
Abstract
Cumulative damage caused by oxidative stress results in diverse pathological conditions. Therefore, elucidating the molecular mechanisms underlying cell death following oxidative stress is important. Here, we describe a novel role for Fas-associated factor 1 (FAF1) as a crucial regulator of necrotic cell death elicited by hydrogen peroxide. Upon oxidative insult, FAF1 translocated from the cytoplasm to the nucleus and promoted the catalytic activation of poly(ADP-ribose) polymerase 1 (PARP1) through physical interaction. Moreover, FAF1 depletion prevented PARP1-linked downstream events involved in the triggering of cell death, including energetic collapse, mitochondrial depolarization and nuclear translocation of apoptosis-inducing factor (AIF), implying that FAF1 has a key role in PARP1-dependent necrosis in response to oxidative stress. We further investigated whether FAF1 might contribute to the pathogenesis of Parkinson’s disease through excessive PARP1 activation. Indeed, the overexpression of FAF1 using a recombinant adeno-associated virus system in the mouse ventral midbrain promoted PARP1 activation and dopaminergic neurodegeneration in a 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease. Collectively, our data demonstrate the presence of an FAF1–PARP1 axis that is involved in oxidative stress-induced necrosis and in the pathology of Parkinson’s disease.
Similar content being viewed by others


Main
Oxidative stress results from an imbalance between the production of reactive oxygen species (ROS) and the rate at which the antioxidant system scavenges cellular ROS.1 During oxidative stress, excessive ROS damage biomolecules and eventually lead to aberrant cell death, which is implicated in the pathogenesis of diverse diseases, such as stroke, Alzheimer’s disease and Parkinson’s disease (PD).1, 2, 3, 4 Hence, understanding the molecular mechanism of cell death in response to oxidative stress is important for the treatment of oxidative stress-related diseases. Although oxidative stress can trigger cell death via multiple cellular pathways, several lines of evidence have suggested that poly(ADP-ribose) polymerase 1 (PARP1) is the key component in the process.5, 6, 7
PARP1 belongs to the family of ADP-ribosyltransferases, which transfer ADP-ribose groups from nicotinamide adenine dinucleotides (NAD+) to their target proteins.8 PARP1 has an N-terminal DNA-binding domain, a central automodification domain and a C-terminal catalytic domain.9 PARP1 has a crucial role in the DNA damage surveillance network after oxidative stress. In response to mild DNA damage, PARP1 recognizes and binds to breaks in the DNA and catalyzes the covalent attachment of poly(ADP-ribose) (PAR) chains to acceptor proteins, including histones, DNA repair proteins and PARP1 itself.9, 10 Subsequently, PARP1 recruits the proteins to sites of DNA damage, thereby maintaining genome stability and cellular homeostasis.
In contrast to the cytoprotective role of PARP1, excessive activation of PARP1 has a prominent role in regulated necrosis under pathological conditions.11 Overactivation of PARP1 by extensive DNA damage quickly depletes intracellular NAD+ and inhibits glycolysis through the PAR-dependent inhibition of hexokinase 1, leading to bioenergetic collapse and regulated necrosis.12, 13, 14 In addition, excess PAR polymers generated by PARP1 result in the depolarization of the mitochondrial membrane potential and the translocation of apoptosis-inducing factor (AIF) from the mitochondria to the nucleus, leading to chromatin condensation and large-scale DNA fragmentation.15, 16 Such biochemical events lead to PARP1-mediated necrosis, specifically named parthanatos.17, 18
Fas-associated factor 1 (FAF1) was initially identified as a Fas-binding protein that potentiates Fas-induced apoptosis.19 FAF1 participates in diverse mechanisms that promote cell death.20 FAF1 mediates caspase-8 activation via both intrinsic and extrinsic pathways and suppresses NF-κB activation by interrupting IκB kinase (IKK) complex assembly.21, 22, 23 In addition, FAF1 arrests the cell cycle by negatively regulating Aurora-A.24 FAF1 also interacts with polyubiquitinated proteins and valosin-containing protein (VCP), inhibiting ubiquitin-dependent protein degradation.25 Consistently with its cell death-promoting roles, FAF1 downregulation has been observed in gastric and uterine cervix carcinomas.26, 27 Hence, FAF1 is thought to act as a tumor suppressor through its cell death-promoting activity.
In addition to its tumor suppressive role, FAF1 is involved in the pathogenesis of PD. FAF1 expression is significantly increased in the frontal cortex and midbrain of PD patients, and it potentiates the toxic effects of stressors associated with PD, including oxidative stress.28 Moreover, FAF1 is a pathogenic substrate of parkin, a ubiquitin E3 ligase.29 The inactivation of parkin by PD-linked mutations or by genetic deletion results in the accumulation of FAF1 and the induction of FAF1-mediated biochemical events, including caspase-3 activation, c-jun-N-terminal kinase (JNK) activation and cell death upon oxidative stress.29 These observations indicate that FAF1 has a pivotal role in oxidative stress-induced cell death and PD pathogenesis through its actions on the apoptotic machinery.
Necrosis as well as apoptosis has been implicated in the PD pathogenesis.30 Certain death-promoting proteins such as p53 and JNK mediate both apoptosis and necrosis upon oxidative stress insults.31, 32, 33 Therefore, we questioned in this study whether FAF1 mediates both types of cell death. Here, we demonstrate that FAF1 also participates in the necrotic machinery via activation of PARP1 upon oxidative stress both at cellular level and in a mouse model of PD. These results have important implications for understanding of the FAF1-mediated pathogenic mechanisms associated with PD.
Results
H2O2 induces PARP1-dependent necrosis in MEFs
Treatment with hydrogen peroxide (H2O2) is commonly used to induce oxidative stress in cells.34 Oxidative stress elicited by H2O2 causes apoptosis and/or necrosis depending on its concentration and on the cell type.1 In the present study, we aimed to investigate the role of FAF1 in necrotic machinery in response to oxidative stress. To this end, we used mouse embryonic fibroblast cells (MEFs) based on the findings that H2O2 mainly induces necrotic cell death in MEFs.7, 35 We first attempted to further define the phase of necrotic cell death in MEFs in response to exposure to 10–1000 μM H2O2, which is a pathophysiologically relevant concentration range for exogenous H2O2.36 As shown in Figure 1a, exposure to H2O2 (250–1000 μM) triggered the death of MEFs in a dose- and time-dependent manner. In contrast, exposure to lower concentrations of H2O2 (10–100 μM) did not induce cell death in MEFs (Supplementary Figure S1). Next, we investigated whether caspase-3 activation, a hallmark of apoptosis, was involved in H2O2-induced cell death. Caspase-3 activity was not detected in MEFs at any concentration of H2O2 (250–1000 μM) that caused cell death (Figure 1b and Supplementary Figure S2). However, caspase-3 was activated when cells were treated with apoptosis inducer TNFα plus cycloheximide (CHX), demonstrating that the apoptotic machinery was intact in the MEFs (Figure 1b and Supplementary Figure S2). To further confirm the absence of caspase activation in H2O2-induced cell death, we used a pan-caspase inhibitor (zVAD-fmk). zVAD-fmk failed to inhibit H2O2-induced cell death, indicating that exposure to H2O2 induces non-apoptotic cell death in MEFs (Figure 1c).

H2O2 induces PARP1-dependent necrosis in MEFs. (a) Left panel: wild-type (WT) MEFs were treated with the indicated concentrations of H2O2 for 6 h. Right panel: WT MEFs were treated for indicated times with 500 μM H2O2. Cell death was determined on the basis of LDH release (n=3). (b) Left panel: WT MEFs were treated with the indicated concentrations of H2O2 or TNFα (50 ng/ml) plus CHX (20 μg/ml) for 4 h and were then analyzed for caspase-3 activity (n=3). Right panel: WT MEFs were treated with 500 μM H2O2 or TNFα (50 ng/ml) plus CHX (20 μg/ml) for the indicated times and were analyzed for caspase-3 activity. Graphs represent the relative activity of caspase-3 compared with the respective controls (n=3). (c) WT MEFs were pretreated with zVAD-fmk (100 μM), Nec-1 (50 μM) or DPQ (30 μM) for 1 h and were then treated with 500 μM H2O2 for 6 h in the presence of individual compounds. Cell death was determined by measuring LDH release (n=3). (d) WT MEFs were pretreated with 3AB (2 mM), PJ-34 (20 μM) or Olaparib (10 μM) for 1 h and were then treated with 500 μM H2O2 for 6 h in the presence of individual compounds. Cell death was determined on the basis of LDH release (n=3). (e) Left panel: Parp1+/+ and Parp1−/− MEFs were treated with 500 μM H2O2 for 6 h. Cell death was determined on the basis of LDH release. Right panel: representative immunoblots show the levels of PARP1 and β-actin expression (n=3). All the data are expressed as the mean±S.E.M. from three independent experiments. Statistical comparisons were evaluated by ANOVA test followed by Games–Howell (Left panel a) or Tukey HSD (Right panel a, b–e) post hoc analysis. ***P<0.001, **P<0.01 and *P<0.05
Various types of non-apoptotic cell death exist, including receptor-interacting protein kinase 1 (RIPK1)-dependent necrosis (necroptosis) and PARP1-dependent necrosis (parthanatos).17 We next examined which type of non-apoptotic cell death occurs in MEFs after H2O2 exposure. We observed that PARP1 inhibitors (DPQ, 3AB, PJ-34 and Olaparib) significantly blocked H2O2-induced cell death (Figure 1c and d), whereas the necroptosis inhibitor necrostatin-1 (Nec-1) was unable to prevent the cell death elicited by H2O2 (Figure 1c). Moreover, PARP1-knockout (Parp1−/−) MEFs were more resistant to H2O2 than were the Parp1+/+ MEFs (Figure 1e). We next examined whether PARP2 was involved in H2O2-induced cell death, because oxidative stress can activate PARP2 as well as PARP1.37 We found that UPF-1069 (a selective PARP2 inhibitor) failed to block cell death and PAR formation induced by H2O2 (Supplementary Figure S3a and S3b). Similarly, siRNA-mediated knockdown of PARP2 did not attenuate H2O2-induced cell death and PAR formation (Supplementary Figure S3c and S3d). Together, our data demonstrate that H2O2 induces PARP1-dependent necrosis in MEFs.
FAF1 is required for PARP1-dependent necrosis induced by H2O2
Next, we examined whether FAF1 is involved in PARP1-dependent necrosis induced by H2O2. Overexpression of FAF1 enhanced H2O2-induced cell death in MEFs in the absence or presence of zVAD-fmk, indicating that FAF1 also functions in non-apoptotic cell death (Figure 2a). In contrast, DPQ prevented the FAF1-mediated cell death elicited by H2O2, indicating that cell death via FAF1 depends on PARP1 activity (Figure 2a). To further investigate the function of FAF1 in PARP1-dependent necrosis, we used the MEFs derived from FAF1 hypomorphic mice.29 The level of FAF1 in MEFs derived from FAF1 hypomorphic mice, Faf1gt/gt, was markedly reduced compared with that in Faf1+/+ MEFs (Figure 2b). Next, we examined whether FAF1 depletion affected the level of PARP1-dependent necrosis induced by H2O2. We found that Faf1gt/gt MEFs exhibited a robust resistance to H2O2 (Figure 2c and d). Furthermore, the restoration of FAF1 in Faf1gt/gt MEFs also regained their sensitivity to H2O2 (Figure 2e). Thus, our data show that FAF1 is required for PARP1-dependent necrosis induced by H2O2.

FAF1 is required for PARP1-dependent necrosis after H2O2 treatment. (a) Left panel: WT MEFs were transfected with the vector control (VC) or Flag-FAF1 plasmids. At 36 h after transfection, cells were pretreated with vehicle (DMSO), zVAD-fmk (100 μM) or DPQ (30 μM) for 1 h and were then treated with 500 μM H2O2 for 6 h in presence of individual compounds. Cell death was determined on the basis of LDH release (n=3). Right panel: representative immunoblots show the levels of FAF1, Flag and β-actin expression. (b) Immunoblot analysis of FAF1 expression in immortalized Faf1+/+ and Faf1gt/gt MEFs. β-actin expression was used as an endogenous control. (c) Faf1+/+ and Faf1gt/gt MEFs were treated with the indicated concentrations of H2O2 for 6 h. Cell death was determined on the basis of LDH release (n=3). (d) Faf1+/+ and Faf1gt/gt MEFs were treated with 500 μM H2O2 for the indicated times. Cell death was determined on the basis of LDH release (n=3). (e) Left panel: Faf1gt/gt MEFs were transfected with the VC or Flag-FAF1 plasmids. Thirty-six hours after transfection, the cells were treated with 500 μM H2O2 for 6 h. Cell death was determined on the basis of LDH release (n=3). Right panel: representative immunoblots show the levels of FAF1, Flag and β-actin expression. Data (a, c–e) are expressed as the mean±S.E.M. from three independent experiments. Statistical comparisons were evaluated by ANOVA test followed by Tukey HSD (a, c–e) post hoc analysis. ***P<0.001, **P<0.01 and *P<0.05
FAF1 translocates to the nucleus and interacts with PARP1 upon oxidative stress
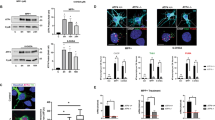
The subcellular localization of proteins often provides insight into their functions. FAF1 contains a nuclear localization sequence and can translocate from the cytoplasm to the nucleus.38 Therefore, we examined the subcellular localization of FAF1 upon oxidative stress. Subcellular fractions and confocal microscopy images of immunofluorescence staining revealed that a fraction of FAF1 translocates from the cytoplasm to the nucleus in MEFs upon treatment with H2O2 (Figure 3a and b).

FAF1 translocates to the nucleus and interacts with PARP1 during oxidative stress. (a) WT MEFs were treated with 500 μM H2O2 for the indicated times and were then fractionated into cytoplasmic and nuclear fractions. The fractions were analyzed by immunoblotting with anti-FAF1, anti-PARP1 (nuclear marker) and anti-β-actin (cytoplasmic marker) antibodies. (b) WT MEFs were treated with 500 μM H2O2 for the indicated times and were then immunostained with the anti-FAF1 antibody. The nuclei were stained using propidium iodide (PI) and analyzed by confocal microscopy. (c) WT MEFs were transfected with the indicated combination of V5-PARP1 and Flag-FAF1 plasmids. At 36 h after transfection, the cells were untreated or treated with 500 μM H2O2 for 30 min. The cell lysates were immunoprecipitated with anti-Flag antibody, followed by immunoblotting with the indicated antibodies. (d) WT MEFs were treated with 500 μM H2O2 for the indicated times. The cell lysates then were immunoprecipitated with the anti-FAF1 antibody. The interactions were determined by immunoblotting with the indicated antibodies
Phosphorylation status of FAF1 is known to regulate its subcellular localization.38 Therefore, we transfected phosphorylation-mimicking form (DD; S289•291D) and phosphorylation-deficient form (AA; S289•291A) of FAF1 to Faf1gt/gt MEFs. The level of FAF1-DD was significantly higher than those of FAF1-AA and FAF1-WT in the nuclei of Faf1gt/gt MEFs (Supplementary Figure S4c). Moreover, phosphorylated FAF1 was found in the nucleus and increased upon oxidative stress in MEFs (Supplementary Figure S4a and S4b). Our data implicate that phosphorylation of FAF1 helps its nuclear presence.
Next, we examined whether FAF1 interacts with PARP1 in the nucleus, because FAF1 is required for PARP1-dependent necrosis induced by H2O2 (Figure 2) and preliminary data obtained by Q-TOF analysis of immunoprecipitates using the FAF1 antibody in HEK293 cells showed that PARP1 is a member of the FAF1-interacting protein group (data not shown). In a co-immunoprecipitation assay, Flag-FAF1 interacted with V5-PARP1 in H2O2-treated MEFs but not in untreated MEFs (Figure 3c). We further confirmed the interaction between endogenous FAF1 and PARP1 in H2O2-treated MEFs, which suggests that FAF1 binds to PARP1 in an oxidative stress-dependent manner (Figure 3d). Furthermore, we found that the FAF1 interacted with the DNA-binding domain of PARP1 and the PARP1 interacted with the death effector domain-interacting domain or c-terminus of FAF1 (Supplementary Figure S5). Collectively, these results imply that oxidative stress triggers the translocation of FAF1 to the nucleus and promotes its binding to PARP1.
FAF1 positively regulates PARP1 activity during oxidative stress
The PARP1 activity can be regulated by the physical interaction of PARP1 with its binding partner.39, 40 Given that FAF1 interacts with PARP1, we examined whether the PARP1 activity might be regulated by FAF1. We found that FAF1 overexpression induced a dose- and time-dependent increase in the PAR polymer in response to H2O2 exposure (Figure 4a and b). However, the level of the PAR polymer was lower in Faf1gt/gt MEFs than in Faf1+/+ MEFs (Figure 4c and d). Moreover, the restoration of FAF1 in Faf1gt/gt MEFs recovered the enzymatic activity of PARP1 during H2O2 exposure (Figure 4e). To elucidate the mechanism how FAF1 activates PARP1, we performed an in vitro PARP1 activity assay. The PAR formation was significantly increased in the presence of GST-FAF1, indicating that FAF1 sufficiently potentiated PARP1 activity without other cellular components (Figure 4f). To further examine the role of FAF1 as positive regulator of PARP1, we treated MEFs with 1-methyl-3-nitro-1-nitrosoguanidine (MNNG), a well-known PARP1 activator. Upon MNNG treatment, nuclear translocation of FAF1 was also observed in MEFs (Supplementary Figure S6a). Moreover, we found that PAR polymers in Faf1gt/gt MEFs were less than those in Faf1+/+ upon MNNG treatment, indicating that FAF1 also participates in the MNNG-induced PARP1 activation (Supplementary Figure S6b). Together, these results indicate that FAF1 acts upstream of PARP1 and that it positively regulates the enzymatic activity of PARP1.

FAF1 promotes the catalytic activity of PARP1 during oxidative stress. (a) Left panel: WT MEFs were transfected with the indicated concentration of Flag-FAF1 plasmids. At 36 h after transfection, the cells were treated with 500 μM H2O2 for 30 min. The cell lysates were immunoblotted with the indicated antibodies. Right panel: the graph shows the result of densitometric analysis of PAR immunoblots (n=3). (b) Left panel: WT MEFs were transfected with the VC or Flag-FAF1 plasmids. At 36 h after transfection, the cells were treated with 500 μM H2O2 for the indicated times. The cell lysates were immunoblotted with the indicated antibodies. Right panel: the graph shows the result of densitometric analysis of PAR immunoblots (n=3). (c) Left panel: Faf1+/+ and Faf1gt/gt MEFs were treated with 500 μM H2O2 for the indicated times. The cell lysates were subjected to immunoblot analysis with the indicated antibodies. Right panel: the graph shows the result of densitometric analysis of PAR immunoblots (n=3). (d) Faf1+/+ and Faf1gt/gt MEFs were treated with 500 μM H2O2 for 30 min and were then immunostained with the anti-PAR antibody. The nuclei were stained using PI and the cells were analyzed by confocal microscopy. (e) Left panel: Faf1gt/gt MEFs were transfected with the VC or Flag-FAF1 plasmids. Thirty-six hours after transfection, the cells were treated with 500 μM H2O2 for the indicated times. The cell lysates were subjected to immunoblot analysis with the indicated antibodies. Right panel: the graph shows the result of densitometric analysis of PAR immunoblot (n=3). (f) Left panel: GST-FAF1 or GST was incubated with recombinant PARP1 (1 unit), β-NAD+ (100 μM) and damaged DNA for 10 min at room temperature. After the in vitro poly(ADP-ribosyl)ation reactions, the samples were subjected to immunoblot analysis. Right panel: the graph shows the results of densitometric analysis of PAR immunoblots (n=3). Quantified data (a–c, e, f) are expressed as the mean±S.E.M. from three independent experiments. Statistical comparisons were evaluated by ANOVA test followed by Tukey HSD (a–c, e, f) post hoc analysis. **P<0.01 and *P<0.05
FAF1-mediated PARP1 activation triggers bioenergetic collapse, mitochondrial depolarization and AIF translocation
Excessive PARP1 activation triggers cell death through the depletion of NAD+ and ATP, the depolarization of the mitochondrial membrane potential and the nuclear translocation of AIF.16 Given that FAF1 promotes the PARP1 activity in PARP1-dependent necrosis during oxidative stress, we examined whether FAF1 regulates the biochemical events downstream of PARP1 activation. First, we measured the levels of intracellular NAD+ and ATP in H2O2-treated Faf1+/+ and Faf1gt/gt MEFs in the presence or absence of DPQ. In response to H2O2 exposure, the consumption of NAD+ and ATP were markedly attenuated in Faf1gt/gt MEFs compared with Faf1+/+ MEFs (Figure 5a and b). Moreover, PARP1 inhibition by DPQ prevented the depletion of intracellular NAD+ and ATP levels in H2O2-treated Faf1+/+ MEFs. These data indicate that FAF1 is required for PARP1-mediated bioenergetic collapse (Figure 5a and b). Next, we measured the mitochondrial membrane potential in Faf1+/+ and Faf1gt/gt MEFs. As shown in Figure 5c, H2O2 treatment led to a significant increase in the mitochondrial depolarization in Faf1+/+ MEFs, whereas the FAF1 depletion significantly attenuated mitochondrial depolarization. The treatment of Faf1+/+ MEFs with DPQ also attenuated mitochondrial depolarization, indicating that FAF1 has a crucial role in PARP1-mediated mitochondrial membrane depolarization (Figure 5c). Finally, we investigated whether FAF1 was also required for nuclear translocation of AIF. Confocal microscopy images of immunofluorescence staining revealed that AIF translocated to the nucleus in Faf1+/+ MEFs upon oxidative stress, but not in Faf1gt/gt MEFs, showing that FAF1 is required for nuclear translocation of AIF after H2O2 exposure (Figure 5d). Collectively, these findings clearly demonstrate that the FAF1 is involved in the biochemical events downstream of PARP1 activation, such as bioenergetic collapse, mitochondrial depolarization and AIF translocation, which testifies the presence of FAF1–PARP1 axis.

FAF1-mediated PARP1 activation induces energy collapse, mitochondrial depolarization and AIF translocation during oxidative stress. (a and b) Faf1+/+ and Faf1gt/gt MEFs were pretreated with vehicle (DMSO) or DPQ (30 μM) for 1 h and then treated with 500 μM H2O2 for 1 h. Depletion of intracellular energy was determined by measuring the levels of NAD+ (a; n=3) and ATP (b; n=3). (c) Faf1+/+ and Faf1gt/gt MEFs were pretreated with vehicle (DMSO) or DPQ (30 μM) for 1 h and then treated with 500 μM H2O2 for 4 h. The cells were analyzed for mitochondrial membrane depolarization with a Muse analyzer (n=3). (d) Faf1+/+ and Faf1gt/gt MEFs were treated with 500 μM H2O2 for 4 h and subsequently immunostained with the anti-AIF antibody. The nuclei were stained using 4',6-diamidino-2-phenylindole (DAPI) and the cells were analyzed by confocal microscopy. Data (a–c) are expressed as the mean±S.E.M. from three independent experiments. Statistical comparisons were evaluated by ANOVA test followed by Tukey HSD post hoc analysis. ***P<0.001, **P<0.01 and *P<0.05
FAF1 regulates PARP1-dependent necrosis in SH-SY5Y cells
PARP1-dependent necrosis has been linked to the pathogenesis of PD.41 Given that FAF1 is a key regulator of PARP1-dependent necrosis, we examined whether FAF1 might be relevant in the pathological aspects of PD. To this end, we used SH-SY5Y human neuroblastoma cells as cellular model of PD.42 First, we investigated whether exposure to H2O2 induces PARP1-dependent necrosis in SH-SY5Y cells. We found that DPQ prevented H2O2-induced cell death in SH-SY5Y cells whereas zVAD-fmk and Nec-1 did not (Supplementary Figure S7a). Similarly, the knockdown of PARP1 by siRNA reduced H2O2-induced cell death in SH-SY5Y cells (Supplementary Figure S7b). These data indicate that exposure to H2O2 induces PARP1-dependent cell death in SH-SY5Y cells. Next, we investigated the effect of FAF1 under these conditions. FAF1 overexpression potentiated H2O2-induced cell death in SH-SY5Y cells, whereas knockdown of FAF1 by siRNA rendered the cells resistant to H2O2, indicating that FAF1 mediates death in SH-SY5Y cells in response to H2O2 treatment (Figure 6a and b). In addition, PARP1 inhibition by DPQ protected cells against FAF1-mediated cell death induced by H2O2, showing that FAF1 mediates PARP1-dependent necrosis in SH-SY5Y cells (Figure 6a). Considering that FAF1 is predominantly present in the cytoplasm and that PARP1 is a nuclear protein in SH-SY5Y cells,29 we then examined whether FAF1 translocates to the nucleus and subsequently interacts with PARP1 in SH-SY5Y cells in response to H2O2. H2O2 induced the translocation of FAF1 to the nucleus and promoted its binding to PARP1 in SH-SY5Y cells (Figure 6c and d). Next, we investigated whether FAF1 regulates the PARP1 activity. We observed that FAF1 overexpression promoted H2O2-induced PARP1 activity in SH-SY5Y cells (Figure 6e). Together, these findings show that FAF1 can regulate PARP1-dependent necrosis in SH-SY5Y cells through the regulation of the PARP1 activity. This result is consistent with our observations in MEFs.

FAF1 mediates PARP1-dependent necrosis in SH-SY5Y cells during oxidative stress. (a) Left panel: SH-SY5Y cells were transfected with the VC or FAF1 plasmids. Thirty-six hours after transfection, the cells were pretreated with vehicle (DMSO) or DPQ (30 μM) for 1 h and then treated with 500 μM H2O2 for 6 h. Cell death was determined on the basis of LDH release (n=3). Right panel: representative immunoblots show the levels of FAF1 and β-actin expression. (b) Left panel: SH-SY5Y cells were transfected with scRNA or FAF1 siRNA. At 48 h after transfection, the cells were treated with 500 μM H2O2 for 6 h. Cell death was determined on the basis of LDH release (n=3). Right panel: representative immunoblots show the levels of FAF1 and β-actin expression. (c) SH-SY5Y cells were treated with 500 μM H2O2 for the indicated times and were then fractionated into cytoplasmic and nuclear fractions. The fractions were analyzed by immunoblot analysis with anti-FAF1, anti-PARP1 (nuclear marker) and anti-β-actin (cytosolic marker) antibodies. (d) SH-SY5Y cells were treated with 500 μM H2O2 for the indicated times and then the cell lysates were immunoprecipitated with the anti-FAF1 antibody followed by immunoblotting with the indicated antibodies. (e) Left panel: SH-SY5Y cells were transfected with the VC or Flag-FAF1 plasmids. At 36 h after transfection, the cells were treated with 500 μM H2O2 for the indicated times. The cell lysates were immunoblotted with the indicated antibodies. Right panel: the graph shows the result of densitometric analysis of PAR immunoblot (n=3). Quantified data (a, b, e) are expressed as the mean±S.E.M. from three independent experiments. Statistical comparisons were evaluated by ANOVA test followed by Tukey HSD post hoc analysis. ***P<0.001, **P<0.01 and *P<0.05
FAF1 promotes dopaminergic neurodegeneration through PARP1-dependent necrosis in an MPTP mouse model of PD
It is known that PARP1 mediates 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurodegeneration.43 Therefore, we chose to use an MPTP-induced model of PD in mice to further study the role of FAF1 in PARP1-mediated neurodegeneration in vivo. We first investigated the translocation of FAF1 in tyrosine hydroxylase (TH)-positive dopaminergic neurons of the ventral midbrain upon oxidative stress induced by MPTP. Following MPTP injection, FAF1 translocated from the cytoplasm to the nucleus in TH-positive neurons (Figure 7a). To determine whether nuclear FAF1 interacts with PARP1 in the ventral midbrain after MPTP injection, we performed a co-immunoprecipitation assay. We found that there was a presence of physical interaction between FAF1 and PARP1 in the midbrain tissue of the mice injected with MPTP (Figure 7b). These data imply that FAF1 translocates into the nucleus and interacts with PARP1 in dopaminergic neuronal cells treated with MPTP. To further confirm the in vivo role of FAF1, we overexpressed FAF1 in the mouse ventral midbrain using a recombinant adeno-associated virus (AAV) system (Figure 7c). FAF1 protein expression in the midbrain tissues collected from mice injected with AAV1-FAF1 viruses was significantly increased compared with that of the controls (Figure 7d). Next, we investigated whether FAF1 overexpression promotes dopaminergic neurodegeneration via PARP1 activation. First, PARP1 activation was determined by western blot analysis for the presence of the PAR polymer in the ventral midbrain. After MPTP treatment, we found that the PAR formation was elevated even further in the ventral midbrain of AAV1-FAF1 injected side compared with that of the controls (Figure 7d). And, caspase-3 was not activated in ventral midbrain of saline- and MPTP-treated AAV1-FAF1 injected mice, indicating that FAF1 activates PARP1 without triggering apoptosis (Figure 7d). Furthermore, the forced expression of FAF1 increased the extent of TH-positive neuronal loss caused by MPTP treatment (Figure 7e–h). Next, we examined whether FAF1 depletion affects PAR formation and dopaminergic neuronal loss induced by MPTP. The PAR formation and dopaminergic neuronal loss in the ventral midbrain of Faf1gt/gt mice were markedly reduced when compared with those of Faf1+/+ mice (Supplementary Figure S8). Collectively, our data indicate that FAF1 overexpression promotes the enzymatic activity of PARP1 and dopaminergic neurodegeneration during the pathological processes associated with PD.

FAF1 promotes dopaminergic neuronal cell death via PARP1 activation in an MPTP mouse model of PD. (a and b) The mice were administered four intraperitoneal injections of MPTP-HCl or saline at 2 h intervals. The mice were killed 24 h after the last injection. (a) Left panel: the subcellular localization of FAF1 was measured by immunofluorescence staining in TH-positive neurons of substantia nigra of mice that were treated with saline or MPTP. The nuclei were stained using DAPI and the tissues were analyzed by confocal microscopy. The white arrows indicate the presence of nuclear FAF1 in TH-positive neurons after MPTP treatment. Right panel: enlarged images of TH-positive neurons were taken from the white box in the merged images. (b) Brain lysates were prepared from the ventral midbrain of saline and MPTP-treated mice and were then subjected to immunoprecipitation with the anti-FAF1 antibody followed by immunoblotting with the indicated antibodies (n=3 per group). (c) The experimental scheme for panel d–f. The mice were injected unilaterally into the substantia nigra (SN) with adeno-associated virus type 1 (AAV1)-FAF1. Two weeks after AAV1-FAF1 injection, the mice were administered MPTP-HCl or saline, at 2 h intervals. The mice were killed 1 day and 7 days after the last MPTP injection and their brain tissues were prepared for western blot (WB) analysis or immunohistochemistry. (d) Left panel: at day 1 after the last MPTP injection, brain lysates were prepared from the ventral midbrain of the AAV1-FAF1-injected (inj.) or non-injected (non-inj.) side of saline- and MPTP-treated mice and were then subjected to immunoblot analysis using the indicated antibodies. Right panel: the graphs show the results of densitometric analysis of PAR and FAF1 immunoblots in left panel (n=3 per group). (e and g) At day 7 after the last MPTP injection, the dopaminergic neurodegeneration was measured by histological analysis for TH-positive (e) and Nissl-positive cells (g) in SN of the AAV1-FAF1 injected (inj.) or non-injected (non-inj.) side of saline- and MPTP-treated mice. Dashed lines represent a region of SN. (f) The graph shows the results of densitometric analysis of TH-stained neurons in e (n=5 per group). (h) The graph shows the counts of Nissl-stained cells in g (n=5 per group). Quantified data (d, f, h) are expressed as the mean±S.E.M. Statistical comparisons were evaluated by ANOVA test followed by Tukey HSD post hoc analysis. ***P<0.001, **P<0.01
Discussion
In this study, we investigated the molecular mechanisms underlying the cell death associated with FAF1 during oxidative stress. To date, FAF1 has been shown to be an apoptotic protein that activates caspase-8 and caspase-3 in the cytoplasm.19, 21, 22 We identified a novel role for nuclear FAF1: the induction of regulated necrosis through PARP1 activation, which consequently leads to dopaminergic neurodegeneration. Our data show that FAF1-mediated cell death includes the process of regulated necrosis in addition to apoptosis.
Regulated necrosis is important in many pathological conditions.11 Extensive studies have uncovered several forms of regulated necrosis that are distinct from classical necrosis, including RIPK1-dependent necrosis (necroptosis) and PARP1-dependent necrosis (parthanatos).17 Our findings show that PARP1, not RIPK1, is a major factor in necrotic cell death induced by H2O2 and that inhibition of PARP1 might provide a therapeutic benefit to patients with oxidative stress-related diseases, such as PD.
Previously, it has been suggested that PARP1 has a role as a downstream effector of RIPK1 and RIPK3 in TNF- or TRAIL-induced necroptosis.44, 45 However, genetic ablation or pharmacological inhibition of RIPK1 failed to block PARP1-mediated cell death induced by DNA alkylating agents.46 Our data also showed that the kinase activity of RIPK1 did not affect PARP1-dependent necrosis in response to H2O2. Collectively, RIPK1 and PARP1 can act either cooperatively within one core process or independently of each other in a cell stress- and cell type-dependent context.
FAF1 was initially identified as a cytoplasmic regulator of apoptosis.19 Translocation of FAF1 to the nucleus may compromise its role as a caspase activator. This finding led us to speculate that FAF1 might serve as a molecular switch in controlling the cell death machinery involved in either apoptosis or regulated necrosis depending on its subcellular localization. Such a dual role in promoting different types of cell death has been shown for p53, which can trigger both apoptosis and necrosis in response to multiple cellular insults.47 However, the subcellular localization of p53 does not appear to determine the type of cell death that it promotes.31, 48, 49 In contrast, Bcl-2/adenovirus E1B 19-kDa interacting protein 3 (BNIP3) appears to act in a manner similar to that of FAF1.50 BNIP3 triggers apoptosis in the mitochondria through mitochondrial pore transition and also initiates a caspase-independent process of cell death in the endoplasmic reticulum through the release of calcium.50
Phosphorylation often serves as an important regulatory tool for the subcellular localization of a protein.51 Casein kinase 2 (CK2) is known to phosphorylate FAF1 at Ser 289 and 291, and CK2-mediated phosphorylation of FAF1 regulates its nuclear translocation.38 Interestingly, CK2 has been linked to oxidative stress-related pathological conditions, such as Alzheimer’s disease and PD, and inhibitors of CK2 have been shown to be promising therapeutic agents for the treatment of these diseases.52 Considering that FAF1 is a substrate of CK2 and nuclear translocation of FAF1 mediates oxidative stress-induced cell death, it is tempting to speculate that the functional interplay between CK2 and FAF1 might have an important role in cell death related to oxidative stress. To address this possibility, further studies are necessary to determine the detailed molecular mechanism of the interaction between CK2 and FAF1 induced by oxidative stress.
This study adds FAF1 to the list of positive regulators of PARP1 activity. PARP1 activators are typically enzymes, including kinases and acetyltransferases.9 The post-translational modifications of PARP1 by these enzymes often promote its catalytic activity. However, some proteins, such as aminoacyl tRNA synthetase complex-interacting multifunctional protein 2 (AIMP2) and glyceraldehyde-3-phosphate dehydrogenase (GAPDH), activate PARP1 through physical interactions.39, 40 FAF1 also promotes the catalytic activity of PARP1 through physical interactions. FAF1 has several protein interaction domains that would facilitate the formation of a multiprotein complex.20 This fact led us to hypothesize that FAF1 might function as a scaffold protein for PARP1 and its other activators.
PARP1-dependent necrosis is thought to have an important role in the pathogenesis of PD.30, 39, 43 This study shows that FAF1 is essential for PARP1-dependent necrosis in response to oxidative stress, in which caspases are not activated. However, caspase-mediated apoptosis has also been found to occur in relation to the pathogenesis of PD, and the oxidative stress-induced damage in a mouse model of PD has been found to be reduced by treatment with caspase inhibitors, suggesting that caspase inhibition is also a plausible strategy for PD treatment.53 FAF1 has a key role in the induction of apoptosis as well.21, 22 Therapeutic interventions that target death proteins involved in cell death, such as FAF1, which participates in both apoptosis and regulated necrosis would thus appear to be an effective strategy for the treatment of PD.
Materials and Methods
Reagents and antibodies
H2O2, Nec-1, DPQ, PJ-34, cycloheximide and MPTP were purchased from Sigma-Aldrich (St. Louis, MO, USA). zVAD-fmk and the protease inhibitor cocktail were from Calbiochem (Darmstadt, Germany). Olaparib was from Selleckchem (Houston, TX, USA). UPF-1069 was from Tocris Bioscience (Minneapolis, MN, USA). TNFα was from AbFrontier (Seoul, South Korea). The mouse anti-FAF1 and rabbit anti-P-FAF1 antibodies have been described previously.24, 29 Rabbit anti-FAF1 antibody was from Proteintech (Chicago, IL, USA). Rabbit anti-caspase-3 antibody was from Cell Signaling Technology (Danvers, MA, USA). Rabbit anti-PAR antibody was from Trevigen (Gaithersburg, MD, USA). Mouse anti-PAR antibody was from Enzo Life Sciences (Farmingdale, NY, USA). Mouse anti-PARP1 antibody was from BD Biosciences (San Jose, CA, USA). Rabbit anti-PARP2 antibody was from Abcam (Cambridge, UK). Mouse anti-Flag, anti-β-actin and anti-TH antibodies were from Sigma-Aldrich. Rabbit anti-TH antibody was from Novus Biologicals (Littleton, CO, USA). Rabbit anti-AIF antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Mouse anti-COX IV, anti-V5, fluorescein-conjugated goat anti-mouse IgG (H+L), Alexa flour 488 or 594-conjugated goat anti-rabbit IgG (H+L) and horseradish peroxidase (HRP)-conjugated secondary antibodies were obtained from Thermo Fisher Scientific (Waltham, MA, USA).
Plasmids
Human full-length FAF1 and its truncated mutants were cloned into a pcDNA3.1-3x Flag vector using standard PCR techniques and custom-designed primers containing the appropriate restriction enzyme sites. The V5-tagged PARP1 construct was kindly provided by Dr. KS Kwon (Korea Research Institute of Bioscience and Biotechnology, Daejeon, South Korea). The PARP1 truncated constructs were generated by PCR and cloned into the pcDNA-V5 vector.
Cell culture and transfections
MEF and human neuroblastoma SH-SY5Y cells were cultured in Dulbecco’s modified Eagle’s medium (WelGENE, Daegu, South Korea) supplemented with 10% fetal bovine serum and a 1 × antibiotic-antimycotic solution (Thermo Fisher Scientific) at 37 °C in a humidified incubator containing 5% CO2. MEF and SH-SY5Y cells were transfected with various plasmids using the Amaxa Nucleofector system (Lonza, Basel, Switzerland) and Lipofectamine 2000 (Thermo Fisher Scientific) according to the manufacturer’s protocol.
Cell death and cell viability assay
The cells were seeded into 96-well plates (MEFs, 15 000 cells per well; SH-SY5Y cells, 50 000 cells per well) and incubated for 12 h. The cells then were pretreated with individual compounds at the indicated concentrations for 1 h, followed by exposure to H2O2. Cell death was assessed by the release of lactate dehydrogenase (LDH) into the extracellular medium, which was measured with the Cytotoxicity Detection Kit (Roche, Basel, Switzerland). The cell viability was determined with the CellTiter96 Aqueous One Solution Cell Proliferation Assay (Promega, Fitchburg, WI, USA).
Caspase-3 activity assay
MEFs were treated with H2O2 or TNFα plus CHX for the indicated times and at the indicated concentrations. Then, caspase-3 activity was measured using a caspase-3 colorimetric assay kit (Biovision, Milpitas, CA, USA) according to the manufacturer’s protocol. Absorbance at 450 nm was measured with the use of a VICTOR microplate reader (PerkinElmer, Norwalk, CT, USA).
Immunofluorescence
MEFs were seeded on poly-d-lysine (Sigma-Aldrich)-coated coverslips in a 12-well culture plate. After cell attachment, the cells were treated with H2O2 for the indicated times. The cells were fixed with 4% paraformaldehyde in PBS for 15 min and subsequently permeabilized with 0.1% Triton X-100 in PBS for 10 min. The cells were incubated with the indicated primary antibodies overnight at 4 °C and then washed three times with cold PBS for 10 min each. The cells were added to solutions with secondary antibodies conjugated with fluorescent dyes for 2 h at room temperature and the nuclei were stained with propidium iodide (Sigma-Aldrich) or 4′,6-diamidino-2-phenylindole (Thermo Fisher Scientific) for 10 min. The coverslips were mounted onto microscope slides using fluorescence-mounting medium (Dako, Carpinteria, CA, USA). For FAF1 immunostaining in mouse brains, the mice were anesthetized and then perfused with PBS followed by 4% paraformaldehyde. The brains were then post-fixed overnight in 4% paraformaldehyde at 4 °C. The brains were cryoprotected by equilibration with 30% sucrose in PBS (pH 7.4). Coronal sections (20 μm thick) were cut with a microtome (Leica, Wetzlar, Germany). The sections were blocked using 10% fetal bovine serum in PBS for 30 min, then incubated with the indicated primary antibodies overnight at 4 °C and then with the secondary antibodies for 2 h at room temperature. All the samples were analyzed using a Zeiss LSM 510 laser scanning confocal microscope (Carl Zeiss, Oberkochen, Germany).
Subcellular fractionation
Subcellular fractionation was performed as described by Lee et al.,39 with some modifications. In brief, MEF or SH-SY5Y cells were suspended in a hypotonic buffer (10 mM HEPES, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 0.1% NP-40 and a protease inhibitor cocktail) and homogenized by passage through a 26 gauge syringe needle 20 times. After incubation on ice for 20 min, the lysates were centrifuged at 720 × g for 5 min. The supernatant was used as the cytoplasmic fraction, and the pellet was washed with the same buffer twice and used as the nuclear fraction.
Co-immunoprecipitation and immunoblot analysis
The mouse brains or cells were lysed in RIPA buffer (Thermo Fisher Scientific) or mammalian lysis buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% Nonidet P-40, 0.4 mM phenylmethylsulfonyl fluoride) supplemented with protease cocktail inhibitor (Calbiochem). The protein levels were quantified using a Bio-Rad protein assay kit (Bio-Rad, Hercules, CA, USA). Co-immunoprecipitation was performed with the indicated antibodies and protein A/G Sepharose (Santa Cruz Biotechnology). The immunocomplexes were then washed with RIPA buffer or mammalian lysis buffer three times, and the samples were prepared by adding a sample-loading buffer. The samples then were electrophoresed on SDS-PAGE and transferred to nitrocellulose membrane. The membranes were blocked with 5% skim milk in PBS-T and incubated with primary antibodies. After incubation with HRP-conjugated secondary antibodies, the immunoblot signal was detected using a chemiluminescent detection kit (AbFrontier).
In vitro PARP1 activity assay
For the in vitro PARP1 activity assay, GST-FAF1 or GST was incubated with recombinant PARP1 (1 Unit, Trevigen), β-NAD+ (100 μM, Sigma-Aldrich) and damaged DNA (Trevigen) for 10 min at room temperature. After the in vitro poly(ADP-ribosyl)ation reactions, the samples were subjected to immunoblot analysis.
Measurement of intracellular NAD+ and ATP levels
MEFs were seeded into 96-well plates (15 000 cells per well) and incubated for 12 h. The cells then were pretreated with vehicle (dimethyl sulfoxide, DMSO) or 30 μM DPQ for 1 h, followed by exposure to H2O2. Intracellular NAD+ and ATP levels were measured using the NAD/NADH-Glo assay kit (Promega) and CellTiter-Glo viability assay kit (Promega) as described by the manufacturers.
Mitochondrial membrane potential assay
Mitochondrial membrane depolarization was measured using the Muse MitoPotential kit (Millipore, Darmstadt, Germany). MEFs were pretreated with vehicle (DMSO) or 30 μM DPQ for 1 h. Then, the cells were treated with H2O2 for 4 h, and then detached by trypsin-EDTA (Thermo Fisher Scientific). The cells were incubated with Muse MitoPotential dye, a cationic lipophilic dye, for 20 min in a 37 °C CO2 incubator. Then, mitochondrial membrane potential changes were determined with a Muse cell analyzer (Millipore).
RNA interference
siRNAs for FAF1, PARP1, PARP2 and scrambled RNA (scRNA) were purchased from Bioneer (Daejeon, South Korea). The target sequences for the siRNA against FAF1, PARP1 and PARP2 were siFAF1 (5'-CAGUAGAUGAGUUAAUGAU-3'), siPARP1 (5'-GGAGGGUCUGAUGAUAGCA-3') and siPARP2 (5'-CUGGAAAAUACGACAUGUU-3'). MEFs or SH-SY5Y cells were transfected with scRNA or siRNAs against FAF1, PARP1 and PARP2 using the Lipofectamine RNAi MAX (Thermo Fisher Scientific) according to the manufacturer's instruction.
MPTP-induced mouse model of PD
All the mice were maintained in the animal facility of Chungnam National University (Daejeon, South Korea), and all animal studies were conducted in accordance with the institutional guidelines for the care and use of laboratory animals. The mice were randomly assigned to either MPTP or saline-treated groups. To generate the MPTP-induced mouse model of PD, 10-week-old male C57BL/6J mice were administered four intraperitoneal injections of 21.06 mg/kg MPTP-HCl or saline at 2 h intervals. The mice were killed 7 days after the last injection, and the brains were processed for further analysis.
Stereological virus injection
Stereotaxic AAV injections were performed as described by Shin et al.,54 with some modifications. In brief, the adeno-associated virus type 1 expressing FAF1 (AAV1-FAF1) was purchased from Vector Biolabs (Malvern, PA, USA). For stereotaxic injection of AAV1-FAF1 into the substantia nigra of the mouse midbrain, 8-week-old male C57BL/6J mice were anesthetized with an intraperitoneal injection of a mixture of Zoletil 50 (Virbac, Fort Worth, TX, USA) and Rompun (Bayer Korea, Seoul, South Korea). A 33 gauge injection needle was used to stereotaxically inject AAV1-FAF1 into the right substantia nigra (anteroposterior, −3.0 mm; mediolateral, 1.2 mm; dorsoventral, 4.3 mm from bregma). The infusion into the substantia nigra was performed at a rate of 0.1 μl/min, and 1.25 μl of AAV1-FAF1 (4.3 × 1013 GC/ml) was injected. After the injection, the needle was left in the substantia nigra for an additional 5 min and then slowly withdrawn. The skin over the injection site was closed by suturing. Surgical wounds were frequently monitored to ensure proper healing.
Immunohistochemistry
The mice were anesthetized and perfused with PBS followed by 4% paraformaldehyde. The brains were then post-fixed overnight in the 4% paraformaldehyde at 4 °C. The brains were cryoprotected by equilibration with 30% sucrose in PBS (pH 7.4). The coronal sections (20 μm thick) were cut with a microtome (Leica). The sections were treated with 3% H2O2 in methanol for 30 min and blocked using 10% fetal bovine serum for 30 min at room temperature. For TH immunostaining, the sections were incubated with a 1 : 1000 dilution of rabbit anti-TH (Novus) for 2 h and visualized with the addition of biotinylated goat anti-rabbit IgG followed by streptavidin-conjugated HRP (Vectastain ABC kit, Vector Laboratories, Burlingame, CA, USA). TH-positive immunostaining was visualized using 3,3′-diaminobenzidine (DAB kit, Sigma-Aldrich). For Nissl staining, the sections were stained with 0.1% cresyl violet solution (Sigma-Aldrich) for 20 min and were dehydrated in 100% ethanol. The sections were cleared in xylene and mounted with Canada balsam (Sigma-Aldrich). Experimenters were blinded for information of mice during histological analyses of TH-positive and Nissl-positive cells. The histological analyses of TH-positive and Nissl-positive cells were measured using Image J software (United States National Institutes of Health).
Statistical analysis
All the data are expressed as the mean±S.E.M. Statistical comparisons were made using Student’s t-test or one-way analysis of variance (ANOVA) followed by Tukey post hoc analysis using SPSS software (Statistic version 22; IBM Inc., Chicago, IL, USA). A P-value lower than 0.05 was considered to indicate significant difference among groups.
Abbreviations
- AAV:
-
adeno-associated virus
- AIF:
-
apoptosis-inducing factor
- CHX:
-
cycloheximide
- DAPI:
-
4′,6-diamidino-2-phenylindole
- FAF1:
-
Fas-associated factor 1
- IP:
-
immunoprecipitation
- LDH:
-
lactate dehydrogenase
- MEFs:
-
mouse embryonic fibroblast cells
- MNNG:
-
1-methyl-3-nitro-1-nitrosoguanidine
- MPTP:
-
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- NAD+:
-
nicotinamide adenine dinucleotides
- Nec-1:
-
necrostatin-1
- PD:
-
Parkinson’s disease
- PAR:
-
poly(ADP-ribose)
- PARP1:
-
poly(ADP-ribose) polymerase 1
- PARP2:
-
poly(ADP-ribose) polymerase 2
- PI:
-
propidium iodide
- TH:
-
tyrosine hydroxylase
- TNFα:
-
tumor necrosis factor α
- VC:
-
vector control
- WT:
-
wild type
References
Ryter SW, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X et al. Mechanisms of cell death in oxidative stress. Antioxid Redox Signal 2007; 9: 49–89.
Allen CL, Bayraktutan U . Oxidative stress and its role in the pathogenesis of ischaemic stroke. Int J Stroke 2009; 4: 461–470.
Perry G, Cash AD, Smith MA . Alzheimer disease and oxidative stress. J Biomed Biotechnol 2002; 2: 120–123.
Hwang O . Role of oxidative stress in Parkinson's disease. Exp Neurobiol 2013; 22: 11–17.
Zhang S, Lin Y, Kim YS, Hande MP, Liu ZG, Shen HM . c-Jun N-terminal kinase mediates hydrogen peroxide-induced cell death via sustained poly(ADP-ribose) polymerase-1 activation. Cell Death Differ 2007; 14: 1001–1010.
Mashimo M, Kato J, Moss J . ADP-ribosyl-acceptor hydrolase 3 regulates poly (ADP-ribose) degradation and cell death during oxidative stress. Proc Natl Acad Sci USA 2013; 110: 18964–18969.
Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science 2002; 297: 259–263.
Gibson BA, Kraus WL . New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 2012; 13: 411–424.
Luo X, Kraus WL . On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev 2012; 26: 417–432.
Swindall AF, Stanley JA, Yang ES . PARP-1: friend or foe of DNA damage and repair in tumorigenesis? Cancers 2013; 5: 943–958.
Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P . Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 2014; 15: 135–147.
Andrabi SA, Umanah GK, Chang C, Stevens DA, Karuppagounder SS, Gagne JP et al. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci USA 2014; 111: 10209–10214.
Fouquerel E, Goellner EM, Yu Z, Gagne JP, Barbi de Moura M, Feinstein T et al. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep 2014; 8: 1819–1831.
Ying W, Garnier P, Swanson RA . NAD+ repletion prevents PARP-1-induced glycolytic blockade and cell death in cultured mouse astrocytes. Biochem Biophys Res Commun 2003; 308: 809–813.
Baek SH, Bae ON, Kim EK, Yu SW . Induction of mitochondrial dysfunction by poly(ADP-ribose) polymer: implication for neuronal cell death. Mol Cells 2013; 36: 258–266.
Fatokun AA, Dawson VL, Dawson TM . Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br J Pharmacol 2014; 171: 2000–2016.
Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death Differ 2012; 19: 107–120.
Andrabi SA, Dawson TM, Dawson VL . Mitochondrial and nuclear cross talk in cell death: parthanatos. Ann NY Acad Sci 2008; 1147: 233–241.
Chu K, Niu X, Williams LT . A Fas-associated protein factor, FAF1, potentiates Fas-mediated apoptosis. Proc Natl Acad Sci USA 1995; 92: 11894–11898.
Menges CW, Altomare DA, Testa JR . FAS-associated factor 1 (FAF1): diverse functions and implications for oncogenesis. Cell Cycle 2009; 8: 2528–2534.
Ryu SW, Lee SJ, Park MY, Jun JI, Jung YK, Kim E . Fas-associated factor 1, FAF1, is a member of Fas death-inducing signaling complex. J Biol Chem 2003; 278: 24003–24010.
Park MY, Ryu SW, Kim KD, Lim JS, Lee ZW, Kim E . Fas-associated factor-1 mediates chemotherapeutic-induced apoptosis via death effector filament formation. Int J Cancer 2005; 115: 412–418.
Park MY, Moon JH, Lee KS, Choi HI, Chung J, Hong HJ et al. FAF1 suppresses IkappaB kinase (IKK) activation by disrupting the IKK complex assembly. sJ Biol Chem 2007; 282: 27572–27577.
Jang MS, Sul JW, Choi BJ, Lee SJ, Suh JH, Kim NS et al. Negative feedback regulation of Aurora-A via phosphorylation of Fas-associated factor-1. J Biol Chem 2008; 283: 32344–32351.
Song EJ, Yim SH, Kim E, Kim NS, Lee KJ . Human Fas-associated factor 1, interacting with ubiquitinated proteins and valosin-containing protein, is involved in the ubiquitin-proteasome pathway. Mol Cell Biol 2005; 25: 2511–2524.
Bjorling-Poulsen M, Seitz G, Guerra B, Issinger OG . The pro-apoptotic FAS-associated factor 1 is specifically reduced in human gastric carcinomas. Int J Oncol 2003; 23: 1015–1023.
Hidalgo A, Baudis M, Petersen I, Arreola H, Pina P, Vazquez-Ortiz G et al. Microarray comparative genomic hybridization detection of chromosomal imbalances in uterine cervix carcinoma. BMC Cancer 2005; 5: 77.
Betarbet R, Anderson LR, Gearing M, Hodges TR, Fritz JJ, Lah JJ et al. Fas-associated factor 1 and Parkinson's disease. Neurobiol Dis 2008; 31: 309–315.
Sul JW, Park MY, Shin J, Kim YR, Yoo SE, Kong YY et al. Accumulation of the parkin substrate, FAF1, plays a key role in the dopaminergic neurodegeneration. Hum Mol Genet 2013; 22: 1558–1573.
Venderova K, Park DS . Programmed cell death in Parkinson's disease. Cold Spring Harbor Perspect Med 2012; 2: a009365.
Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM . p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012; 149: 1536–1548.
Yamamoto H, Ozaki T, Nakanishi M, Kikuchi H, Yoshida K, Horie H et al. Oxidative stress induces p53-dependent apoptosis in hepatoblastoma cell through its nuclear translocation. Genes Cells 2007; 12: 461–471.
Shen HM, Liu ZG . JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med 2006; 40: 928–939.
Gough DR, Cotter TG . Hydrogen peroxide: a Jekyll and Hyde signalling molecule. Cell Death Dis 2011; 2: e213.
Shen HM, Lin Y, Choksi S, Tran J, Jin T, Chang L et al. Essential roles of receptor-interacting protein and TRAF2 in oxidative stress-induced cell death. Mol Cell Biol 2004; 24: 5914–5922.
Schroder E, Eaton P . Hydrogen peroxide as an endogenous mediator and exogenous tool in cardiovascular research: issues and considerations. Curr Opin Pharmacol 2008; 8: 153–159.
Wyrsch P, Blenn C, Bader J, Althaus FR . Cell death and autophagy under oxidative stress: roles of poly(ADP-Ribose) polymerases and Ca(2+). Mol Cell Biol 2012; 32: 3541–3553.
Olsen BB, Jessen V, Hojrup P, Issinger OG, Boldyreff B . Protein kinase CK2 phosphorylates the Fas-associated factor FAF1 in vivo and influences its transport into the nucleus. FEBS Lett 2003; 546: 218–222.
Lee Y, Karuppagounder SS, Shin JH, Lee YI, Ko HS, Swing D et al. Parthanatos mediates AIMP2-activated age-dependent dopaminergic neuronal loss. Nat Neurosci 2013; 16: 1392–1400.
Nakajima H, Kubo T, Ihara H, Hikida T, Danjo T, Nakatsuji M et al. Nuclear-translocated glyceraldehyde-3-phosphate dehydrogenase promotes poly(ADP-ribose) polymerase-1 activation during oxidative/nitrosative stress in stroke. J Biol Chem 2015; 290: 14493–14503.
Lee Y, Kang HC, Lee BD, Lee YI, Kim YP, Shin JH . Poly (ADP-ribose) in the pathogenesis of Parkinson's disease. BMB Rep 2014; 47: 424–432.
Krishna A, Biryukov M, Trefois C, Antony PM, Hussong R, Lin J et al. Systems genomics evaluation of the SH-SY5Y neuroblastoma cell line as a model for Parkinson's disease. BMC Genomics 2014; 15: 1154.
Mandir AS, Przedborski S, Jackson-Lewis V, Wang ZQ, Simbulan-Rosenthal CM, Smulson ME et al. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc Natl Acad Sci USA 1999; 96: 5774–5779.
Jouan-Lanhouet S, Arshad MI, Piquet-Pellorce C, Martin-Chouly C, Le Moigne-Muller G, Van Herreweghe F et al. TRAIL induces necroptosis involving RIPK1/RIPK3-dependent PARP-1 activation. Cell Death Differ 2012; 19: 2003–2014.
Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G . Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 2010; 11: 700–714.
Sosna J, Voigt S, Mathieu S, Lange A, Thon L, Davarnia P et al. TNF-induced necroptosis and PARP-1-mediated necrosis represent distinct routes to programmed necrotic cell death. Cell Mol Life Sci 2014; 71: 331–348.
Nikoletopoulou V, Markaki M, Palikaras K, Tavernarakis N . Crosstalk between apoptosis, necrosis and autophagy. Biochim Biophys Acta 2013; 1833: 3448–3459.
Amaral JD, Xavier JM, Steer CJ, Rodrigues CM . The role of p53 in apoptosis. Discov Med 2010; 9: 145–152.
Montero J, Dutta C, van Bodegom D, Weinstock D, Letai A . p53 regulates a non-apoptotic death induced by ROS. Cell Death Differ 2013; 20: 1465–1474.
Zhang L, Li L, Liu H, Borowitz JL, Isom GE . BNIP3 mediates cell death by different pathways following localization to endoplasmic reticulum and mitochondrion. FASEB J 2009; 23: 3405–3414.
Nardozzi JD, Lott K, Cingolani G . Phosphorylation meets nuclear import: a review. Cell Commun Signal 2010; 8: 32.
Perez DI, Gil C, Martinez A . Protein kinases CK1 and CK2 as new targets for neurodegenerative diseases. Med Res Rev 2011; 31: 924–954.
Yang L, Sugama S, Mischak RP, Kiaei M, Bizat N, Brouillet E et al. A novel systemically active caspase inhibitor attenuates the toxicities of MPTP, malonate, and 3NP in vivo. Neurobiol Dis 2004; 17: 250–259.
Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O et al. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson's disease. Cell 2011; 144: 689–702.
Acknowledgements
This research was supported by a grant of the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI16C0947).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by RA Knight
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article

Cite this article
Yu, C., Kim, Bs. & Kim, E. FAF1 mediates regulated necrosis through PARP1 activation upon oxidative stress leading to dopaminergic neurodegeneration. Cell Death Differ 23, 1873–1885 (2016). https://doi.org/10.1038/cdd.2016.99
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2016.99
This article is cited by
-
Sumoylation of SAP130 regulates its interaction with FAF1 as well as its protein stability and transcriptional repressor function
BMC Molecular and Cell Biology (2024)
-
Substance abuse and neurodegenerative diseases: focus on ferroptosis
Archives of Toxicology (2023)
-
Non-coding RNAs in necroptosis, pyroptosis and ferroptosis in cancer metastasis
Cell Death Discovery (2021)
-
PARP overactivation in neurological disorders
Molecular Biology Reports (2021)
-
A novel function of FAF1, which induces dopaminergic neuronal death through cell-to-cell transmission
Cell Communication and Signaling (2020)
