
Abstract
DNA damage induced by reactive oxygen species and several chemotherapeutic agents promotes both p53 and poly (ADP-ribose) polymerase (PARP) activation. p53 activation is well known to regulate apoptotic cell death, whereas robust activation of PARP-1 has been shown to promote a necrotic cell death associated with energetic collapse. Here we identify a novel role for p53 in modulating PARP enzymatic activity to regulate necrotic cell death. In mouse embryonic fibroblasts, human colorectal and human breast cancer cell lines, loss of p53 function promotes resistance to necrotic, PARP-mediated cell death. We therefore demonstrate that p53 can regulate both necrotic and apoptotic cell death, mutations or deletions in this tumor-suppressor protein may be selected by cancer cells to provide not only their resistance to apoptosis but also to necrosis, and explain resistance to chemotherapy and radiation even when it kills via non-apoptotic mechanisms.
Similar content being viewed by others
Main
Mutations in the p53 protein (encoded by TP53 in humans) are detected in approximately half of human cancers.1, 2, 3 This tumor-suppressor protein regulates apoptosis, cell cycle and metabolism. Numerous signaling pathways converge in p53 after cellular insults, including DNA damage, oncogene activation or hypoxia. For instance, after DNA injury, p53 is activated promoting cell cycle arrest, transactivation of repairing enzymes and, if the damage cannot be repaired, apoptosis.4 Under these conditions, p53 is posttranscriptionally modified, assisting in the expression of several proteins including p21, GADD45/PCNA or pro-apoptotic genes like BAX, PUMA, NOXA, BID and APAF-1.5 Moreover, a directly pro-apoptotic cytosolic function has been described, directly binding to anti-apoptotic proteins (BCL-2 and BCL-XL) and directly activating BAX and BAK to induce mitochondrial outer membrane permeabilization (MOMP) and apoptosis in a transcriptional-independent manner.6 Because of its broad effects in the cell, loss of p53 activity confers growth advantage and ensures survival in cancer cells by inhibiting the apoptotic response necessary for tumor suppression.7, 8, 9
Following DNA damage, poly(ADP-ribose)polymerase-1 (PARP-1), the founding member of the PARP superfamily of proteins, is activated by DNA nicks and rapidly uses nicotinamide adenine dinucleotide (NAD) to polymerize adenosine diphosphate (ADP)-ribose and covalently modify target proteins to orchestrate DNA repair. These include chromatin structure regulators and DNA modulators like histones, HMG proteins, topoisomerases I and II, DNA helicases, single-strand break repair and base-excision repair enzymes, and different transcription factors.10, 11 Briefly, PARP activation promotes chromatin relaxation and the recruitment of base-excision repair proteins, including XRCC1, DNA ligase III and DNA polimerase β, that will ensure the DNA repair. For instance, several studies using either PARP inhibitors or PARP knockout (KO) mice demonstrate the important role of this protein in maintaining DNA integrity,12 and several benzamide-derived PARP inhibitors are being tested to treat cancer patients.13, 14 One of the targets of PARP-1 is p53,15, 16, 17, 18 promoting its poly(ADP-ribosyl)ation and accumulation in the nucleus, to control its transcriptional activity. However, hyperactivation of PARP-1 after severe DNA damage can cause NAD and ATP depletion leading to a unique form of necrotic cell death named parthanatos.19, 20 In this process, apoptosis-inducing factor translocates from the mitochondrial intermembrane space to the nucleus, triggering chromatin condensation and DNA fragmentation.21, 22 This regulation is still not completely understood, but the activation of the poly(ADP-ribose) glycohydrolase (PARG) enzyme, with both exo- and endoglycosidase activities to release ADP-ribose monomers and polymers (poly(ADP-ribose) seems to have an important role as secondary messenger in parthanatos cell death.19, 23 Inhibition of PARP-1 activity using PARP inhibitors or in KO models confers resistance to necrosis induced by DNA-damaging agents.24
Reactive oxygen species (ROS) induce DNA damage causing cleavage of DNA strands, DNA–protein cross-linking and oxidation of purines, leading to DNA-damage response and the activation of PARP-1.25 Depending on the impact, can either repair the damage or activate cell death pathways.26 In PARP-mediated cell death, NAD is rapidly depleted and, because this cofactor is necessary for different metabolic processes like glycolysis or the Krebs cycle, leads to ATP loss and a necrotic cell death. Upon p53 activation, the apoptotic process can be induced, leading to MOMP, caspase activation, apoptosome formation and programmed cell death. We show that apoptosis blockade is insufficient to maintain cell survival following ROS stress, but loss of p53 does maintain survival. To our knowledge, this is the first observation that p53 regulates PARP-mediated cell death.
Results
p53 loss protects against cell death induced by DNA damage
Hydrogen peroxide (H2O2) is a ROS that has been extensively used to induce DNA damage because it can easily diffuse in all cell compartments. To assess how ROS induces cell death, we used different genotypes of mouse embryonic fibroblast (MEF) cells: WT MEF, Bax Bak double KO (DKO) MEF and p53 KO MEF. DKO MEF cannot undergo MOMP and caspase-mediated intrinsic apoptosis, thus were employed to interrogate non-apoptotic cell death.27, 28
In order to determine the extent of DNA damage induced by H2O2 treatment (1 mM), we analyzed by confocal microscopy staining for histone H2AX phosphorylation, which occurs in the chromatin microenvironment surrounding a DNA double-strand break. As shown, short-term exposure to H2O2 rapidly induced DNA damage and appearance of phosphorylated H2AX foci in all three cell lines with similar extent (Figure 1a). However, using the same concentration of H2O2 for 24 h, we observed that WT and DKO cells were completely killed by this treatment, as assessed by Annexin V/ propidium iodide (PI) fluorescence-activated cell sorting (FACS) analysis (Figure 1b and c), indicating a MOMP-independent cell death induction. However, around 50% of the p53 KO cells survived (Figure 1b and c), pointing to a non-apoptotic, p53-regulated cell death. Similar results were observed in WT and p53 KO primary fibroblasts (Supplementary Figure 1). When cells were treated with lower doses of H2O2 (0.5 and 0.75 mM) for 24 h, we did not observe significant cell death in DKO nor p53 KO, suggesting that a different type of cell death, like apoptosis, is occurring in WT under these conditions (Supplementary Figure 2A).

p53 KO MEF cells, but not WT or Bax Bak DKO, survive to DNA damage induced by H2O2. (a) To assess DNA damage extent, cells were exposed to 1 mM H2O2 for 30 min and immunostained using anti-phospho Histone H2AX antibody (red). Nuclei were stained using DAPI (blue) and observed under a confocal microscope. Similar DNA damage extent was observed after treatment in all three cell lines. (b) WT MEF, Bax Bak DKO and p53 KO were treated for 24 h with 1 mM H2O2. Cells were stained with fluorescent conjugates of annexin-V and propidium iodide (PI) and analyzed by FACS. Viable cells are annexin-V negative and PI negative, and cell death is expressed as 100%-viable cells. Values indicate mean values±S.E.M. All experiments were performed independently at least three times (N≥3). **P<0.01 (compared with WT MEF H2O2-treated). (c) Cell death is expressed as 100% - viable cells. Representative FACS plots are shown
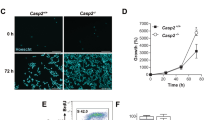
Under the microscope, WT and DKO cells showed morphological changes becoming round shaped and detached from the plate, whereas the p53 KO cells conserved their morphology, although their proliferation decreased (Figure 2a). Moreover, p53 KO cells, but not the other MEFs, were able to form colonies after H2O2 exposure meaning that viable cells survived to this treatment (Figure 2b).

After DNA damage, p53 KO cells survive conserving their clonogenic capacities. (a) Images under an optical microscope were obtained before and after 1 mM H2O2 treatment for 24 h. WT MEF and DKO showed necrotic characteristics, whereas p53 KO conserved their morphology. (b) Colony formation assay. Cells were treated for 4 h with 1 mM H2O2 counted and 200 cell/well were plated. After 18 days, cells were fixed and stained with a crystal violet solution. Only p53 KO cells were able to form colonies after treatment. All experiments were performed three times (N=3)
H2O2-induced DNA damage kills through PARP-mediated necrotic cell death
We wanted to determine whether PARP-1 was implicated in H2O2-induced cell death. When there is an extensive DNA damage, PARP-1 starts polymerizing ADP-ribose, consuming NAD and leading to a non-apoptotic cell death form known as parthanatos. This type of cell death shows NAD depletion that also leads to an ATP drop and the energetic collapse of the cell. PARP-1 KO mice are protected against transient cerebral ischemia reperfusion, and PARP-1 KO MEFs are protected against H2O2-induced cell death and DNA alkylating agents.21, 22, 29 When WT, DKO and p53 KO cells were treated with H2O2 (1 mM) for 8 h, we observed that the first two showed extensive cell death, whereas p53 KO cells were protected against this treatment (Figure 3a). A time course was performed following cell death response in the first 6 h after H2O2 treatment, showing a gradual increase in WT and DKO cells and a significant protection in the p53 KO cells (Supplementary Figure 2B). We observed a rapid induction of necrosis as assessed by PI incorporation30 (Supplementary Figure 2C) in all three cell lines, correlating with the observed cell death.

H2O2 induces PARP-mediated cell death, with NAD loss and ATP collapse in WT MEF and DKO, but not in p53 KO cells. (a) WT MEF, DKO and p53 KO cells were treated for 8 h with 1 mM H2O2, with or without PARP inhibitor 2 μM 4-amino-1,8-naphthalimide (4-ANI) 16 h preincubation. Cells were stained with fluorescent conjugates of annexin-V and propidium iodide (PI) and analyzed by FACS. (b) NAD was measured using a fluorimetric assay. All three cell lines were treated for 1 or 4 h with 1 mM H2O2. Results are normalized to basal NAD levels. (c) ATP was measured after the same treatment at different time points (0, 5, 15, 30, 60, 120, 240 and 480 min), with or without 2 μM 4-ANI preincubation, using CellTiter-Glo Luminescent Cell Viability Assay. All values are expressed as percentage to basal ATP levels. Values indicate mean values±S.E.M. *P<0.05, **P<0.01 (compared with H2O2 treated). All experiments were performed independently at least three times (N≥3)
In order to better understand this cell death mechanism, we used two inhibitors: the pan-caspase inhibitor qVD-OPH, which would rescue from an apoptotic cell death, and necrostatin-1, which would inhibit necroptosis. Neither of them were sufficient to rescue these cells from H2O2-induced cell death (Supplementary Figure 3). Interestingly, when all three cell lines were preincubated with the PARP inhibitor 4-amino-1,8-naphthalimide (4-ANI), they were protected from H2O2-induced death (Figure 3a) at 8 h, indicating that this process is PARP dependent.
Prior studies have demonstrated that NAD depletion can accompany non-apoptotic, PARP-dependent cell death.20 As expected, all three cell lines demonstrated a rapid decline in NAD concentration in the first hour after treatment, but p53 KO cells were able to recover, ensuring their survival (Figure 3b). When we analyzed ATP levels, we observed a rapid decay in WT and DKO cells, which could be prevented by the 4-ANI preincubation. In contrast, the p53 KO cells showed just a partial drop of ATP, and were able to recover and restore their initial levels, with or without PARP inhibitor preincubation (Figure 3c). These observations correlate with a PARP-dependent cell death that promotes the energy collapse of the cell.19, 20 Intriguingly, we find that loss of p53 protects against the NAD and ATP depletion associated with this cell death.
When PARP-dependent necrosis is inhibited, apoptosis is observed
Because both necrosis and apoptosis coexist in the cell after DNA damage, we wanted to assess whether there was any implication of the latter in H2O2-induced cell death. As commented previously, at 8 h WT MEF, DKO and, in less extent, p53 KO cells treated with H2O2 in the presence of the PARP inhibitor showed protection (Figure 3a). At shorter time points, we observed protection against necrosis in the presence of 4-ANI as assessed by PI incorporation30 (Supplementary Figure 2C) in all three cell lines, correlating with the observed cell death protection (Supplementary Figure 2B). But by 24 h cell death could be observed in WT MEF under the same conditions, suggesting that the apoptotic process was engaged (Figure 4a). Treatment with H2O2 as a single agent showed no caspase activity in WT, but when preincubated with 4-ANI, caspase activation was observed in the first 8 h, reaching a plateau at 24 h (Figure 4b). Thus, when WT are treated with 4-ANI, there is apoptotic activation, a process that is ATP dependent.31 In DKO cells, no increase in caspase activity was detected in any of these treatments as they cannot undergo MOMP to permit the apoptosome formation. In p53 KO cells, H2O2 induced caspase activation, although less in extent compared with WT, and PARP inhibition slightly increased this observed activity (Figure 4b). Thus, we pretreated with 4-ANI and the pancaspase inhibitor qVD-OPH before a 24-h H2O2 treatment, and observed that this combination was protecting WT against ROS-induced cell death (Figure 4a). For instance, p53 KO cells were already protected against H2O2 and the preincubation with either 4-ANI alone or especially in combination with qVD-OPH slightly improved the observed survival (Figure 4a). Similar results were observed in WT and p53 KO primary fibroblasts (Supplementary Figure 1). In order to confirm these results, we performed a western blot to detect caspase-3 cleavage that would indicate the ongoing apoptotic process. We observed that WT treated with H2O2 and 4-ANI showed caspase-3 cleavage and in a lower degree in p53 KO cells (Figure 4c), correlating with the previous results. As expected, caspase-3 cleavage was not detected in DKO cells. As p53 KO cells are partially protected against the rapid H2O2 activation of PARP-mediated necrosis, we believe that some of the cells that overcome NAD and ATP depletion undergo apoptosis because of the cellular damage. Therefore, by inhibiting PARP-1 in p53 KO cells, we increase the percentage of necrosis-surviving cells, increasing the number of apoptotic cells. However, because p53 is a key regulator of apoptosis after DNA damage,5 these cells also revealed a better survival to this process, as assessed by Bcl-2 homology domain 3 (BH3) profiling. Briefly, BH3 profiling, can measure a cell’s proximity to the threshold of the mitochondrial pathway of apoptosis, measuring mitochondrial dysfunction in response to a panel of peptides derived from the BH3 domains of pro-apoptotic BH3-only proteins, defining how ‘primed’ a cell is for death.32, 33, 34 When we analyzed these cell lines, we observed that WT were more primed for apoptosis compared to p53 KO; DKO cannot undergo MOMP, consequently no response to Bim peptide was seen (Figure 4d).

PARP inhibition induces time-delayed apoptosis in H2O2-treated cells. (a) MEFs were treated for 24 h with 1 mM H2O2, with or without a 16 h preincubation with PARP inhibitor 2 μM 4-ANI and/or the pan-caspase inhibitor 10 μM qVD-OPH. Cells were stained with fluorescent conjugates of annexin-V and propidium iodide (PI) and analyzed by FACS. **P<0.01 (compared to WT H2O2+4-ANI treated). (b) Caspase activation was measured after 1 mM H2O2 treatment, with or without a 2 μM 4-ANI, 16 h preincubation, at different time points (0, 15, 30, 60, 120, 240, 480 and 1440 min) using Caspase-Glo 3/7 Assay. All values are expressed as percentage to basal caspase 3/7 activity. (c) Caspase-3 activation by cleavage was detected by western blot in all three cell lines treated for 8 h with H2O2 with or without 4-ANI preincubation. Actin was used as loading control. (d) Apoptotic sensitivity measured by BH3 profiling using low concentrations of Bim peptide. WT MEF are more primed, showed more sensitivity, compared to p53 KO for apoptosis. DKO showed no priming, are resistant to apoptotic cell death. *P<0.05 (compared with WT MEF). Values indicate mean values±S.E.M. All experiments were performed independently at least three times (N≥3)
To validate these results observed with caspase activation, we checked if we could detect a subG1 population, resulting from apoptotic DNA fragmentation, performing a cell cycle analysis. Correlating with the observed caspase activation, we detected subG1 cells in WT treated with 4-ANI in combination with H2O2, but not with H2O2 alone. DKO cells did not show any apoptotic feature with any of the treatments, indicating that apoptotic trigger in this context needs MOMP, cytochrome c release, the apoptosome formation and caspase activation (Supplementary Figure 4A). However, p53 KO cell analysis indicated that H2O2 alone induced a small subG1 population, and when pretreated with 4-ANI the percentage of apoptotic cells increased, but in any case with the same extent as observed in WT, correlating with survival and caspase activation (Figures 1b and 3a). Treatment with the PARP inhibitor efficiently protected DKO cells against H2O2-induced cell death, but was not sufficient to rescue WT as they were undergoing apoptosis. When exposed to different chemotherapy agents, p53 KO showed protection against paclitaxel and etoposide (Supplementary Figures 4B and C). Correlating with the BH3 profiling data, DKO were resistant to all these treatments, as they cannot undergo instrinsic apoptosis (Supplementary Figures 4B and C). Thus, p53 loss protects against apoptosis, as had been previously reported by several groups.35, 36
These results demonstrate that both PARP-mediated cell death and apoptosis are two different processes that occur after DNA damage, in different timeframes. When necrosis is inhibited, the apoptotic response is triggered, but as p53 also regulates the apoptotic cell death after DNA damage, transactivating several proapoptotic genes and downregulating other antiapoptotic BCL-2 family members,5, 35, 36 p53 loss inhibits apoptotic cell death.
p53 loss protects against PARP-mediated cell death by decreasing PARP activity
In order to better understand how p53 protects not only against apoptosis but also, especially, to PARP-mediated necrosis, we used a p53 knockdown (KD) approach. We tested several short hairpin RNA (shRNA) constructs37 to efficiently KD this protein (Table 1). p53 KD was successfully achieved using several constructs; and the shRNA4 construct was the most effective in knocking down p53, so it was selected to continue this study (Supplementary Figure 5A).
In order to answer how p53 loss protects against DNA damage-induced cell death, we performed several experiments to analyze the PARP-1 protein. Its expression was first analyzed by immunoblotting and we observed that the shRNA against p53 effectively decreased its levels without decreasing PARP-1 expression (Figure 5a). Then we analyzed PARP activity and we saw that p53 KD and KO showed decreased enzymatic activity (Figure 5b). Furthermore, when we checked protein poly(ADP-ribosyl)ation (PARylation), we observed that similarly to PARP activity, p53 KD cells showed less PARylation compared with WT MEF cells, and that could be prevented by preincubating with the PARP inhibitor 4-ANI (Figure 5c). Consequently, p53 KD cells were protected against H2O2 (1 mM) induced cell death (Supplementary Figure 5B). In a similar manner, p53 KO had decreased PARylation when exposed to ROS treatment (Figure 5c) correlating with the observed resistance, but when PARP-1 was overexpressed in p53 KO cells (Supplementary Figure 6C), H2O2 sensitivity was restored (Figure 5d).

p53 loss impairs PARP activity and protects against PARP-mediated cell death. (a) Western blotting analysis of PARP-1 and p53 was performed in WT MEF, MEF shRNA p53 knockdown (KD) and p53 KO cells. Actin was used as loading control. (b) PARP activity was analyzed in all three cell lines using a colorimetric PARP ELISA assay kit with Histones-coated strip wells. Results are normalized to basal WT MEF PARP activity. **P<0.01. (c) Cells were treated for 1 h with 1 mM H2O2, with or without 4-ANI preincubation, and lysates (in the presence of PARG inhibitor ADP-HPD) were obtained and analyzed by western blot, immunostaining against protein-bound poly(ADP-ribose). Actin was used as loading control. p53 KD and KO showed a decreased PARP activity and protein PARylation, protecting against PARP-mediated cell death. (d) WT MEF and p53 KO, transfected with an empty or a PARP-1 overexpressing vector, were treated for 24 h with 1 mM H2O2. PARP-1 overexpression restored p53 sensitivity to H2O2. Cells were stained with fluorescent conjugates of annexin-V and propidium iodide (PI) and analyzed by FACS. Values indicate mean values±S.E.M. Densitometry analysis represent average±S.E.M. from at least three different western blots; KD and KO results were normalized to WT MEF. *P<0.05, **P<0.01. All experiments were performed independently at least three times (N≥3)
In summary, p53 loss partially protects against DNA damaging agents and non-apoptotic cell death by impairing PARP activity. Thus, p53 loss protects against both PARP-mediated necrotic cell death and apoptosis, ensuring cell survival in MEFs.
p53 loss protects against PARP-mediated cell death in other cellular models
We next wanted to determine whether loss of p53 had similar effects in other cell types. For this purpose, we used a human carcinoma cell line, HCT116, and a human breast cancer, MCF7, both expressing wild-type p53. When we compared HCT116 WT and HCT116 p53 KO, we observed that loss of p53 (Supplementary Figure 6A) impairs PARP activity and protein PARylation (Figure 6a), protecting against H2O2-induced cell death (Figure 6b), as observed previously with MEFs. With MCF7 we used another approach, inactivating p53 function using a dominant negative (DD) form of p53. Briefly, we infected the cells with a retrovirus containing a plasmid that encodes for a 89 residues short form of wt mouse p53 that dimerizes with the WT p53 present in MCF7, impairing its function.38 To check p53 DD transduction efficiency, we treated parental MCF7 and MCF7 DD with the DNA intercalating drug daunorubicin, which activates p53-inducing cell cycle arrest, through p21. After 24 h of daunorubicin treatment, MCF7 overexpressed p53 and p21, whereas in MCF7 DD, although p53 was expressed at similar levels as seen in WT MCF7, p21 expression was reduced indicating that p53 function is inhibited (Supplementary Figure 6B). MCF7 DD also showed a reduction in PARylation (Figure 6c), in a similar manner as seen in MEF p53 KD. Furthermore, MCF7 were sensitive to H2O2 (1 mM), but could be prevented by preincubating with the PARP inhibitor, 4-ANI; and the functional inactivation of p53 in MCF7 DD showed a similar protection as the one observed in MEF p53 KD (Figure 6d).

p53 loss protects against PARP-mediated cell death in human colon carcinoma cells. (a) HCT 116 WT and p53 KO cells, and (b) WT MCF7 or p53 dominant-negative (DD) cells were treated for 1 h with 1 mM H2O2, with or without 4-ANI preincubation, and lysates (in the presence of PARG inhibitor ADP-HPD) were obtained and analyzed by western blot to detect protein-bound poly(ADP-ribose). Actin was used as loading control. (b and d) Cell death was analyzed at 24 h under the same treatments. Cells were stained with fluorescent conjugates of annexin-V and propidium iodide (PI) and analyzed by FACS. HCT116 p53 KO and MCF7 DD cells showed less PARylation compared with their respective WT, and were protected against H2O2-induced cell death. (a) and (c) belong to the same blot and immunodetection. Error bars indicate mean values±S.E.M. *P<0.05, **P<0.01 (compared with control cells for each cellular type). Densitometry analysis represent average±S.E.M. from at least three different western blots; results were normalized to untreated cells (ctrl) for each cellular type. All experiments were performed independently at least three times (N≥3)
Our observations suggest that also in human carcinoma and breast cancer cells, loss of p53, or its functionality, impairs PARP activity protecting against H2O2-induced cell death.
Discussion
An important finding of this paper is that p53 can also control non-apoptotic cell death. p53 has, of course, long been understood to participate in control of apoptosis by the mitochondrial pathway.8 Several studies support p53 to perform this function by transcriptional upregulation of pro-apoptotic BCL-2 family proteins like PUMA, NOXA and BAX.5, 39 In addition, some have posited that p53 can directly interact with BCL-2 family proteins at the mitochondria to induce apoptosis in a manner that is independent of transcription.6 Both of these mechanisms of programmed cell death are dependent on BAX and BAK, like all deaths that occur via the mitochondrial apoptotic pathway. In contrast, we find that loss of BAX and BAK does not prevent death by ROS, whereas loss of p53 does, implying that p53 regulates a death that is independent of BAX and BAK, and hence, independent of apoptosis. This does not rule out, of course, the participation of p53 in apoptotic cell death induced by other agents.
A key feature of the death induced by ROS is the extremely rapid loss of ATP and NAD. This energy loss is prevented by inhibition of PARP-1, demonstrating that the energy loss and death are PARP dependent.20, 21 The combination of the p53 dependence and PARP dependence of this necrotic cell death led us to ask whether PARP activity depended on p53. Indeed, we found that PARP activity at baseline and after ROS stimulation was reduced in the absence of p53. Although it was previously understood that p53 could control an apoptotic death, and PARP a necrotic death following ROS and DNA damage, this is the demonstration that there is cross-regulation of the two pathways, with p53 controlling the PARP-dependent necrotic death (Figure 7). In our studies, when there is a DNA damage like the one induced by ROS (including H2O2), PARP-1 is rapidly activated promoting NAD and ATP depletion leading to a necrotic cell death. PARP activity is indirectly activated by p53. If this PARP-mediated necrosis is inhibited, and the DNA damage cannot be repaired, p53 induces caspase-mediated apoptosis. One consequence is that in the absence or loss of function of p53, there is a double protection against PARP-mediated necrosis as well as apoptosis.

PARP-1 and p53 regulate each other activity. One of the many targets of PARP-1 is p53, covalently modifying the latter through poly(ADP-ribosyl)ation. On the other hand, p53 also regulates PARP-1 by modulating its activity. PARP-1 controls necrosis and p53 apoptosis after DNA damage, thus both types of cell death are well regulated
The mechanism by which p53 regulates PARP activity it is not well understood, but we detected an increase in the expression of SIRT1 in p53 KD and p53 KO (data not shown), that has been implicated in the modulation of PARP-1 activity by acetylation,40 but further experiments are required to demonstrate this regulation. It has also been described that p53 modulates necrosis through cathepsin Q regulation.36 However, this work used DNA damaging agents instead of ROS to induce cell death. Furthermore, commitment to death apparently occurred over days, as opposed to the 15 min at which we observed catastrophic energetic collapse in these experiments. Another recent study, explores the role of p53 promoting Permeability Transition Pore (PTP) by its interaction with cyclophilin D.29 Most of the experiments described in that work use lower concentrations of H2O2 that have a distinct effect on the cell death mechanism (as shown in Supplementary Figure 2), as previously described for Bax Bak DKO.36 As shown in Supplementary Figure 7, Cyp D KO cells showed a modest protection compared with p53 KO cells when treated with H2O2 1 mM. Moreover, when preincubated with cyclosporin A, we did not observe a significant protection in WT MEF and DKO (data not shown). Therefore, it seems likely that we are studying a mechanism of cell death quite distinct from the ones described in those reports.
To our knowledge, p53 regulation of PARP activity and PARP-dependent necrotic cell death has not been described before. In our opinion, this provides an additional reason why p53 mutations are selected by cancer cells, as they not only would protect tumor cells against apoptosis but also to PARP-mediated cell death, contributing to DNA-damaging therapies resistance, as observed in chemotherapy or radiation therapy35, 39, 41
There is considerable interest in combining DNA-damaging agents with PARP inhibitors for treatment of cancer. Our results suggest that some caution should be taken into consideration when using this combination in contexts where DNA damage induces death mainly by necrosis. In this situation, inhibition of PARP may actually inhibit cell death induced by the DNA-damaging agent. Cells that are particularly resistant to apoptosis may fall into this category. We would suggest that clinical results where PARP inhibitors under-perform expectations may in part be explained by this mechanism.
Materials and Methods
Cell lines and treatments
The media used was supplemented with 10% heat-inactivated fetal bovine serum (Gibco, Carlsbad, CA, USA), 10 mM L-glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin. DMEM was used for SV40 immortalized MEFs (MEF, Bax Bak DKO and p53 KO, or Cyp D KO); RPMI 1640 was used for the culture of the following breast cancer cell lines (MCF-7, MCF7 DD), and McCoy’s media for human colorectal cancer cells (HCT116 WT, HCT116 p53 KO). Primary fibroblasts were cultured in IMDM supplemented with 20% heat-inactivated fetal bovine serum (Gibco), 10 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin and 10 mM of non-essential amino acids. The cells were cultured at 37 °C in a humidified atmosphere of 5% CO2.
The shRNA to perform Trp53 KD in WT MEF were obtained from William Hahn laboratory. Five different constructs were used and the most effective knocking down Trp53 was selected: 5′-CCGGCCACTACAAGTACATGTGTAACTCGAGTTACACATGTACTTGTAGTGGTTTTT-3′. The lentiviral production and infection were performed as previously described.37 Selection of positive p53 KD cells was assessed with puromycin (Sigma, St. Louis, MO, USA).
To transduce the MCF7 cell line with the 89 residues miniprotein p53 DD encoding plasmid we used a retroviral vector as previously described,38 kindly offered by Dr. DeCaprio laboratory. Selection was performed using G418 (Cellgro, Manassas, VA, USA, 0.2–0.8 mg/ml).
Full-length PARP-1 overexpression in WT and p53 KO MEF cells was performed as previously described.42 Selection of positive PARP-overexpressing cells was assessed with puromycin (Sigma).
Ψm BH3 profile using whole cells (WC-JC-1)
A total of 2 × 104 cells/well of adherent MEF cells were used; and 15 μl of 0.3 or 1 μM peptides in T-EB (300 mM Trehalose, 10 mM HEPES-KOH pH 7.7, 80 mM KCl, 1 mM ethyleneglycoltetraacetic acid, 1 mM EDTA, 0.1% bovine serum albumin, 5 mM succinate) were deposited per well in a black 384-well plate (BD Biosciences, San Jose, CA, USA, #3573). Single-cell suspensions were washed in T-EB before being resuspended at 4 × their final density. One volume of the 4x cell suspension was added to one volume of a 4x dye solution containing 4 μM JC-1, 40 μg/ml oligomycin, 0.02% digitonin, 20 mM 2-mercaptoethanol in T-EB. This 2x cell/dye solution stood at RT for 5–10 min to allow permeabilization and dye equilibration. A total of 15 μl of the 2x cell/dye mix was then added to each treatment well of the plate, shaken for 15 s inside the reader, and the fluorescence at 590 nm monitored every 5 min at RT. Percentage loss of Ψm for the peptides is calculated by normalization to the solvent-only control dimethyl sulfoxide (0%) and the positive control trifluorocarbonylcyanide phenylhydrazone (100%).32
Cell viability assays
MEFs, breast cancer and human carcinoma cell lines, when they reached 50–70% confluency, were treated with H2O2 (1 mM or the indicated concentration) for 8 and 24 h, or otherwise indicated. When specified, a pretreatment for 16 h with the PARP inhibitor 2 μM 4-ANI and/or the 10 μM pan-caspase inhibitor qVD-OPH (Mp Biomedicals, Santa Ana, CA, USA, # OPH109) or 10 μM Necrostatin-1 (Enzo Life Sciences, Ann Arbor, MI, USA, BML-AP309) was perfomed. Cells were also treated with other chemotherapy agents: etoposide (100 μM), staurosporine (1 μM) and paclitaxel (1 μM). Cells were stained with fluorescent conjugates of annexin-V and/or PI (1 μg/ml final concentration) and analyzed on a FACSCanto machine (BD, Franklin Lakes, NJ, USA). Annexin V was prepared are previously described.43 Viable cells are annexin-V negative and PI negative, and cell death is expressed as 100% - viable cells.
Immunoblotting
Total cell lysates were prepared in 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) buffer (5 mM MgCl2, 137 mM NaCl, 1 mM EDTA, 1 mM ethyleneglycoltetraacetic acid, 1% CHAPS, 20 mM Tris-HCl (pH 7.5) and protease inhibitors (Complete, Roche, MAnnkin, Germany, EU)). Cells were washed twice, resuspended with 50–100 μl of CHAPS lysis buffer, and kept on ice for 30 min. Then, the cellular suspension was centrifuged at 16 100 × g for 5 min, and the supernatant used to perform the immunoblotting analysis. For poly(ADP-ribose) detection, PARG inhibitor 0.5 μM ADP-HPD (EMD Biosciences, Billerica, MA, USA, #118415) was used in the CHAPS lysis buffer to avoid poly(ADP-ribose) degradation.
Twenty microgram of protein was loaded on NuPAGE 10% Bis-Tris polyacrylamide gels (Invitrogen, Grand Island, NY, USA). The following antibodies were used to detect proteins on the membrane (dilution 1 : 1000): actin (Chemicon, Billerica, MA, USA, MAB1501); cleaved caspase 3 (Cell Signaling #9661, Danvers, MA, USA, ); PARP-1 (Cell Signaling, #9542); p53 (for MEFs, Calbiochem, PAb421); p53 (for HCT116/MCF7, Santa Cruz Biotechnology, Dallas, TX, USA, sc-126); anti-poly(ADP-ribose) (Trevigen, Gaithersburg, MD, USA, 4336-BPC-100); and p21 (Santa Cruz Biotechnology, sc-397).
Immunocytochemistry and laser confocal imaging
Cells were fixed for 10 min with 3.7% paraformaldehyde in 0.1 mol/l phosphate buffer before permeabilization with 0.1% saponin in 0.5% bovine serum albumin/phosphate-buffered saline (PBS) buffer for 5 min. Cells were incubated for 1 h with mouse monoclonal antibody anti-phospho Histone H2AX (ser 139; 1 : 200; Millipore, Billerica, MA, USA, 05-636), rinsed with PBS, and incubated for 45 min with the secondary antibody. Images were obtained by confocal microscopy. Confocal images were collected using a Leica SP5X Laser Scanning Confocal Microscope (Leica, Buffalo Grove, IL, USA) equipped with a 63 × Plan Apo oil immersion objective and a confocal pinhole set at 1 Airy unit. All the confocal images shown were single optical sections.
ATP and caspase activity
In a 96-well plate, 15 000 cells/well were plated and pretreated when indicated for 16 h with the PARP inhibitor 4-ANI 2 μM. Measurement of ATP and caspase 3/7 activity were performed using CellTiter-Glo Luminescent Cell Viability Assay (Promega, Madison, WI, USA, #G7573) and Caspase-Glo 3/7 Assay (Promega, #G8093), as described by the manufacturer.
NAD measurement
NAD was determined using a fluorimetric assay (Fluoro NAD/NADH, Cell technology, Mountain View, CA, USA, FLNADH 100-2). MEFs were treated for 1 or 4 h with 1 mM H2O2, and 2 × 106 cells were used for each NAD measurement as described by the manufacturer.
PARP activity
PARP activity was determined using a colorimetric PARP assay kit with Histones-coated strip wells (Trevigen, # 4677-096-K). WT MEF, MEF Bax Bak DKO and MEF p53 KO cells were lysated as described by the manufacturer and the protein measured. A measure of 40–100 μg of protein were used for each determination.
Colony-forming assay
WT MEF, MEF Bax Bak DKO or MEF p53 KO cells were treated for 4 h with 1 mM H2O2. Then washed with fresh media and plated in 60-mm tissue culture dishes at 200 cells per plate. After 18 days, the plates were stained with crystal violet (0.5% crystal violet in a 3 : 1 (v/v) mixture of distilled water/methanol).
Cell cycle assay
Cell cycle analysis was performed using PI staining. Briefly, 105–106 cells were first washed with PBS and fixed with 100% ETOH, added slowly into the cells in a drop wise manner while vortexing. Cells were then kept at 4 °C for 30 min and washed with PBS. 5 μg/ml of RNAseA was added to each sample and incubated 37 °C for 30 min. Finally, 50 μg/ml of PI was added to each sample and analyzed on a FACSCanto machine (BD).
Densitometry
Densitometric analysis for western blots was assessed using ImageJ (Java-based image processing program, NIH).
Statistical analysis
Statistical significance of the results was analyzed using two-tailed Student’s t-test using GraphPad Prism 5.0 software (San Diego, CA, USA). *P<0.05 and **P<0.01 were considered significant. S.E.M. stands for standard error of the mean.
Abbreviations
- 4-ANI:
-
4-amino-1,8-naphthalimide
- ADP:
-
adenosine diphosphate
- BH3:
-
Bcl-2 homology domain 3
- CHAPS:
-
3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate
- DD:
-
dominant negative
- DKO:
-
double knockout
- FACS:
-
fluorescence-activated cell sorting
- H2O2:
-
hydrogen peroxide
- KD:
-
knockdown
- KO:
-
knockout
- MEF:
-
mouse embryonic fibroblast
- MOMP:
-
mitochondrial outer membrane permeabilization
- NAD:
-
nicotinamide adenine dinucleotide
- PARG:
-
poly (ADP-ribose) glycohydrolase
- PARP:
-
poly (ADP-ribose) polymerase
- PBS:
-
phosphate-buffered saline
- ROS:
-
reactive oxygen species
- ShRNA:
-
short hairpin RNA
References
Ikediobi ON, Davies H, Bignell G, Edkins S, Stevens C, O'Meara S et al. Mutation analysis of 24 known cancer genes in the NCI-60 cell line set. Mol Cancer Ther 2006; 5: 2606–2612.
Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012; 483: 603–607.
Greenblatt MS, Bennett WP, Hollstein M, Harris CC . Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer research 1994; 54: 4855–4878.
Riley T, Sontag E, Chen P, Levine A . Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9: 402–412.
Chipuk JE, Green DR . Dissecting p53-dependent apoptosis. Cell death and differentiation 2006; 13: 994–1002.
Chipuk JE, Kuwana T, Bouchier-Hayes L, Droin NM, Newmeyer DD et al. Direct activation of Bax by p53 mediates mitochondrial membrane permeabilization and apoptosis. Science (New York, N.Y 2004; 303: 1010–1014.
Schmitt CA, Fridman JS, Yang M, Baranov E, Hoffman RM, Lowe SW . Dissecting p53 tumor suppressor functions in vivo. Cancer Cell 2002; 1: 289–298.
Villunger A, Michalak EM, Coultas L, Müllauer F, Böck G, Ausserlechner MJ et al. p53- and drug-induced apoptotic responses mediated by BH3-only proteins puma and noxa. Science (New York) NY 2003; 302: 1036–1038.
Macip S, Kosoy A, Lee SW, O'Connell MJ, Aaronson SA . Oxidative stress induces a prolonged but reversible arrest in p53-null cancer cells, involving a Chk1-dependent G2 checkpoint. Oncogene 2006; 25: 6037–6047.
Schreiber V, Dantzer F, Ame JC, de Murcia G . Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol 2006; 7: 517–528.
Chang P, Jacobson MK, Mitchison TJ . Poly(ADP-ribose) is required for spindle assembly and structure. Nature 2004; 432: 645–649.
Yelamos J, Farres J, Llacuna L, Ampurdanes C, Martin-Caballero J . PARP-1 and PARP-2: New players in tumour development. Am J Cancer Res 2011; 1: 328–346.
Leung M, Rosen D, Fields S, Cesano A, Budman DR . Poly(ADP-ribose) polymerase-1 inhibition: preclinical and clinical development of synthetic lethality. Mol Med (Cambridge) Mass 2011; 17: 854–862.
Munoz-Gamez JA, Martín-Oliva D, Aguilar-Quesada R, Cañuelo A, Nuñez MI, Valenzuela MT et al. PARP inhibition sensitizes p53-deficient breast cancer cells to doxorubicin-induced apoptosis. Biochem J 2005; 386: 119–125.
Malanga M, Althaus FR . The role of poly(ADP-ribose) in the DNA damage signaling network. Biochem Cell Biol 2005; 83: 354–364.
Kumari SR, Mendoza-Alvarez H, Alvarez-Gonzalez R . Functional interactions of p53 with poly(ADP-ribose) polymerase (PARP) during apoptosis following DNA damage: covalent poly(ADP-ribosyl)ation of p53 by exogenous PARP and noncovalent binding of p53 to the M(r) 85,000 proteolytic fragment. Cancer Res 1998; 58: 5075–5078.
Valenzuela MT, Guerrero R, Núñez MI, Ruiz De Almodóvar JM, Sarker M, de Murcia G et al. PARP-1 modifies the effectiveness of p53-mediated DNA damage response. Oncogene 2002; 21: 1108–1116.
Alvarez-Gonzalez R . Genomic maintenance: the p53 poly(ADP-ribosyl)ation connection. Sci STKE 2007; 2007: pe68.
David KK, Andrabi SA, Dawson TM, Dawson VL . Parthanatos, a messenger of death. Front Biosci 2009; 14: 1116–1128.
Zong WX, Ditsworth D, Bauer DE, Wang ZQ, Thompson CB . Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev 2004; 18: 1272–1282.
Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science (New York) NY 2002; 297: 259–263.
Moubarak RS, Yuste VJ, Artus C, Bouharrour A, Greer PA, Menissier-de Murcia J et al. Sequential activation of poly(ADP-ribose) polymerase 1, calpains, and Bax is essential in apoptosis-inducing factor-mediated programmed necrosis. Mol Cell Biol 2007; 27: 4844–4862.
Blenn C, Wyrsch P, Bader J, Bollhalder M, Althaus FR . Poly(ADP-ribose)glycohydrolase is an upstream regulator of Ca2+ fluxes in oxidative cell death. Cell Mol Life Sci 2011; 68: 1455–1466.
Ha HC, Snyder SH . Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc Natl Acad Sci USA 1999; 96: 13978–13982.
Zong WX, Thompson CB . Necrotic death as a cell fate. Genes Dev 2006; 20: 1–15.
Tong WM, Hande MP, Lansdorp PM, Wang ZQ . DNA strand break-sensing molecule poly(ADP-Ribose) polymerase cooperates with p53 in telomere function, chromosome stability, and tumor suppression. Mol Cell Biol 2001; 21: 4046–4054.
Zong WX, Lindsten T, Ross AJ, MacGregor GR, Thompson CB . BH3-only proteins that bind pro-survival Bcl-2 family members fail to induce apoptosis in the absence of Bax and Bak. Genes Dev 2001; 15: 1481–1486.
Korsmeyer SJ, Wei MC, Saito M, Weiler S, Oh KJ, Schlesinger PH . Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ 2000; 7: 1166–1173.
Vaseva AV, Marchenko ND, Ji K, Tsirka SE, Holzmann S, Moll UM . p53 opens the mitochondrial permeability transition pore to trigger necrosis. Cell 2012; 149: 1536–1548.
Vanden Berghe T, Grootjans S, Goossens V, Dondelinger Y, Krysko DV, Takahashi N et al. Determination of apoptotic and necrotic cell death in vitro and in vivo. Methods 2013; S1046-2023(13)00030-3.
Eguchi Y, Shimizu S, Tsujimoto Y . Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res 1997; 57: 1835–1840.
Ryan JA, Brunelle JK, Letai A . Heightened mitochondrial priming is the basis for apoptotic hypersensitivity of CD4+ CD8+ thymocytes. Proc Natl Acad Sci USA 2010; 107: 12895–12900.
Chonghaile TN, Letai A . Who put the ‘A’ in Atg12: autophagy or apoptosis? Mol Cell 2011; 44: 844–845.
Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science (New York) NY 2011; 334: 1129–1133.
Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE et al. p53 status and the efficacy of cancer therapy in vivo. Science (New York) NY 1994; 266: 807–810.
Tu HC, Ren D, Wang GX, Chen DY, Westergard TD, Kim H et al. The p53-cathepsin axis cooperates with ROS to activate programmed necrotic death upon DNA damage. Proc Natl Acad Sci USA 2009; 106: 1093–1098.
Mani M, Carrasco DE, Zhang Y, Takada K, Gatt ME, Dutta-Simmons J et al. BCL9 promotes tumor progression by conferring enhanced proliferative, metastatic, and angiogenic properties to cancer cells. Cancer Res 2009; 69: 7577–7586.
Gottlieb E, Haffner R, von Ruden T, Wagner EF, Oren M . Down-regulation of wild-type p53 activity interferes with apoptosis of IL-3-dependent hematopoietic cells following IL-3 withdrawal. EMBO J 1994; 13: 1368–1374.
Lowe SW, Ruley HE, Jacks T, Housman DE . p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell 1993; 74: 957–967.
Rajamohan SB, Pillai VB, Gupta M, Sundaresan NR, Birukov KG, Samant S et al. SIRT1 promotes cell survival under stress by deacetylation-dependent deactivation of poly(ADP-ribose) polymerase 1. Mol Cell Biol 2009; 29: 4116–4129.
Symonds H, Krall L, Remington L, Saenz-Robles M, Lowe S, Jacks T et al. p53-dependent apoptosis suppresses tumor growth and progression in vivo. Cell 1994; 78: 703–711.
Dutta C, Day T, Kopp N, van Bodegom D, Davids MS, Ryan J et al. BCL2 suppresses PARP1 function and nonapoptotic cell death. Cancer Res 2012; 72: 4193–4203.
Brumatti G, Sheridan C, Martin SJ . Expression and purification of recombinant annexin V for the detection of membrane alterations on apoptotic cells. Methods 2008; 44: 235–240.
Acknowledgements
We gratefully acknowledge funding from the Beatriu de Pinós program from la Generalitat de Catalunya in Spain (JM) and NIH grant P01CA139980. AL is a Leukemia and Lymphoma Society Scholar. We thank Dr. James DeCarpio’s lab, Dr. Ruben Carrasco’s lab, Nadja Kopp and Jeremy Ryan for technical assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by M Oren
Supplementary Information accompanies this paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Montero, J., Dutta, C., van Bodegom, D. et al. p53 regulates a non-apoptotic death induced by ROS. Cell Death Differ 20, 1465–1474 (2013). https://doi.org/10.1038/cdd.2013.52
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2013.52
Keywords
This article is cited by
-
Comparison of the effects of oxidative and inflammatory stresses on rat chondrocyte senescence
Scientific Reports (2023)
-
Inhibition of AKT induces p53/SIRT6/PARP1-dependent parthanatos to suppress tumor growth
Cell Communication and Signaling (2022)
-
Combined inhibition of BADSer99 phosphorylation and PARP ablates models of recurrent ovarian carcinoma
Communications Medicine (2022)
-
Derepression of the USP22-FASN axis by p53 loss under oxidative stress drives lipogenesis and tumorigenesis
Cell Death Discovery (2022)
-
NAMPT-dependent NAD+ salvage is crucial for the decision between apoptotic and necrotic cell death under oxidative stress
Cell Death Discovery (2022)
