogen-derived noxious stimuli. Assembly of the NLRs or PYHINs, ASC, and pro-caspase-1 leads to the formation of a multi-protein structure: the inflammasome. Activation of the inflammasome leads to maturation and secretion of IL-1β and IL-18 as well as inflammatory cell death (pyroptosis). ASC, apoptosis-associated speck-like protein containing a caspase recruitment domain; MDP, muramyl dipeptide; MSU, monosodium urate; NLR, Nod-like receptor.
Regulation of Mucosal Immune Responses by Inflammasomes
Inflammasome assembly and activation depend on the simultaneous expression of all components within the same cell type. Caspase-1 and ASC are usually expressed in many tissues and cell types, whereas the inflammasome sensors feature a more restricted pattern of expression, suggesting cell type-specific mechanisms for sensing tissue perturbations. In fact, in recent years it has become evident that multiple inflammasomes within different cell-types (e.g., epithelial, hematopoietic cells) accomplish different, but often complementary functions during mucosal immune responses. This is particularly evident in the gastrointestinal tract, a region where multiple cell types come in intimate contact with an immense load of commensal and potentially pathogenic microbes. In the following sections, we discuss how activation of intestinal inflammasomes in different lineages of cells regulates physiological reactions and how their hyper-activation or absence can lead to deleterious consequences such as inflammation or cancer progression.
Inflammasome Regulation of Intestinal Infection
Acute and chronic infectious disorders of the gastrointestinal tract constitute common causes of morbidity and mortality in humans. Multiple defense mechanisms are aimed at minimizing and containing such infectious, including physical barriers such as tightly adherent epithelial cells, geographical and nutritional competition by a community of commensal microbes, a variety of constitutive and inducible anti-microbial peptides, and complex mucosal innate and adaptive immune arms aimed at containing and eliminating invasive infections. Nevertheless, very little is known about host-derived sensing systems and ways by which they differentiate between harmless commensal microbes and nomadic pathogens that pose threat of infection.
The enormous repertoire of commensal microbes that includes bacteria, viruses, fungi, parasites, and archaea, potentially harmful commensals (pathobionts) and occasionally invading microbial pathogens necessitates host “friend–foe” recognition based on general motifs rather than that of individual microbial species. The innate immune response has evolved several classes of innate receptors to recognize such molecular patterns, including PAMPs and DAMPs, which include TLRs, Nod-like receptor (NLRs) and C lectin-like receptors (CLRs), and Rig-I-like receptors (RLRs). Of these, inflammasome-forming NLRs possess an ability to respond to microbial antigens but also to factors released during tissue stress or damage. In the gut, such capability may enable molecular sensing platforms to identify microbial pathogenicity based on both unique microbial-related activators and factors released during tissue damage after invasion of the host mucosa. The later may elicit an innate immune response that is related to the damage inflicted rather than to the identity of the invading microbe.
Indeed, the most widely studied inflammasomes, the NLRC4 and NLRP3 inflammasomes have been shown to regulate a number of common intestinal mucosal infections. The enteric pathogen Salmonella typhimurium activates the NLRC4 inflammasome in a type III secretion system (T3SS)-dependent manner. Leakage of flagellin through the T3SS is necessary for NLRC4 activation in macrophages, although direct binding of flagellin to NLRC4 has not been documented.17 Flagellin-induced activation of the NLRC4 inflammasome is independent of flagellin-induced TLR5 signaling, indicating that innate sensing of intestinal pathogens may be optimized by integration of signals from complementary sensing modules. Such integration may provide a fail-safe mechanism in which the potentially harmful immune response may be triggered only upon full-blown bacterial invasion. In vivo, oral inoculation of Caspase-1/11 knockout (KO) mice with Salmonella was associated with increased levels of infection, demonstrating that inflammasome activation plays a protective anti-microbial role in this infection.18 The T3SS effector protein SopE was found to be required for epithelial Caspase-1/11-mediated inflammation.19 Both IL-1β, IL-18, and macrophage pyroptosis were shown to participate in this anti-microbial response.20, 21 Surprisingly, neither Nlrc4 KO mice18 nor Nlrp3 KO mice22 featured enhanced susceptibility to Salmonella infection, at least on the C57Bl/6 background.23 This discrepancy with the in vivo Caspase-1/11 results was recently resolved when the two inflammasomes were shown to feature redundant roles, by reacting to distinct Salmonella signals.24 The NLRC4 inflammasome activates Caspase-1 in response to flagellin released by the SPI-2 T3SS, while NLRP3 responds to a yet unknown T3SS-independent signal. Thus, deficiency of either sensor alone does not result in significant susceptibility to infection, yet Nlrc4/Nlrp3 double KO mice feature susceptibility that is indistinguishable that that of Caspase-1/11 KO mice.
In contrast to Salmonella, the enteric pathogen Shigella flexneri lacks flagellin yet activates the same NLRC4 inflammasome through the conserved T3SS component PrgJ.25 Shigella-infected inflammasome-deficient macrophages feature a marked reduction in pyroptosis coupled with a substantial enhancement in autophagy, suggesting that inflammasome signaling may regulate the interplay between these distinct cellular death/repair mechanisms.26 As both flagellin and PrgJ are not believed to directly interact with NLRC4, distinct members of the Naip family of proteins were recently suggested to provide a physical link between either flagellin (Naip5 and Naip6) or PrgJ (naip2) with NLRC4, establishing a specific NLRC4 inflammasome activation platform.13, 14, 27, 28 However, the human Naip family only consists of a single member, suggesting that complementary mechanisms providing specificity to NLRC4 inflammasome activation may exist in humans.
Another recent example of inflammasome regulation of enteric infection is Citrobacter rodentium, a member of the attaching and effacing bacterial group, which is a mouse model for entero-hemorrhagic and entero-pathogenic Echerichia coli infection in humans.29 Nlrp3, Nlrc4 and Caspase-1/11 KO mice featured enhanced susceptibility to Citrobacter infection, which was phenocopied by both Il1b and Il18 KO mice and was independent of T3SS. Thus, it is possible that other enteric infections are also sensed and regulated by one or more inflammasome signaling pathways.
Nevertheless, inflammasome signaling in response to enteric enterobacteriaceae infection may not always play protective roles. As recently demonstrated by Ayres et al.,30 expansion of an antibiotic-resistant pathobiont strain of E. coli following administration of wide-spectrum antibiotics coupled with colonic epithelial disruption by dextran sodium sulfate (DSS), resulted in a lethal sepsis syndrome mediated by systemic bacterial dissemination. Systemic disease severity in this model was highly dependent upon the Naip5-NLRC4 inflammasome pathway and mediated by IL-1β, suggesting that NLRC4 signaling is also involved with downstream systemic consequences of disseminated bacterial infection. In contrast, NLRC4 expressed in lamina propria macrophages featured a protective role against an orally administered external pathogen (Salmonella or Pseudomonas), eliciting activation by cleavage of macrophage pre-formed IL-1β upon infection that induced a protective innate immune response.31 Altogether NLRC4, possibly in conjunction with other yet unidentified innate sensors, emerges as a key sensor orchestrating host differentiation between pathogens and commensals, resulting in pro-inflammatory or tolerogenic mucosal immune response in different physiological contexts.
Inflammasome Regulation of Intestinal Auto-inflammation
Inflammatory caspases: early players in intestinal inflammation
As inflammasome activation can lead to a potent pro-inflammatory immune response, aberrant inflammasome signaling may elicit a break in homeostasis culminating in auto-inflammation, in which disproportional and poorly regulated inflammation imposes immuno-pathology to host tissues. Indeed, the downstream inflammasome effector molecules Caspase-1 and Caspase-11 were shown to be central in the maintenance of mucosal homeostasis in mice, even prior to the discovery of the inflammasome as the major activating platform for these two inflammatory caspases. In work published over a decade ago by the Dinarello group,32 intestinal auto-inflammation was induced in wild-type (WT) and Caspase-1/11 KO mice by oral administration of DSS, an agent that induces colonic epithelial cell apoptosis, resulting in mucosal influx of normal commensal microbes, leading to an auto-inflammatory reaction. Caspase-1/11 KO mice (thought at the time to be Caspase-1 KO mice, prior to the recent identification of a spontaneous Caspase-11 mutation in this lineage33) featured reduced colitis severity as compared with WT mice, accompanied by reduced local levels of IL-1β, IL-18, and IFN-γ. Administration of IL-1 receptor antagonist yielded a similar protective response, suggesting that reduced IL-1 production was responsible for the amelioration of colitis in these mice. Similar results were obtained when using a Caspase-1 antagonist, Pralnacasan, that induced an amelioration of DSS colitis accompanied by reduced levels of IL-18 and IFN-γ in both Balb/C and C57Bl mice.34, 35 Of note, WT mice employed in these works were not littermate controls, thus the possible contribution of the intestinal microflora was not assessed at the time.
As with IL-1β, IL-18 has been suggested to be involved in the pathogenesis of inflammatory bowel disease (IBD) in animal models and in humans. In mice induced with either 2,4,6-trinitrobenzene sulfonic acid (TNBS) colitis or DSS colitis, colonic IL-18 levels of both macrophage and epithelial cell origin were markedly elevated. Transgenic over-expression of IL-18 was associated with exacerbated colitis,36 while administration of IL-18 neutralizing antibodies resulted in significantly reduced inflammation.37, 38 Similarly, local administration by an adenovirus vector of IL-18 anti-sense mRNA to mice induced with T cell transfer colitis was associated with reduced disease severity.39 In contrast, Takagi et al.40 demonstrated that IL-18 and IL-18 receptor deficient mice featured exacerbated DSS colitis, suggesting a more complex role for IL-18 signaling in auto-inflammation. In human Crohn’s disease (CD), elevated levels of active cleaved IL-18 were shown to be expressed in colonic inflammatory lesions, where IL-18 was found to be secreted from both intestinal epithelial cells, macrophages and dendritic cells.41, 42 Most important, polymorphisms in IL-18 or IL-18 accessory protein were suggested to be linked to a susceptibility to IBD.43, 44, 45 It is unclear, however, whether gain or loss of function is responsible for the observed phenotypes in humans. Altogether, these early results indicated that Caspase-1 signaling is a central mediator of the pathogenesis of auto-inflammation, possibly through secretion of mature active IL-1β and IL-18 from epithelial and hematopoietic cells during the inflammatory process.
The confusing roles of inflammasomes in intestinal auto-inflammation
The discovery of inflammasomes as the molecular multi-protein platform that mediate Caspase-1 activation and resultant cleavage of IL-1β and IL-18 led to intense research of their possible regulatory roles in intestinal homeostasis and auto-inflammation (Figure 2). While polymorphisms in NLRP3 were suggested (at least in some human cohorts) to be linked to a susceptibility to IBD,46, 47 their role in the pathogenesis of colonic auto-inflammation in mice became confusing and often conflicting. In a study by Dupaul-Chicoine et al.48 Caspase-1/11 KO mice featured enhanced severity of acute DSS colitis, measured by mortality, weight loss, microscopic parameters, and colonic NF-κB activity. Disease severity was attributed to defective tissue regeneration, leading to enhanced intestinal permeability that in turn resulted in microbial influx into the lamina propria. Administration of recombinant IL-18 ameliorated colitis severity in Caspase-1/11 KO mice, leading to the conclusion that local IL-18 production, mainly of epithelial origin, was protective from auto-inflammation. Similarly, Nlrp3, Asc and Caspase-1/11 KO mice were shown to be hypersensitive to acute DSS colitis, with low colonic IL-18 levels being associated with this disease susceptibility, while administration of exogenous IL-18 was shown to ameliorate colitis severity.49 Supporting these results, susceptibility of Nlrp3-inflammasome-deficient mice was also noticeable during induction of both acute TNBS colitis50 and chronic DSS colitis.51 In contrast, Bauer et al.52 demonstrated that Nlrp3 KO mice are protected from DSS colitis, thus supporting the earlier study by Siegmund et al., and suggested that DSS itself may activate the NLRP3 inflammasome.

Regulation of intestinal homeostasis and immune response by inflammasomes. Expression of multiple inflammasomes, including NLRC4, NLRP3, and NLRP6 is noted in cell lineages of the hematopoietic and non-hematopoietic gastrointestinal compartments. In steady state (left panel), inflammasomes regulate tissue repair and proliferation, composition of the microflora, and host–microflora interactions. Dysregulation of these processes by aberrant functions of inflammasomes (middle panel) leads to auto-inflammation of the intestine, mediated by alterations in the microflora, local elevation of IL-18 and IL-1β leading to aberrant tissue repair and an uncontrolled inflammatory response. Chronic inflammasome dysregulation may eventually lead to inflammation-induced tumorogenesis (right panel), mediated by the dysbiotic microflora, chronically impaired tissue repair mechanisms, and altered local tumor surveillance. Inflammasome regulation of these processes involves epithelial cells (NLRC4 and NLRP6 inflammasomes, possibly NLRP12), hematopoietic lineages (NLRP3 inflammasomes, possibly NLRP12), and stromal cells such as myofibroblasts (NLRP6).
One possible explanation for the opposing and conflicting results in these multiple studies is that different inflammasomes exert differential, at times redundant effects on development and progression of auto-inflammation that may be additive or contrasting, resulting in a hierarchical combinatorial net effects on intestinal auto-inflammation.53 As such, activation of a particular inflammasome in hematopoietic cells such as dendritic cells and macrophages may result in local release of IL-1β and/or IL-18 that induces inflammatory changes such as secretion of IFN-γ, while IL-18 secretion from epithelial cells may play a local role in tissue regeneration and proliferation in response to injury. Furthermore, inflammasome-induced pyroptosis in damaged or infected epithelial cells may affect tissue regeneration and consequently the level of microbial influx into the lamina propria and its effect on inflammation severity. These effects may be induced by different inflammasomes and introduce a complex integrative net result based on temporal and bio-geographical variations. Such differential cell-specific contribution of inflammasome signaling remains to be demonstrated experimentally. An alternative explanation for the many discrepancies in this field is that the specific effects of mutations in the inflammasome pathway on inflammation in the intestine may differ depending on the composition of the commensal microbiota that is present in each individual mouse colony. Indeed, our recent work (see below) suggests that the inflammasome is a critical regulator of colonic microbial ecology.
NLRP6 inflammasome is a key regulator of the colonic microbial ecology
The recently described NLRP6 inflammasome was shown to participate in colonic homeostasis and prevention of auto-inflammation, and demonstrates a prime example of the complexity and hierarchical contributions of different inflammasomes in intestinal homeostasis. In three concurrent publications, Nlrp6 KO mice were shown to feature enhanced colonic auto-inflammation induced by DSS. Chen et al.54 showed that this enhanced inflammatory reaction was accompanied by increased colonic mucosal permeability and pro-inflammatory cytokine production. Similar findings were reported by Normand et al.55 Likewise, we found NLRP6 to be pivotal in colonic homeostasis, with Nlrp6 KO mice featuring mild chronic spontaneous colitis and dramatically exacerbated inflammation following induction of acute DSS colitis.3 Notably, while high colonic expression levels of NLRP6 were identified during steady state in all studies, the relative contribution of different intestinal cellular subsets to NLRP6 expression remain debatable. Chen et al. suggested, through creation of WT or Nlrp6 KO bone marrow chimeric mice, that absence of NLRP6 in the hematopoietic compartment was accountable for the observed inflammation-induced tumorogenesis phenotype (see below). Normand et al. noted expression of NLRP6 mRNA in mouse myofibroblasts and detected protein NLRP6 expression in human myofibroblasts, as demonstrated by immunohistochemistry. Our results suggested that NLRP6 mRNA and protein are expressed in colonic epithelial cells, as demonstrated by mRNA and protein expression quantified in sorted epithelial cells and mixed bone marrow chimeric mice. To further assess a potential epithelial source of NLRP6 expression in steady state, we recently cross-bred flox-stop-flox td-tomato mice to mice transgenically expressing the deleter Cre protein under control of the intestinal epithelial promoter Villin. The resultant mice highly expressed the fluorescent td-tomato marker in intestinal epithelial cells, allowing for high-purity sorting of colonic epithelial cells. The resultant mRNA (Figure 3) clearly demonstrated the predominant expression of all NLRP6 inflammasome components in epithelial cells.

Colonic epithelial expression of the NLRP6 inflammasome. Intestinal epithelial cells from C57bl colons were sorted based on high expression of the td-tomato fluorescent protein in villin-positive cells. NLRP6 mRNA expression in sorted epithelial cells (a) was high and comparable to that of unsorted total colons. Likewise, sorted epithelial cells featured high mRNA expression levels of the inflammasome components ASC and Caspase-1 (b). Data are representative of two independent experiments, N=2–5 mice per group. Error bars represent the s.e.m. of samples within a group.
Importantly, our results showed that enhanced auto-inflammation in Nlrp6 KO mice was mediated by dysregulation of the intestinal microflora. Colitis exacerbation in Nlrp6 KO mice was fully transferable to WT mice upon prolonged cohousing, which fully transmitted the NLPR6 KO microbiota configuration into cohabitated WT mice. As a result, colitis in fully cohoused Nlrp6 KO and WT mice was comparably severe when compared with singly housed WT mice. This transferable phenotype was suggested to be mediated by an inflammasome, as both Caspase-1/11 KO mice and Asc KO mice phenocopied the transferable severe DSS colitis phenotype, with Asc KO mice also featuring vertical transferability of the phenotype upon cross-fostering of newborn Asc KO and WT mice. Furthermore, colonic IL-18 levels were reduced in Nlrp6 KO mice both in steady state and during auto-inflammation. Moreover, Il18 KO mice phenocopied the transferable phenotype and WT/IL-18 KO mixed bone marrow chimeric mice demonstrated that the transferable phenotype was mediated by IL-18 originating from the non-hematopoietic compartment. One must note, though, that while formation of an ASC-dependent Nlrp6 inflammasome was suggested in previous in vitro studies,56 no direct in vivo biochemical evidence has so far been provided for formation of such an inflammasome in colonic epithelial cells.
16S rDNA taxonomic analysis of the microbiota of WT, Asc KO, Nlrp12 KO, and Il18 KO mice in singly and cohoused conditions in our facility revealed profound alteration in NLRP6 inflammasome-deficient mice, that include over-representation of the strict anaerobic taxa Prevotellacae and TM7, with reduced representation of Lactobacillus. This configuration was transferable to WT mice upon cohousing and was reversible upon wide-spectrum antibiotic treatment, negating the transferable DSS colitis phenotype. The over-representation of Prevotellacae was even more pronounced when examined in the bio-geographical region adjacent to the colonic epithelial layer, suggesting that aberrant regulation of the host–microbiota interaction in the NLRP6-deficient state may be accountable for the observed differences. The mechanisms of these alterations may involve defects in host regulatory pathways such as anti-microbial peptide production, mucus secretion, or epithelial adhesion molecules, and remain to be explored. Interestingly, an aberrant microflora configuration was also recently reported for Nlrp3 KO mice, coupled with altered anti-microbial peptide secretion pattern,50 leading to enhanced susceptibility to DSS- and TNBS-induced intestinal auto-inflammation. These effects were suggested to be mainly mediated by cellular subsets of the hematopoietic compartments. These results suggest that inflammasome-mediated regulation of the intestinal microflora may involve the activation of multiple inflammasomes in diverse cellular compartments, acting together to promote innate regulation of the microflora and its interactions with the mucosal immune system. Thus, the differential effects observed in previous studies may, in fact, be explained by defective inflammasome regulation of the composition of the microflora coupled with differences in native microflora in different facilities.
Systemic consequences of inflammasome-mediated dysbiosis
The downstream mechanisms by which the altered inflammasome-deficient microflora exerts its pro-inflammatory effects remain an intriguing open question. The chemokine CCL5 (also known as Rantes) was found to be induced in colonic epithelial cells by the “inflammasome-deficient” microflora, where it induced pro-inflammatory cell influx into the lamina propria.3 Ccl5 KO mice cohoused with Nlrp6 KO mice failed to feature the transferable DSS phenotype despite full acquisition of the colitogenic microflora configuration. Further downstream mechanisms by which the inflammasome-deficient microflora exert systemic effects were recently demonstrated with non-alcoholic fatty liver disease (NAFLD), a disease of epidemic proportions among obese individuals, whose progressive inflammatory variant (non-alcoholic steatohepatitis, NASH) leads to cirrhosis, portal hypertension and hepatocellular carcinoma.57 Cohousing of WT mice or leptin receptor deficient mice with mice deficient in inflammasome components ASC, Caspase-1/11, NLRP3, and NLRP6 induced a transferable exacerbated NAFLD/NASH progression in multiple dietary or genetically induced models (methionine choline-deficient diet (MCDD), high-fat diet (HFD), and leptin receptor deficiency-induced NASH). This intriguing transferability of a metabolic phenotype was abrogated by wide spectrum oral antibiotic treatment. Taxonomic analysis revealed that, in addition to Prevotellacae, the combination of diet (MCDD and HFD) and host genetic alterations (inflammasome deficiency) resulted in emergence of Porphyromonadaceae, a family of strict anaerobe commensal bacteria whose several members have been implicated in the pathogenesis of components of the metabolic syndrome in mice and humans58, 59
Interestingly, non-hepatic manifestations of the metabolic syndrome, such as obesity, were also transferable upon prolonged cohousing, while glucose intolerance was affected by the microflora yet non-transferable, indicating that the downstream systemic effects of representatives of the microflora may extend far beyond those of local inflammatory changes. Thus, inflammasome regulation of intestinal mucosal homeostasis may carry consequences that reach far beyond the gut itself. The mechanism by which altered components of the microflora exert effects on a distal, seemingly unrelated organ such as the liver were shown to involve a cooperative signaling by two sensing systems—the NLRs (mediating the primary effects on the microflora) and TLRs. TLR4 and TLR9 agonists from the locally invasive inflammasome-deficient microflora accumulate in the portal venous system, and induce, in predisposed fatty livers, local induction of TNF that promotes inflammation and hepatocyte damage culminating in NASH. This integrative activity of two innate sensory systems supports our notion that divergence of innate sensing signals may regulate both steady state and the anti-microbial inflammatory systemic responses.
Inflammasomes and Colitis-Associated Tumorigenesis
Colorectal cancer (CRC) is the third most frequent cause of cancer-associated mortality. The development of CRC is determined by complex processes such as genetic mutations in tumor suppressor genes, rapid proliferation of neoplastic cells, and the aberrant induction of angiogenesis. Importantly, genetic mutations in the tumor suppressor gene APC and IBD are important predisposing factors for CRC. Mice that carry a mutation in the Apc gene (ApcMin) develop spontaneous intestinal tumors with a predilection for the small intestine.60 Signaling from the innate immune system seems to play a role in the ApcMin mouse model, as Myd88 KO and germ-free mice have decreased tumorigenic potential.61, 62 Nevertheless, the role of inflammasomes in Apc-driven spontaneous tumorigenesis seems to be minimal, as ApcMin mice treated with Anakinra (IL-1 receptor antagonist) or deficient in either IL-1R or Caspase-1/11 featured a comparable rate of adenomatous polyp formation to control ApcMin mice.63
In addition to idiopathic and familial genetic risk factors, a major risk factor for the development of CRC is prolonged IBD, with the prevalence reaching up to 18% after 30 years of disease.64, 65 The pathogenesis of inflammation-induced CRC involves chronic epithelial cell exposure to inflammatory stimuli such as IL-6 and TNF-α.66, 67, 68, 69 In addition, activation of NF-κB by the classical inhibitor of nuclear factor κB kinase (IKKb)-dependent pathway is crucial for tumor growth and progression.70 In recent years it was realized that, in contrast to idiopathic CRC, inflammasomes play a critical role in the progression of colitis-associated CRC.
IL-18: a pivotal cytokine in colitis-associated tumorigenesis
Prolonged epithelial cell exposure to the noxious microenvironment associated with chronic inflammation, that includes among others elevated levels of cytokines, chemokines, and ROS production, is strongly associated with increased rates of epithelial proliferation, angiogenesis, and tissue remodeling, elevating the risk of malignant transformation. The most commonly employed mouse model for inflammation-induced CRC is the Azoxymethane-chronic DSS model (AOM-DSS). Mice administered a combination of the procarcinogen AOM and repeated rounds of DSS develop chronic colitis and associated genomic mutations leading to formation of inflammation-induced CRC. Recent reports suggest that IL-18 is a key protective cytokine in this model. Il-18- and Il18r-deficient mice were shown to be hyper-susceptible to DSS-induced colitis and colorectal tumorigenesis.71 Moreover, administration of exogenous IL-18 alleviated the severity of colitis and colitis-induced tumorigenesis in Caspase-1/11- and Nlrp3-deficient mice.72 In addition, Myd88 KO mice, which are defective in IL-1β and IL-18 production as well as for signaling downstream of their cognate receptors, exhibited increased colonic epithelial proliferation, damage and colorectal tumorigenesis.71 The underlying mechanism of this striking phenotype seems to be associated with reduced levels of DNA repair factors and increased expression of mitogenic and angiogenic genes. In contrast, Il1r KO mice showed equal number of colorectal tumors in the CAC AOM/DSS model, highlighting the unique and essential role of IL-18 during intestinal tumor progression.
Hematopoietic NLRP3 suppresses colitis-associated tumorigenesis
Consistent with the protective role of IL-18 in inflammation-induced CRC, Nlrp3-, Caspase-1-, and Asc-deficient exposed to AOM/DSS have also been reported to have augmented tumor burden by unclear mechanisms51, 72 (Figure 2). Zaki et al.72 proposed that NLRP3-dependent IL-18 secretion is required for STAT1 activation and IFN-γ induction, leading to decreased immune tumor surveillance in the absence of IL-18. Allen et al.51 also demonstrated that Nlrp3−/− mice had increased disease outcome, morbidity, histopathology, and polyp formation. Interestingly, through bone marrow reconstitution experiments they showed that the NLRP3 inflammasome in hematopoietic cells, rather than intestinal epithelial cells or stromal cells, is responsible for protection against increased tumorigenesis. The cell types sensitive to NLRP3-mediated IL-18 secretion within the tumor microenvironment and the molecular pathways affected by IL-18 remain to be fully elucidated and deserve further characterization in the near future.
NLRP6: an epithelial suppressor of intestinal tumorigenesis
In a similar fashion to NLRP3, NLRP6 was recently shown to play a protective role against the development of colitis-associated tumors in the AOM/DSS model.73, 74 Although it is possible that NLRP6 expression is induced in hematopoietic cells upon activation, NLRP6 seems to be primarily expressed in the non-hematopoietic compartment, including intestinal epithelial cells (Figure 3). One possible explanation of these data is that there are compartmentalized functions of different inflammasomes that lead to complementary and appropriate anti-tumor immune responses. The increased tumorigenesis in Nlrp6-deficient mice was associated with elevated inflammatory responses and defective production of IL-18 within the colon.73 Furthermore, gene expression analysis of tumors from Nlrp6 mice showed alterations in pathways involved in epithelial proliferation and transformation (mainly of the Notch and Wnt pathways).74 While the exact mechanism of NLRP6-mediated protection against tumor progression remains to be elucidated, it is tempting to speculate that NLRP6 regulation of microbial communities might be a pivotal event in the prevention of inflammation and CRC.
NLRP12: a new player in colonic inflammation and tumorigenesis
NLRP12 has been shown to be a negative regulator of NF-κB activity and potentially to form an inflammasome.75 Two recent reports indicate that Nlrp12-deficient mice are highly susceptible to colitis and colitis-associated CRC, which was associated with an increased production of inflammatory cytokines and chemokines.76, 77 In concordance with previous reports, the molecular mechanism through which NLRP12 regulates tumorigenesis seems to be associated with its ability to dampen the NF-κB and ERK pathways.76, 77 Importantly, whereas Zaki et al.77 showed that NLRP12 tumor suppressor activity was restricted to macrophages, Allen et al.76 demonstrated that the non-hematopoietic compartment contributed dominantly to this phenotype. The exact molecular mechanism of NLRP12 in each cellular compartment, its role in mucosal homeostasis, its potential effect on microflora ecology, and whether the assembly of an inflammasome is required during NLRP12-mediated mucosal processes is the topic of current intense research. Further evidence implicating inflammatory caspases in inflammation-induced CAC was demonstrated in studies in Caspase-12 KO mice, a dominant-negative regulator of the NF-κB pathway. The absence of Caspase-12 leads to severe colitis; moreover, Caspase-12-deficient mice exhibited an increased inflammatory response after injury, resulting, in turn, to compensatory proliferation and cell survival, which eventually led to enhanced intestinal tumorigenesis.48
Inflammation-Independent Regulation of Colitis-Associated Cancer by NLRC4
Nlrc4-deficient mice have been reported to develop more tumors in the AOM/DSS model.78 Remarkably, this protective effect was independent of inflammation since the severity of colitis was not significantly different between Nlrc4 KO and WT mice. Furthermore, through bone marrow chimera experiments, Hu et al.78 demonstrated that the tumor suppressor function of NLRC4 was required in the non-hematopoietic compartment and proposed that NLRC4 regulates epithelial proliferation and apoptosis in advanced tumors. Importantly, NLRC4 tumor suppressor activity was shown to be required in both compartments (hematopoietic and non-hematopoietic), which points towards an upstream influence of multiple inflammasome components during inflammation-induced tumorigenesis.79 Interestingly, Allen et al.51 found no difference in tumor potential between Nlrc4−/− and WT mice using the AOM/DSS model, indicating that differences in the gut microbiota in different facilities could affect the outcomes of NLRC4 deficiency. Altogether, integrated signaling from multiple inflammasomes, some regulating the intestinal inflammatory process and host–microbiota interactions and others directly regulating the neoplastic process, seem to be governing inflammasome-dependent regulation of inflammation-induced CRC.
Concluding Remarks
A decade has passed since the discovery of the inflammasomes. Since then, the biochemical characterization of the different inflammasomes has led to a more complete understanding of innate immune responses in the context of infection and tissue damage. As expected, these fascinating structures have proven to be critical for immune responses in mucosal surfaces. Moreover, in recent years their roles have been extended to tumorigenesis, regulation of microbial ecology, and metabolic syndrome. Despite this wealth of information, it is still unclear how some of these platforms are activated; furthermore, it is also unknown how cells “decide” to engage death pathways after inflammasome activation and what are the consequences of this phenomenon in tissue homeostasis and repair, underlining the necessity of differential activation of distinct inflammasome platforms in different cellular compartments. It is expected that studies of mice with conditional inactivation of the various inflammasome components will identify the specific functions of inflammasomes in epithelial and hematopoietic cells in the context of mucosal–microbial interactions. Improved understanding of cross-talk between NLRs and PRRs will help better understand these issues, for example, PRR-dependent recognition of microbes in required to prime for activation of at least some inflammasomes; on the other hand, inflammasome-dependent regulation/restriction of the microbial community in the gut is required to prevent aberrant PRR signaling in normally sterile organs/microenvironments. Finally, studying the roles of inflammasomes and their downstream effector molecules in human gastrointestinal infection, auto-inflammation, and tumorigenesis should add important mechanistic insight pertinent to the pathogenesis and treatment of human disease.
References
Rakoff-Nahoum, S., Paglino, J., Eslami-Varzaneh, F., Edberg, S. & Medzhitov, R. Recognition of commensal microflora by toll-like receptors is required for intestinal homeostasis. Cell 118, 229–241 (2004).
Strowig, T., Henao-Mejia, J., Elinav, E. & Flavell, R. Inflammasomes in health and disease. Nature 481, 278–286 (2012).
Elinav, E. et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757 (2011).
Henao-Mejia, J. et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 482, 179–185 (2012).
Schroder, K. & Tschopp, J. The inflammasomes. Cell 140, 821–832 (2010).
Fernandes-Alnemri, T., Yu, J.W., Datta, P., Wu, J. & Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513 (2009).
Kerur, N. et al. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe. 9, 363–375 (2011).
Martinon, F., Burns, K. & Tschopp, J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell 10, 417–426 (2002).
Hornung, V. et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518 (2009).
Roberts, T.L. et al. HIN-200 proteins regulate caspase activation in response to foreign cytoplasmic DNA. Science 323, 1057–1060 (2009).
Hsu, L.C. et al. A NOD2-NALP1 complex mediates caspase-1-dependent IL-1beta secretion in response to Bacillus anthracis infection and muramyl dipeptide. Proc. Natl. Acad. Sci. USA 105, 7803–7808 (2008).
Shimada, K. et al. Oxidized mitochondrial DNA activates the NLRP3 inflammasome during apoptosis. Immunity 36, 401–414 (2012).
Zhao, Y. et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477, 596–600 (2011).
Kofoed, E.M. & Vance, R.E. Innate immune recognition of bacterial ligands by NAIPs determines inflammasome specificity. Nature 477, 592–595 (2011).
Zhou, R., Tardivel, A., Thorens, B., Choi, I. & Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 11, 136–140 (2010).
Hornung, V. et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat. Immunol. 9, 847–856 (2008).
Franchi, L. et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 7, 576–582 (2006).
Lara-Tejero, M. et al. Role of the caspase-1 inflammasome in Salmonella typhimurium pathogenesis. J. Exp. Med. 203, 1407–1412 (2006).
Muller, A.J. et al. The S. Typhimurium effector SopE induces caspase-1 activation in stromal cells to initiate gut inflammation. Cell Host Microbe. 6, 125–136 (2009).
Raupach, B., Peuschel, S.K., Monack, D.M. & Zychlinsky, A. Caspase-1-mediated activation of interleukin-1beta (IL-1beta) and IL-18 contributes to innate immune defenses against Salmonella enterica serovar Typhimurium infection. Infect. Immun. 74, 4922–4926 (2006).
Miao, E.A. et al. Caspase-1-induced pyroptosis is an innate immune effector mechanism against intracellular bacteria. Nat. Immunol. 11, 1136–1142 (2010).
Sutterwala, F.S. et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 24, 317–327 (2006).
Franchi, L., Munoz-Planillo, R. & Nunez, G. Sensing and reacting to microbes through the inflammasomes. Nat. Immunol. 13, 325–332 (2012).
Broz, P. et al. Redundant roles for inflammasome receptors NLRP3 and NLRC4 in host defense against Salmonella. J. Exp. Med. 207, 1745–1755 (2010).
Miao, E.A. et al. Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc. Natl. Acad. Sci. USA 107, 3076–3080 (2010).
Suzuki, T. et al. Differential regulation of caspase-1 activation, pyroptosis, and autophagy via Ipaf and ASC in Shigella-infected macrophages. PLoS Pathog. 3, e111 (2007).
Lightfield, K.L. et al. Critical function for Naip5 in inflammasome activation by a conserved carboxy-terminal domain of flagellin. Nat. Immunol. 9, 1171–1178 (2008).
Lightfield, K.L. et al. Differential requirements for NAIP5 in activation of the NLRC4 inflammasome. Infect. Immun. 79, 1606–1614 (2011).
Liu, Z. et al. Role of inflammasomes in host defense against citrobacter rodentium infection. J. Biol. Chem. 287, 16955–16964 (2012).
Ayres, J.S., Trinidad, N.J. & Vance, R.E. Lethal inflammasome activation by a multidrug-resistant pathobiont upon antibiotic disruption of the microbiota. Nat. Med. 18, 799–806 (2012).
Franchi, L. et al. NLRC4-driven production of IL-1beta discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat. Immunol. 13, 449–456 (2012).
Siegmund, B., Lehr, H.A., Fantuzzi, G. & Dinarello, C.A. IL-1 beta -converting enzyme (caspase-1) in intestinal inflammation. Proc. Natl. Acad. Sci. USA 98, 13249–13254 (2001).
Kayagaki, N. et al. Non-canonical inflammasome activation targets caspase-11. Nature 479, 117–121 (2011).
Loher, F. et al. The interleukin-1 beta-converting enzyme inhibitor pralnacasan reduces dextran sulfate sodium-induced murine colitis and T helper 1 T-cell activation. J. Pharmacol. Exp. Ther. 308, 583–590 (2004).
Bauer, C. et al. The ICE inhibitor pralnacasan prevents DSS-induced colitis in C57BL/6 mice and suppresses IP-10 mRNA but not TNF-alpha mRNA expression. Dig. Dis. Sci. 52, 1642–1652 (2007).
Ishikura, T. et al. Interleukin-18 overproduction exacerbates the development of colitis with markedly infiltrated macrophages in interleukin-18 transgenic mice. J. Gastroenterol. Hepatol. 18, 960–969 (2003).
Kanai, T. et al. Macrophage-derived IL-18-mediated intestinal inflammation in the murine model of Crohn’s disease. Gastroenterology 121, 875–888 (2001).
Siegmund, B. et al. Neutralization of interleukin-18 reduces severity in murine colitis and intestinal IFN-gamma and TNF-alpha production. Am. J. Physiol. Regul. Integr. Comp. Physiol. 281, R1264–R1273 (2001).
Wirtz, S., Becker, C., Blumberg, R., Galle, P.R. & Neurath, M.F. Treatment of T cell-dependent experimental colitis in SCID mice by local administration of an adenovirus expressing IL-18 antisense mRNA. J. Immunol. 168, 411–420 (2002).
Takagi, H. et al. Contrasting action of IL-12 and IL-18 in the development of dextran sodium sulphate colitis in mice. Scand. J. Gastroenterol. 38, 837–844 (2003).
Pizarro, T.T. et al. IL-18, a novel immunoregulatory cytokine, is up-regulated in Crohn’s disease: expression and localization in intestinal mucosal cells. J. Immunol. 162, 6829–6835 (1999).
Kanai, T. et al. Interleukin 18 is a potent proliferative factor for intestinal mucosal lymphocytes in Crohn’s disease. Gastroenterology 119, 1514–1523 (2000).
Takagawa, T. et al. Association between IL-18 gene promoter polymorphisms and inflammatory bowel disease in a Japanese population. Inflamm. Bowel Dis. 11, 1038–1043 (2005).
Tamura, K. et al. IL18 polymorphism is associated with an increased risk of Crohn’s disease. J. Gastroenterol. 37 (Suppl 14), 111–116 (2002).
Zhernakova, A. et al. Genetic analysis of innate immunity in Crohn’s disease and ulcerative colitis identifies two susceptibility loci harboring CARD9 and IL18RAP. Am. J. Hum. Genet. 82, 1202–1210 (2008).
Villani, A.C. et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat. Genet. 41, 71–76 (2009).
Lewis, G.J. et al. Genetic association between NLRP3 variants and Crohn’s disease does not replicate in a large UK panel. Inflamm. Bowel Dis. 17, 1387–1391 (2011).
Dupaul-Chicoine, J. et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 32, 367–378 (2010).
Zaki, M.H. et al. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32, 379–391 (2010).
Hirota, S.A. et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm. Bowel Dis. 17, 1359–1372 (2011).
Allen, I.C. et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J. Exp. Med. 207, 1045–1056 (2010).
Bauer, C. et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 59, 1192–1199 (2010).
Siegmund, B. Interleukin-18 in intestinal inflammation: friend and foe? Immunity 32, 300–302 (2010).
Chen, G.Y., Liu, M., Wang, F., Bertin, J. & Nunez, G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J. Immunol. 186, 7187–7194 (2011).
Normand, S. et al. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc. Natl. Acad. Sci. USA 108, 9601–9606 (2011).
Grenier, J.M. et al. Functional screening of five PYPAF family members identifies PYPAF5 as a novel regulator of NF-kappaB and caspase-1. FEBS Lett. 530, 73–78 (2002).
Henao-Mejia, J., Elinav, E., Strowig, T. & Flavell, R.A. Inflammasomes: far beyond inflammation. Nat. Immunol. 13, 321–324 (2012).
Bajaj, J.S. et al. Linkage of gut microbiome with cognition in hepatic encephalopathy. Am. J. Physiol. Gastrointest. Liver Physiol. 302, G168–G175 (2012).
Makiura, N. et al. Relationship of Porphyromonas gingivalis with glycemic level in patients with type 2 diabetes following periodontal treatment. Oral Microbiol. Immunol. 23, 348–351 (2008).
Rosenberg, D.W., Giardina, C. & Tanaka, T. Mouse models for the study of colon carcinogenesis. Carcinogenesis 30, 183–196 (2009).
Rakoff-Nahoum, S. & Medzhitov, R. Regulation of spontaneous intestinal tumorigenesis through the adaptor protein MyD88. Science 317, 124–127 (2007).
Dove, W.F. et al. Intestinal neoplasia in the ApcMin mouse: independence from the microbial and natural killer (beige locus) status. Cancer Res. 57, 812–814 (1997).
Lee, S.H. et al. ERK activation drives intestinal tumorigenesis in Apc(min/+) mice. Nat. Med. 16, 665–670 (2010).
Jess, T., Rungoe, C. & Peyrin-Biroulet, L. Risk of colorectal cancer in patients with ulcerative colitis: a meta-analysis of population-based cohort studies. Clin. Gastroenterol. Hepatol. 10, 639–645 (2012).
Eaden, J.A., Abrams, K.R. & Mayberry, J.F. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut 48, 526–535 (2001).
Popivanova, B.K. et al. Blocking TNF-alpha in mice reduces colorectal carcinogenesis associated with chronic colitis. J. Clin. Invest. 118, 560–570 (2008).
Becker, C. et al. TGF-beta suppresses tumor progression in colon cancer by inhibition of IL-6 trans-signaling. Immunity 21, 491–501 (2004).
Bollrath, J. et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell 15, 91–102 (2009).
Grivennikov, S. et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell 15, 103–113 (2009).
Greten, F.R. et al. IKKbeta links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell 118, 285–296 (2004).
Salcedo, R. et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J. Exp. Med. 207, 1625–1636 (2010).
Zaki, M.H., Vogel, P., Body-Malapel, M., Lamkanfi, M. & Kanneganti, T.D. IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J. Immunol. 185, 4912–4920 (2010).
Chen, G.Y., Liu, M., Wang, F., Bertin, J. & Nunez, G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J. Immunol. 186, 7187–7194 (2011).
Normand, S. et al. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc. Natl. Acad. Sci. USA 108, 9601–9606 (2011).
Lich, J.D. et al. Monarch-1 suppresses non-canonical NF-kappaB activation and p52-dependent chemokine expression in monocytes. J. Immunol. 178, 1256–1260 (2007).
Allen, I.C. et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity 36, 742–754 (2012).
Zaki, M.H. et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 20, 649–660 (2011).
Hu, B. et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc. Natl. Acad. Sci. USA 107, 21635–21640 (2010).
Hu, B., Elinav, E. & Flavell, R.A. Inflammasome-mediated suppression of inflammation-induced colorectal cancer progression is mediated by direct regulation of epithelial cell proliferation. Cell Cycle 10, 1936–1939 (2011).
Acknowledgements
We thank C Thaiss, MN Katz, and N Palm for critically reading the manuscript and F Duffy for help with its submission. We apologize to those whose work was not cited owing to space constraints. EE was supported by the Cancer Research Institute (2010–2012) and the United States-Israel Educational Foundation (2009), and is the recipient of the Claire and Emmanuel G. Rosenblatt award from the American Physicians for Medicine in Israel Foundation. JHM was supported by a Postdoctoral Fellowship from the Leukemia and Lymphoma Society. This work was supported in part by the United States-Israel binational Foundation grant (EE and RAF). RAF is an Investigator of the Howard Hughes Medical Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declared no conflict of interest.
Rights and permissions
About this article
Cite this article
Elinav, E., Henao-Mejia, J. & Flavell, R. Integrative inflammasome activity in the regulation of intestinal mucosal immune responses. Mucosal Immunol 6, 4–13 (2013). https://doi.org/10.1038/mi.2012.115
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/mi.2012.115
This article is cited by
-
Epithelial Nlrp10 inflammasome mediates protection against intestinal autoinflammation
Nature Immunology (2023)
-
Uric acid drives intestinal barrier dysfunction through TSPO-mediated NLRP3 inflammasome activation
Inflammation Research (2021)
-
Gain-of-function SNPs in NLRP3 and IL1B genes confer protection against obesity and T2D: undiscovered role of inflammasome genetics in metabolic homeostasis?
Endocrine (2018)
-
Control of lupus nephritis by changes of gut microbiota
Microbiome (2017)
-
The AIM2 inflammasome is a central regulator of intestinal homeostasis through the IL-18/IL-22/STAT3 pathway
Cellular & Molecular Immunology (2017)
