
Abstract
A new polyene macrolide family, closely related to the pentaene macrolide antibiotic roflamycoin, was isolated from the both fermentation broth and biomass of Streptomyces durmitorensis wild-type strain MS405. The main compound was identified by NMR and Fourier transform ion cyclotron resonance mass spectrometry as 32,33-didehydroroflamycoin (1; DDHR). Additional four structurally related compounds were determined solely by MS analysis. DDHR induces cell death by apoptosis in various cancer cell lines as demonstrated by DNA fragmentation. Striking feature of DDHR is its internal fluorescence allowing visualization of labeled plasma membranes and internal membrane structures.
Similar content being viewed by others
Introduction
The main stream of discovery of new bioactive compounds is now concentrated on marine macro and microorganisms and is likely to provide thousands of new compounds with a wide range of bioactivities.1, 2, 3 Actinomycetes in particular Streptomyces species still represent almost inexhaustible source of new compounds with antibiotic, antifungal and other important biological activities.4
Polyketides a highly diverse group of natural products are currently represented with over 10 000 members so far identified.5 The most important subgroup of this family is represented by polyene macrolide antibiotics counting over 200 known compounds isolated and characterized since 1950. Most of these compounds are produced by soil actinomycetes belonging to the Gram-positive bacteria of the genus Streptomyces.6 These natural products have a polyene ranging from three to seven double bonds in length. Only few compounds of this family represent clinically important drugs, most probably because of their high toxicity causing serious side effects as renal failure, hypokalemia and thrombophlebitis.4 Among those with clinical usage are rapamycin (triene),7 nystatins (tetraenes),8 filipins (pentaenes)9 and amphotericin B (heptaene).10 In recent years there has been a considerable progress in the genetic analysis and manipulation of producing strains, which led to engineered biosynthesis of several new polyenes used for detailed studies of structure-activity relationships.11, 12 This strategy together with isolation and characterization of new compounds produced by different microorganisms13, 14, 15 could result in potential applications in therapy of cancer.
Roflamycoin, pentaene macrolide antibiotic originally called flavomycoin was isolated over 20 years ago from Streptomyces roseoflavus JA 5068.16 The flat structure was reported in 1981 by Schlegel,17 whereas the absolute configuration and total synthesis was elucidated by Rychnovsky group.18, 19 Roflamycoin is an unusual member of the polyene macrolide antibiotics that has been shown to form well-defined ion channels.20 In addition to oxo-polyene macrolides, mycosamine-containing polyene macrolide antibiotics such as nystatin and amphotericin B21 also possess well-characterized ion-channel-forming properties. Despite their toxicity and side effects22 they have been used for >40 years as the most effective antifungal agents. For this reason, the finding of new antifungal drugs has become an exciting challenge.
In this work the submerged fermentation, isolation, purification and structure determination of 32,33–didehydroroflamycoin (1, DDHR), a member of new macrolide family produced by Streptomyces durmitorensis MS405 is described. Its biological activity was tested on HeLa cells and different carcinoma mouse and human cell lines.
Materials and methods
General experimental procedures
UV spectra were monitored in methanol in the range of 190–700 nm using Shimadzu multipurpose recording spectrophotometer MPS—2000 (Shimadzu, Kyoto, Japan) equipped with graphic printer PR—3.
HPLC analyses were run on a Waters system (Waters, Milford, MA, USA) equipped with flow controller 600, autosampler 717 and UV detector 486 (Waters). Data were processed with Millennium 32 software (Waters).
Specimens prepared from the biological activity assays were imaged and images were processed using the Olympus IX-81 Cell microscope (Olympus Czech Group, Prague, Czech Republic) equipped with Hamamatsu C4742-80-12AG digital camera (Hamamatsu Photonics, Hamamatsu, Japan).
Optical rotation was measured on AUTOPOL IV polarimeter (Rudolph Research Analytical, Hackettstown, NJ, USA). [α]D20−33.2 (c 0.6, MeOH).
Fermentation
The stock cultures of S. durmitorensis sp. nov., type strain MS405 (DSM41863T; wild type)23, 24 and the mutant strain named as A10 were maintained at 30 °C on petri dishes containing NE medium (1% glucose, 0.2% yeast extract, 0.1% beef extract, 0.2% casamino acids and 2% agar; pH=7.0). The seed culture was made by inoculation of 5 ml NE liquid medium with completely dry white to gray colored colonies with high production of spores, and the growth was performed for 14 days at 28 °C in the dark on a rotary shaker (200 r.p.m.). The seed culture (5 ml) was inoculated into the fermentation (Tryptic Soy Broth; Difco Detroit, MI, USA; 3% Triptic Soy Broth Powder supplemented with 10% mannitol) medium using 500 ml flasks and the same cultivation conditions as mentioned above.
Extraction and isolation
Harvested broth (1.6 l) of S. durmitorensis was centrifugated (12 000 r.p.m.; 20 min; 22 °C), filtered and obtained supernatant was passed through SPE cartridge (STRATA C18-E; giga tubes 20 g per 60 ml, 55 μm, 70 A; Phenomenex, Chromservis, Prague, Czech Republic) first conditioned with MeOH (100 ml) and subsequently with H2O (100 ml). After application of supernatant (150 ml), followed by washing step (3 × 100 ml of H2O), the compounds were eluted with stepwise MeOH gradient (from 10 to 90%; each step 100 ml). Collected fractions were evaporated to dryness, frozen or immediately reconstituted in MeOH. Bioactivity assay was carried out by the same procedure as published previously.25 Active fractions (80 and 90%) were applied on HPLC analysis for further purification and characterization. Mycelium (175 g of wet biomass containing mainly spores and cell debris) was collected from fermentation broth of S. durmitorensis after centrifugation, followed by three times extraction with EtOAc. The pooled extracts were evaporated to dryness under reduced pressure and disolved in MeOH. Semi-preparative HPLC analyses were carried out on Discovery BIO Wide Pore C18 column (10 mm × 150 mm; 10 μm; Supelco, Sigma-Aldrich, St Louis, MO, USA) with a mobile phase A (5% MeOH in H2O) and B (MeOH). Gradient elution started at 100% A (0 min), increasing linearly to 100% B within 45 min, followed by column washing step (100% B, 10 min), and equilibration step (100% A, 15 min) at flow rate 1.5 ml min−1; UV detection at 360 nm; column temperature, 35 °C. The collected fractions maintained in dark were pooled, evaporated to dryness under reduced pressure and stored at −20 °C.
Mass spectrometry
The isolated HPLC fraction and the crude EtOAc extract of the mycelia were dissolved in MeOH/H2O (50:50) and analyzed by direct infusion on an APEX-Ultra FTMS instrument equipped with a 9.4 T. Superconducting magnet and a Dual II ion source (Bruker Daltonics, Billerica, MA, USA). The analysis was performed using electrospray ionization and the spectra were acquired in both positive and negative ion mode. The cell was opened for 1.3 ms, accumulation time was set at 0.2 s for MS experiment (1.0 s for MS/MS experiment), and one experiment consisting of the average of eight spectra was collected for each sample. After MS one MS/MS experiment was performed from the desired ion. The isolation window was set 4 a.m.u. and the collision energy was maintained at −24 V (positive mode) and 16 V (negative mode). The acquisition data set size was set to 1 M points with the mass range starting at m/z 150 a.m.u., resulting in a resolution of 100 000 at m/z 400. The instrument was externally calibrated using clusters of arginine resulting in mass accuracy below 2 p.p.m. The acquired spectra were apodized with a square sine bell function and Fourier transformed with one zero-fill. The interpretation of mass spectra was carried out using DataAnalysis version 4.0 software package (Bruker Daltonics).
NMR spectroscopy
NMR spectra were recorded on a Bruker Avance III (600.23 MHz. for 1H, 150.94 MHz for 13C, Bruker BioSpin GmbH, Rheinstetten, Germany) in CD3OD at 303 K. The residual solvent signal was used as an internal standard (δH 3.330 p.p.m., δC 49.30 p.p.m.). 1H NMR, 13C NMR, gCOSY, 1H-13C gHSQC, 1H-13C gHMBC, ROESY and 1D-TOCSY spectra were measured using standard manufacturers' software. 1H NMR spectrum was zero filled to fourfold data points and multiplied by a window function (two-parameter double-exponential Lorentz–Gauss function) before Fourier transformation to improve the resolution. 13C NMR spectrum was zero filled to twofold data points. Then the line broadening (1 Hz) was used to improve the signal to noise ratio. Protons were assigned by COSY and the assignment was transferred to carbons by HSQC. Chemical shifts are given in δ scale (p.p.m.), coupling constants are given in Hz.
Bioactivity assays on carcinoma cell lines
All carcinoma cell lines were purchased from the American Type Culture Collection (Manassas, VA, USA). Cell lines HL-60 (human promyelocytic leukemia), 4T1 (mouse mammary carcinoma) and CT26.CL25 (mouse colon carcinoma) were maintained at exponential growth in RPMI 1640 medium, HeLa (human cervical carcinoma) and A431 (human epidermoid carcinoma) in Dulbecco's modified Eagle's medium, and MDA-MB-231 (human breast carcinoma) in Leibovitz's L-15 medium with 2 mM L-glutamine, 10 mM 4-(2-hydroxyethylpiperazine-1-ethanesulfonic acid, 0.45% glucose, 100 U ml−1 penicillin, 50 μg ml−1 streptomycin, 1 mM sodium pyruvate and 10% fetal calf serum. Cells were maintained at 37 °C in a humidified 5% CO2 atmosphere. For in vitro toxicity and cell death experiments, 1–1.5 × 105 cells were seeded in 1.8 cm2 well and next day various concentrations of component 1 (10–100 μM) were added to the cultivation media. The cell mortality of treated cultures was determined by the Trypan blue exclusion method after 12, 24 and 48 h. Control experiments were performed in parallel with addition of dimethyl sulfoxide solvent only.
DNA fragmentation was determined in DDHR-treated HL-60 cells. Cells were harvested by centrifugation (700 g for 5 min), incubated with 0.5 ml extraction buffer (10 mM TRIS, 0.1 mM EDTA and 0.5% SDS) containing RNase (10 μg ml−1) at 37 °C for 1 h and digested with proteinase K (final concentration 300 μg ml−1) for 2 h at 50 °C. The DNA was then extracted with phenol/chloroform mixture (1:1), precipitated with ethanol and analyzed by 1.5% agarose gel electrophoresis for 3.5 h (2 V/cm gradient). GeneRuler Express DNA Ladder (Fermentas, St Leon-Rot, Germany) was used to size the DNA fragments. The gel was stained with ethidium bromide and the DNA bands were viewed under 312 nm light.
Bioactivity assays on HeLa cells
Cells were cultivated in D-MEM medium supplemented with 10% fetal calf serum (Gibco, Invitrogen, Carlsbad, CA, USA) grown on the glass cover slips (up to 50% density) in 6-well plates (Nunc, Thermo Fisher Scientific, Waltham, MA, USA), treated with DDHR dissolved in dimethyl sulfoxide (stock solution 10 mM) for various times and concentrations. Wells containing dimethyl sulfoxide only (maxmimum 1%) were assayed as a blank control—no alteration of cellular morphology and physiology was observed. All samples were tested in triplicates. Treated cells (various times and concentrations) grown at 37 °C in a humidified 5% CO2 atmosphere were fixed (3.7% paraformaldehyde in phosphate-buffered saline, 20 min, room temperature), permeabilized (0.1% Triton X-100 in phosphate-buffered saline), blocked (1% bovine serum albumin in phosphate-buffered saline) and stained with Phalloidin-Alexa567 or Phalloidin-Alexa488, anti-tubulin antibody (TUB-1, Exbio, Vestec, Czech Republic) followed by secondary GAM-Alexa488 or anti-Lamp-1 antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), followed by secondary GAM-Alexa594 antibody. Staining of the nuclei was performed mounting specimens in Mowiol-DAPI. DDHR-based fluorescent labeling was performed on fixed cells (3.7% paraformaldehyde in phosphate-buffered saline, 20 min, room temperature) without permeabilization (1 h, room temperature). All fluorescent reagents (including secondary antibodies) were purchased from Molecular Probes, Invitrogen.
Results and discussion
Fermentation and isolation of active compound
During screening of soil samples collected in the area of the Durmitor National Park, Serbia and Montenegro we isolated and identified the actinomycete S. durmitorensis sp. nov., type strain MS405 (DSM41863T; wild type).23 Using bioactivity assay25 on immunosuppressant-sensitive Saccharomyces cerevisiae FAV20 strain we found that this isolate produces compound(s) with a similar mechanism of action like immunosuppressant FK506. The disruption of the gene for the β-keto-synthase domain of the thiostrepton-resistant transformant of S. durmitorensis mutant strain A10 completely abolished the production of the active compounds. The presence of the inactivation cassette in this strain was also confirmed by Southern blot analysis using tsr gene as a probe (data not shown). This experiment confirmed that the modular polyketide synthase is responsible for the production of bioactive compound.
Time-dependent production of active compound was investigated in both S. durmitorensis strains as a function of CFU ml−1 over time and monitored in a bioassay on FK506-sensitive yeast strain S. cerevisiae FAV20. Only the wild-type strain MS405 produced active metabolite upon reaching the stationary phase (72 h) in contrary to the mutant strain A10 (data not shown).
To purify the active compound(s) the culture supernatant (150 ml) of wild-type S. durmitorensis was loaded on SPE cartridge and eluted with stepwise gradient of MeOH in water (10–100%). Obtained 10 fractions and crude EtOAc extracts (1.5 g) prepared from wet mycelia cake (175 g) were tested for bioactivity on FK506-sensitive yeast strain S. cerevisiae FAV20. The crude EtOAc extract and fractions eluted with 80 and 90% of MeOH were found to contain the bioactive compound.
Further HPLC purification was carried out using the Discovery BIO Wide Pore C18 column. Out of the several mobile phases tested (different buffers and organic modifiers) the mobile phase consisting of MeOH–water was selected for further compound purification because of the lowest degree of decomposition observed. It could be seen from the presented chromatogram (Figure 1) that under these conditions only partial separation of about seven closely related compounds was reached. The described procedure allows us to isolate the major active component 1 in sufficient quantity and quality for further structure determination by UV, MS/MS and NMR spectrometry.

HPLC chromatogram of the crude EtOAc extract of the Streptomyces durmitorensis sp. nov., type strain MS405 mycelia. Chromatographic conditions: column, Discovery BIO Wide Pore C18 (Supelco), 150 × 10 mm ID; particle size 10 μm; mobile phase A (5% MeOH in H2O) and B (MeOH). Gradient elution, 100% A (0 min) increasing linearly to 100% B within 45 min, followed by column washing step (100% B, 10 min), and equilibration step (100% A, 15 min); flow rate 1.5 ml min−1; UV detection, 360 nm; column temperature, 35 °C.
Structure elucidation
The combined fractions of semipreparative runs containing active major component 1 were evaporated to dryness under reduces pressure. The resulting yellow amorphous powder was soluble in MeOH, EtOH, Me2CO and dimethyl sulfoxide, but insoluble in water. The UV–VIS spectra in MeOH showed characteristic absorption maxima for polyene macrolide; a broad UV absorption band in the range 243–275 nm with maxima 260 nm and strong UV absorption maxima at 363 nm with a shoulder at 375 nm, thus suggesting the presence of a conjugated pentaene part.
Mass spectrometric analysis of active fraction in positive ion mode revealed sodium and potassium adduct ions at m/z 759.42891 and 775.40317 corresponding to elemental composition C40H64O12 within mass accuracy 0.1 and 0.3 p.p.m., respectively. Also, an extensive family of relating ions with water losses (from one up to nine) was observed. The search for compounds of related composition retrieved roflamycoin containing two more hydrogens in the molecule.18, 19 The MS/MS experiment of the sodium adduct ion at m/z 759.42891 resulted in the prediction of DDHR structure. The analysis was performed in negative ion mode as well. In the negative ion mode only the (M−H+)− and (M+Cl−)− ions of component 1 were observed. There was no evidence for dehydrated forms of the polyene macrolide. The MS/MS experiments of deprotonated ion at m/z 735.43295 and chlorine adduct ion at m/z 771.41001 confirmed the structure of the active substance. Finally, the crude extract was analyzed in negative ion mode in terms to characterize other substances produced. Several macrolides structurally related to component 1 were detected (Figure 2, Table 1).

Negative ion mode FTMS spectrum of the crude EtOAc extract of the Streptomyces durmitorensis sp. nov., type strain MS405 mycelia. All substances were observed as chlorinated adduct ions.
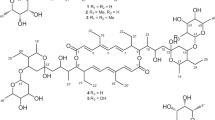
The structure of component 1 (Figure 3) was elucidated by extensive NMR studies. The complete assignment of all proton and carbon resonances listed in Table 2 was achieved using various 2D NMR techniques including COSY, HSQC, HMBC, ROESY and 1D TOCSY experiments.

Structure of 32,33-didehydroroflamycoin component 1.
The 13C NMR spectrum contains 39 signals, which in fact represent 40 carbons due to an overlap of two carbons identified by HSQC. Their multiplicity was sorted by combination of multiplicity-edited HSQC and HMBC spectra. A total of 4 Me, 10 methylenes, 23 methines (11 sp2- and 2 sp3 hybridized and 10 oxymethines) and 3 quaternary carbons were distinguished. The 1H NMR spectrum contains three secondary Me (doublets at 0.911, 1.009 and 1.122 p.p.m.), olefinic Me at 1.993 p.p.m. together with numerous methylenes, oximethines and olefinic protons.
A detailed analysis of the homonuclear and heteronuclear 2D NMR spectra allowed us to identify two main fragments (A and B). The fragment A was deduced as follows. When tracing the COSY correlations starting from methine H-3 at 7.243 p.p.m., a continuous sequence of signals could be followed within structural part A to a terminal aliphatic methylene group resonating at 2.003 and 1.250 p.p.m. The part A can be further extended due to long-range COSY correlation to the methyl 2-Me (1.993 p.p.m.), which indicates that the Δ1,2 double bond is substituted by the methyl group. The HMBC correlation of H-15 and H-16 protons to the carbon signal at 99.13 p.p.m. indicates a quaternary carbon at the end of the fragment A. The heteronuclear long-range coupling between H-13 and C-17 allowed closing the terahydropyrane moiety. The coupling pattern of the proton signals H-13 and H-15 indicated their axial orientation. The configuration of the double bonds in the fragment A can be derived from vicinal coupling constants. Their values are around 15 Hz, which indicates E configuration of the Δ4,5, Δ6,7, Δ8,9 and Δ10,11 double bonds. The configuration on the remaining Δ1,2 double bond was derived from the ROESY spectrum. The detected correlation between 2-Me and H-4 is consistent with E configuration.
The structure elucidation of the part B originated from the oxymethine proton H-35 (δH 4.831) in two directions. It has correlation with a methine H-36, which is further coupled to two methyls (doublets at 0.911 and 1.009 p.p.m.). The chain of the part B can be extended in the opposite direction through the observation of strong vicinal COSY correlation to the aliphatic methine proton at 2.691 p.p.m. The COSY spectrum allowed to identify the fragment from H-34 to H-27 and H-21 to H-18. The remaining part was derived from the HMBC correlations. The coupling constant 3JH−32,H−33=15.8 Hz indicated their E configuration.
Protons H-19, and H-18 has HMBC contact with C-16, which connected fragment B to the fragment A. The correlation between H-35 and C-1 revealed the lactone ring closure.
Biological activity
Compound 1 (DDHR) induces cell death in various cancer cell lines with slightly different effectivity as demonstrated by the range of IC50 (Table 3). The toxicity toward cancer cells was dose dependent and increased with incubation time, but the progression was not linear as most of the cell killing occurred within 12 h (Figure 4). The prevalent mode of DDHR induced cell death was apoptosis as demonstrated by DNA fragmentation (Figure 5).

Toxicity of component 1 (DDHR) against HL60 cells. Cells were incubated with various concentrations of component 1 (a=60 μM, b=70 μM, and c=80 μM) and cell death was determined after 12, 24 and 48 h by trypan blue exclusion method. The averages for three independent experiments are shown with s.d.s.

DNA fragmentation in HL60 cells treated with component 1 (DDHR). Cells exposed to 50 μM of component 1 were harvested after 4 h and isolated DNA was analyzed by 1.5% agarose gel electrophoresis. Control cells (C) were treated with dimethyl sulfoxide only. M, DNA marker.
Biological activity assay performed on HeLa cells shows dramatic modulation of the cellular physiology. Concentrations >25 μM had a cytotoxic effect, cells detached and became permeable for dyes, indicating dead or terminally apoptotic cells (DAPI). Treatment of HeLa cells with component 1 (10 μM, 24 h) resulted in complete loss of F-actin (visualized using fluorescently labeled phalloidin) and transformation of microtubular cytoskeleton into distinct speckles (Figure 6). Staining of the HeLa cells with component 1 after fixation with PFA could be followed by the permeabilization and antibody staining, allowing us to identify the nature of the DDHR-positive internal vesicles as Lamp-1-positive ones (late endosomes and lysosomes; Figure 7). Component 1 obviously quickly entered the cellular interior through membrane flow and vesicular trafficking. This feature together with internal fluorescence could be used for direct visualization of uptake and cellular localization after in vivo treatment and in fixed cells.

Alteration of the actin and tubulin cytoskeleton in HeLa cells treated with 10 μM. DDHR for 24 h. Blue, nuclei stained with DAPI; red, F-actin stained with Phalloidin-Alexa594, green, tubulin stained with the anti-tubulin monoclonal antibody. Scale bar 20 μm. A full color version of this figure is available at The Journal of Antibiotics journal online.

Labeling of PFA-fixed HeLa cells with component 1 (DDHR, 1 h, 300 μM), co-localization of the DDHR-positive vesicles with the late endosomal/lysosomal marker Lamp-1. Red, typical distribution of the late endosomes/lysosomes stained with anti-LAMP-1 antibody; white, fluorescent signal of DDHR localized at the membrane and in the distinct foci highly co-localizing with the Lamp-1 signal. Scale bar 10 μm. A full color version of this figure is available at The Journal of Antibiotics journal online.
References
Hill, L. T. & Fenical, W. Pharmaceuticals from marine natural products: surge or ebb? Curr. Opini. Biotechnol. 21, 777–779 (2010).
Blunt, J. W., Coop, B. R., Munro, M. H. G., Northcote, P. T. & Prinsep, M. R. Marine natural products. Nat. Prod. Rep. 27, 165–237 (2010).
Blunt, J. W. et al. Marine natural products. Nat. Prod. Rep. 26, 170–244 (2009).
Berdy, J. Bioactive microbial metabolites: a personal view. J. Antibiot. 58, 1–26 (2005).
Staunton, J. & Weissman, K. J. Polyketide biosynthesis: a millennium review. Nat. Prod. Rep. 18, 380–416 (2001).
Omura, S. & Tanaka, H. Production and antimicrobial activity of macrolides. In Macrolide antibiotics: chemistry, biology and practice (ed. Omura, S.) 1–19 Academic Press ISBN 0-12-526450-X., Orlando, Fla, (1984).
Findlay, J. A. & Radics, L. On the chemistry and high field nuclear magnetic resonance spectroscopy of rapamycin. Can. J. Chem. 58, 579–590 (1980).
McGarvey, G. J., Mathys, J. A. & Wilson, K. J. Synthesis of amphotericin B. A convergent strategy to the polyol segment of the heptaene macrolide antibiotics. J. Org. Chem. 61, 5704–5705 (1996).
Pandey, R. C. & Rinehart, K. L Jr Polyene antibiotics. V. Characterization of components of the filipin complex by mass spectrometry. J. Antibiot. 23, 414–417 (1970).
Holz, R. W. Polyene antibiotics: nystatin, amphotericin B and filipin. In Antibiotics 5, (ed. Hahn, F. E.) 313–340 Springer-Verlag, New York, (1979).
Caffrey, P., Aparicio, J. F., Malpartida, F. & Zotchev, S. B. Biosynthetic Engineering of polyene macrolides towards generation of improved antifungal and antiparasitic agents. Curr. Topics Med. Chem. 8, 639–653 (2008).
Bruheim, P. et al. Chemical diversity of polyene macrolides produced by Streptomyces noursei ATTC 11455 and recombinant strain ERD44 with genetically altered polyketide synthase NysC. Antimicrob. Agents Chemother. 48, 4120–4129 (2004).
Kwon, H. C., Kauffman, C. A., Jensen, P. R. & Fenical, W. Marinisporolides, polyene-polyol macrolides from marine actinomycete of the new genus Marinispora. J. Org. Chem. 74, 675–684 (2009).
Xiang, W- S., Wang, J- D., Wang, X- J. & Zhang, J. A novel macrolide compound from Streptomyces bingchenggensis: fermentation, isolation, structure elucidation and biological properties. J. Antibiot. 62, 229–231 (2009).
Kozone, I. et al. Novel 24-membered macrolides JBIR-19 and -20 isolated from Metarhizium sp. fE61. J. Antibiot. 62, 159–162 (2009).
Schlegel, R. & Thrum, H. A new polyene antibiotic, flavomycoin. Structural investigations. I. J. Antibiot. 24, 360–374 (1971).
Schlegel, R., Thrum, H., Zielinski, J. & Borowski, E. The structure of roflamycoin, a new polyene macrolide antifungal antibiotic. J. Antibiot. 34, 122–123 (1981).
Rychnovsky, S. D., Griesgraber, G. & Schlegel, R. Stereochemical determination of roflamycoin: 13C acetonide analysis and synthetic correlation. J. Am. Chem. Soc. 117, 197–210 (1995).
Rychnovsky, S. D., Khire, U. R. & Yang, G. Total synthesis of the polyene macrolide roflamycoin. J. Am. Chem. Soc. 119, 2058–2059 (1997).
Grigorjev, P. A. & Bezrukov, S. M. Hofmeister effect in ion transport: reversible binding of halide anions to the roflamycoin channel. Biophys. J. 67, 2265–2271 (1994).
Bolard, J. How do the polyene macrolide antibiotics affect the cellular membrane properties? Biochim. Biophys. Acta 864, 257–304 (1986).
Zotchev, S. B. Polyene macrolide antibiotics and their applications in human therapy. Curr. Med. Chem. 10, 211–223 (2003).
Savic, M., Bratic, I. & Vasiljevic, B. Streptomyces durmitorensis sp. nov., a producer of an FK506-like immunosuppressant. Int. J. Syst. Evol. Microbiol. 57, 2119–2124 (2007).
Savic, M. & Vasiljevic, B. Targeting polyketide synthase gene pool within actinomycetes: new degenerate primers. J. Ind. Microbiol. Biotechnol. 33, 423–430 (2006).
Skoko, N. et al. Construction of Saccharomyces cerevisiae strain FAV20 useful in detection of immunosuppressants produced by soil actinomycetes. J. Microbiol. Methods 61, 137–140 (2005).
Acknowledgements
We thanks the Institutional Research Concept AV 0Z502 00510, Center of Molecular and Cellular Immunology 1M6837805001 nda MSM0021620858, grant no. 2B08064 of the Ministry of Education Youth and Sport, grant no. 143056 of the Ministry of Science of the Republic of Serbia and Bilateral cooperation project between Serbian Academy of Sciences and Academy of Sciences of the Czech Republic for their financial contribution.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Stodůlková, E., Kuzma, M., Hench, I. et al. New polyene macrolide family produced by submerged culture of Streptomyces durmitorensis. J Antibiot 64, 717–722 (2011). https://doi.org/10.1038/ja.2011.81
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ja.2011.81
Keywords
This article is cited by
-
Isolation and identification of endophytic actinobacteria from Citrullus colocynthis (L.) Schrad and their antibacterial properties
Microbial Cell Factories (2022)
-
Disease control efficacy of 32,33-didehydroroflamycoin produced by Streptomyces rectiviolaceus strain DY46 against gray mold of tomato fruit
Scientific Reports (2019)
-
In vitro antifungal activity of antifungalmycin 702, a new polyene macrolide antibiotic, against the rice blast fungus Magnaporthe grisea
Biotechnology Letters (2013)
