
Abstract
Death receptors, such as Fas/CD95 and TRAIL receptors, engage the extrinsic pathway for caspase activation, but also couple to the intrinsic mitochondrial route. In so-called Type II cells, death receptors require the mitochondrial pathway for apoptotic execution, whereas in Type I cells they reportedly do not. For established tumor cell lines, the Type I/Type II distinction is based on short-term apoptosis assays. We report here that the mitochondrial pathway is essential for apoptotic execution of Type I tumor cells by death receptors, when long-term clonogenicity is taken into account. A blockade of the mitochondrial pathway in Type I tumor cells – by RNA interference for Bid or Bcl-2 overexpression – reduced effector caspase activity and mediated significant clonogenic resistance to TRAIL. Downstream from the mitochondria, Caspase-9 did not contribute to clonogenic death of TRAIL-treated Type I cells. Rather, the release of Smac/DIABLO and the inhibition of XIAP activity proved to be crucial for full effector caspase activity and clonogenic execution. Thus, in Type I cells the intrinsic pathway downstream from death receptors is not redundant, but limits clonogenicity by virtue of Smac/DIABLO release and XIAP inhibition. This finding is relevant for cancer therapy using death receptor agonists.
Similar content being viewed by others

Main
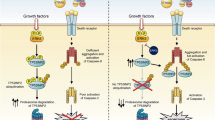
Death receptors, such as Fas/CD95 and the TRAIL receptors, activate the extrinsic apoptotic pathway. This involves assembly of the death-inducing signaling complex (DISC) at their cytoplasmic tail, which results in Caspase-8 and/or -10 activation.1 These inducer caspases subsequently cleave and activate effector caspases and the BH3 domain-only Bcl-2 family member Bid.2 The effector caspases can in principle directly dismantle the cell to generate apoptotic bodies. Bid, however, connects death receptors to the intrinsic pathway for effector caspase activation. Upon Bid cleavage, its carboxy-terminal fragment translocates to the mitochondria, where it triggers mitochondrial outer membrane permeability (MOMP) through the Bcl-2 family members Bak and Bax.2, 3 This releases apoptogenic factors, including Cytochrome c, Smac/DIABLO and HtrA2/Omi into the cytosol. There, Cytochrome c binds to Apaf-1, creating the apoptosome, which is a platform for recruitment and activation of Caspase-9. This inducer caspase in turn cleaves and activates effector caspases.3
Given these scenarios, the mitochondrial pathway is generally viewed as a signal amplifier for effector caspase activation after death receptor stimulation. Moreover, different cell types proved more or less reliant on the mitochondrial pathway for effector caspase activation. In so-called Type I cells, inhibition of MOMP by Bcl-2 overexpression did not block death receptor-induced apoptosis, whereas it did so in Type II cells.4 This distinction was valid for long-term established tumor cell lines in vitro, but also for primary cells in vivo. Specifically, hepatocytes are classified as Type II cells,5 whereas thymocytes and peripheral T cells are classified as Type I cells.6, 7, 8, 9
The efficiency of Caspase-8/10 activation in response to death receptor stimulation is greater in Type I cells than in Type II cells.4 Therefore, it is generally assumed that Type I cells do not require the mitochondrial amplification loop for effector caspase activation, whereas Type II cells do.10 Among solid tumor cell lines, Type I and Type II cells have distinctions, which are connected to the mode of death receptor signaling: Type I cells express mesenchymal-like genes, whereas Type II cells express epithelium-like markers.11 In Type I cells, but not in Type II cells, Fas/CD95 receptors aggregate on ligand binding and efficiently form a DISC with the aid of filamentous actin.10
These earlier studies focused on the strength of the Caspase-8/10 signal in Type I versus Type II cells and essentially viewed the mitochondria as signal amplifier for effector caspase cleavage, by engaging Caspase-9. Recent studies, however, have highlighted another control point in apoptotic execution by death receptors, which is that imposed by XIAP.8, 12 The XIAP protein is an inhibitor of apoptosis protein (IAP) that controls the catalytic activities of Caspase-9, Caspase-3 and Caspase-7 after their initial activation. It uses a baculovirus inhibitor repeat (BIR) domain to bind to an IAP-binding motif (IBM) in the small subunit of these caspases, which is exposed on their proteolytic cleavage. In addition, XIAP shields the substrate-binding site of the effector caspases with a peptide strand preceding the BIR domain.13 Upon their release from the mitochondria, Smac/DIABLO and HtrA2/Omi bind to XIAP through their own IBM, and thereby free the caspases from inhibition.14, 15, 16, 17 The caspases can subsequently process themselves into fully active, non-XIAP inhibitable fragments and propagate the apoptotic signal.18 By XIAP neutralization, mitochondria make a unique contribution to effector caspase activation, which is not offered by the extrinsic pathway.
In Type II cells, the mitochondrial pathway makes two potential contributions to effector caspase activation downstream from death receptors: Caspase-9 activation for initial effector caspase cleavage and XIAP elimination for full effector caspase activity.19 It was recently shown that loss of XIAP rendered hepatocytes independent of Bid for apoptosis induction, that is, allowed for an apparent switch from Type II to Type I signaling.8 This suggests that in Type II cells, death receptors need the mitochondrial pathway for XIAP neutralization rather than for Caspase-9 activity.
In Type I cells, Caspase-8 activity also leads to Bid cleavage, MOMP and the consequent release of Smac/DIABLO and HtrA2/Omi, but this is reportedly not essential for the apoptotic response. We surmised that in Type I cells these factors have a role in death receptor-induced apoptosis that had previously been overlooked. Therefore, we examined the impact of the mitochondrial pathway on clonogenic death of Type I tumor cells. We observed that blockade of the mitochondrial pathway by Bid RNA interference (RNAi) or Bcl-2 overexpression mediated significant clonogenic resistance to death receptor ligand TRAIL. Upon close examination, the mitochondrial pathway did seem to contribute to effector caspase activation. Smac/DIABLO function, but not Caspase-9 activity, was decisive for full effector caspase activation and clonogenic execution of Type I cells. Downregulation of XIAP by RNAi significantly reduced clonogenicity of TRAIL-treated Type I cells, indicating that the inhibition of XIAP activity is an important function of Smac/DIABLO. We conclude that the mitochondrial pathway limits clonogenicity of Type I cells after death receptor stimulation by enabling full effector caspase activation, not with the aid of the apoptosome, but by virtue of IAP antagonism.
Results
Identification of Bid as a regulator of clonogenic death in Type I tumor cells
We performed an RNAi screen in MCF-7 breast carcinoma cells to identify new regulators of TRAIL-induced apoptosis. The MCF-7 cells were chosen because they proved an excellent model system for studying cell biological aspects of death receptor signaling.20 These cells lack Caspase-3, but behave as Type I cells when Caspase-3 has been reconstituted.4 Upon stable reconstitution of Caspase-3, only 1 in 5000 MCF-7Casp-3 cells proved spontaneously resistant to isoleucine zippered (IZ) TRAIL21 in a clonogenic assay (data not shown). The screening approach was validated by the selection of a Caspase-8-targeting shRNA from the retroviral library of about 24 000 shRNAs directed at 8000 genes.22 In addition, a Bid-targeting shRNA emerged from the screen. As the mitochondrial pathway is thought to be redundant for death receptor-induced apoptosis in Type I cells, this was unexpected. Therefore, we decided to look further into the contribution of the mitochondrial pathway to TRAIL-induced apoptosis in MCF-7Casp-3 cells.
A stable Bid RNAi MCF-7Casp-3 cell line was made with the Bid-targeting shRNA (Bid shRNA1) identified in the RNAi screen. In addition, a stable Caspase-8 RNAi version of this cell line was made. The same batch of MCF-7Casp-3 cells was transduced in parallel with empty vector (EV), shRNA for Caspase-8, or shRNA for Bid to exclude background effects. High efficiency of retroviral transduction furthermore excluded clonal selection of transduced cells. Both Bid and Caspase-8 expression were efficiently silenced by their targeting shRNAs (Figure 1a,b). To examine the Type I nature of MCF-7Casp-3 cells, we monitored TRAIL receptor-induced Caspase-3 cleavage in control and Bid RNAi versions of this cell line. Cells were stimulated with 25, 50 or 100 ng/ml IZ-TRAIL for 3 h and Caspase-3 cleavage was monitored by flow cytometry using an antibody that recognizes all cleaved forms of Caspase-3, but not the proform. This analysis validated the effect of the shRNA for Caspase-8, as cleavage of its substrate Caspase-3 was significantly impaired at all IZ-TRAIL doses (Figure 1c). In case of Bid RNAi, Caspase-3 cleavage was not significantly altered (Figure 1c). Furthermore, (apoptotic) cell death, as read out after 16 h by membrane permeability and phosphatidyl serine (PS) exposure, was not evidently affected by Bid RNAi (Figure 1d). These data confirm that MCF-7 cells reconstituted with Caspase-3 are of a Type I nature. However, the recovery of Bid-targeted shRNA from the screen suggested that in these cells, the mitochondrial route contributes to cellular execution by TRAIL.

MCF-7Casp-3 cells are of a Type I nature. MCF-7Casp-3 cells were retrovirally transduced with an empty RNAi vector (EV) or vectors encoding shRNAs targeting Bid or Caspase-8. (a,b) Bid protein expression (a) and Caspase-8 protein expression (b) in cells transduced (Td) with shRNA are reduced as compared with that in EV control cells, as demonstrated by immunoblotting on total cell lysates. Actin served as loading control. (c) The same cells as characterized in (a, b) were treated (TX) with 0, 25, 50 or 100 ng/ml of IZ-TRAIL for 3 h. The percentage of cells with cleaved Caspase-3 was determined by flow cytometry after intracellular staining with an antibody that specifically recognizes cleaved Caspase-3. Data presented are expressed as mean+S.D. values of three independent experiments. Statistically significant differences between values of EV and Casp-8 shRNA samples are indicated for *P<0.05 and **P<0.01. (d) MCF-7Casp-3 cells stably expressing EV or Bid shRNA1 were treated with 0, 25, 50 or 100 ng/ml of IZ-TRAIL for 16 h. The percentage (%) of dead cells was determined by flow cytometry after staining with FITC Annexin-V and PI
The mitochondrial pathway limits clonogenicity of Type I tumor cells after TRAIL treatment
As the Bid RNAi phenotype was revealed at the level of clonogenicity, subsequent assays were performed using this read out. For this purpose, MCF-7Casp-3 cells were treated with 50 ng/ml IZ-TRAIL and examined for clonogenic outgrowth at day 15. The RNAi for Caspase-8 gave rise to clonogenic resistance to TRAIL as compared with EV-transduced cells, serving as a positive control for the assay (Figure 2a,b). In repeated assays, Bid RNAi mediated significant clonogenic resistance to TRAIL (Figure 2a,b, also see 4c,d).

Type I cells require the mitochondrial apoptosis pathway for TRAIL-induced clonogenic cell death. (a,b) MCF-7Casp-3 cells stably expressing EV, Bid shRNA1 or Caspase-8 shRNA were plated at 50 000 or 10 000 cells per 10-cm dish for colony visualization (a) or counting purposes (b), respectively, and treated with 50 ng/ml IZ-TRAIL for 15 days. Resistant colonies were visualized with Coomassie-blue fixing solution and counted. Plates (a) and colony numbers (b) are representative of two independent experiments. The plating efficiency of untreated cells was 100% (data not shown). (c–g) MCF-7Casp-3 cells and SKW6.4 B-lymphoma cells were transduced to stably express an empty vector (EV) or a vector encoding Bcl-2. (c,e) Bcl-2 overexpression in MCF-7Casp-3 and SKW6.4 cells was confirmed by immunoblotting on total cell lysates, in which Actin served as a loading control. (d) Control (EV) or Bcl-2-overexpressing MCF-7Casp-3 cells were plated for the colony assay at 50 000 cells per 10-cm dish and left untreated (−) or treated (TX) with 50 ng/ml IZ-TRAIL for 15 days. Resistant colonies were visualized with Coomassie blue fixing solution. (f) SKW6.4 cells are of a Type I nature. Control (EV) and Bcl-2-overexpressing SKW6.4 cells were treated with 0, 12,5, 25 or 50 ng/ml of IZ-TRAIL for 3 h. The percentage of cells with cleaved Caspase-3 was determined by flow cytometry after intracellular staining with an antibody that specifically recognizes cleaved Caspase-3. Data presented are expressed as mean+S.D. values of three independent experiments. (g) Control (EV) and Bcl-2-overexpressing SKW6.4 cells were plated in 96-well plates at 1 cell per well and were treated with 50 ng/ml IZ-TRAIL for 15 days. Resistant colonies were scored by visual inspection. The percentages of colony-forming cells are depicted, corrected for plating efficiencies. Data are mean+ S.D. values of two independent experiments
To further address the relevance of the mitochondrial apoptosis signaling pathway, it was blocked by Bcl-2 overexpression. The MCF-7Casp-3 cells were retrovirally transduced with a Bcl-2-encoding vector or with EV as control (Figure 2c). The overexpression of Bcl-2 conferred resistance to TRAIL, confirming that the mitochondrial apoptosis pathway contributed to clonogenic death of MCF-7Casp-3 cells after TRAIL treatment (Figure 2d, also see 6b,c). To examine whether this phenotype could be observed in other Type I cells, we used the B-lymphoma cell line SKW6.4 and the breast carcinoma cell line T47D.11 The SKW6.4 cells were transduced with EV or a vector encoding Bcl-2 and T47D cells with EV or a Bid-targeting shRNA (Bid shRNA2). Immunoblotting demonstrated effective Bcl-2 overexpression in SKW6.4 cells (Figure 2e) and silencing of Bid protein expression in T47D cells (Supplementary Figure 1a). The overexpression of Bcl-2 in SKW6.4 cells did not significantly affect TRAIL-induced Caspase-3 cleavage, in line with the Type I classification,11 but it did produce clonogenic resistance to TRAIL (Figure 2f,g). Similarly, Bid RNAi produced clonogenic resistance to TRAIL in T47D cells (Supplementary Figure 1b). These results demonstrate that the intrinsic apoptosis pathway limits clonogenic outgrowth of Type I cells after TRAIL treatment.
Caspase-9 is not required for TRAIL receptor-induced clonogenic cell death in Type I cells
To study the importance of Caspase-9 activity for TRAIL-induced clonogenic cell death in Type I cells, we expressed a catalytically inactive mutant (dnCaspase-9) in MCF-7Casp-3 cells (Figure 3a). To validate the dominant negative effect of this mutant, we tested Caspase-3 cleavage in response to irradiation with UV light, as this is completely dependent on the intrinsic pathway.23 The UV-induced Caspase-3 cleavage was completely abrogated in dnCaspase-9-overexpressing cells, indicating that endogenous Caspase-9 activity was effectively inhibited (Figure 3b).

Caspase-9 is not required for clonogenic elimination of Type I cells by TRAIL. MCF-7Casp-3 cells were transduced (Td) to stably express an empty vector (EV) or dominant-negative Caspase-9 mutant (dnCasp-9). (a) Overexpression of dnCasp-9 protein was confirmed by immunoblotting on total cell lysates, in which Actin served as a loading control. (b,c) EV- and dnCasp-9-expressing cells were exposed to the indicated doses of UV light (b) or IZ-TRAIL (c) and percentages of cells with cleaved Caspase-3 were determined by flow cytometry after 16 h or 3 h, respectively. Data are expressed as mean+S.D. values of three independent experiments. Statistically significant differences between values of EV and dnCasp-9 samples are indicated for *P<0.05 and ***P<0.001. (d,e) EV- and dnCasp-9-expressing cells were plated at 50 000 or 10 000 cells per 10-cm dish for colony visualization (d) and counting (e) purposes, respectively, and left untreated (−) or treated with 50 ng/ml IZ-TRAIL for 15 days. Resistant colonies were visualized with Coomassie blue fixing solution and counted. Colony numbers in (e) are means+S.D. values of three independent experiments and plates in (d) are representative of these. Differences were statistically non-significant (NS)
To study the effect of Caspase-9 on TRAIL-induced Caspase-3 cleavage, EV-transduced and dnCaspase-9-transduced cells were exposed to different dosages of TRAIL for 3 h. Noticeably, dnCapase-9 did not inhibit TRAIL-induced Caspase-3 cleavage at any dose (Figure 3c). In contrast to previous effects of Bid downregulation or Bcl-2 overexpression, Caspase-9 inhibition did not significantly rescue MCF-7Casp-3 cells from TRAIL-induced clonogenic death, as documented by visual inspection (Figure 3d) and by the quantification of colonies (Figure 3e). Similar results were obtained when Caspase-9 expression was silenced in MCF-7Casp-3 cells by RNAi (Supplementary Figure 2). We conclude from these findings that Caspase-9 activity is not required for TRAIL-induced Caspase-3 cleavage and clonogenic death in Type I cells.
Smac/DIABLO is essential for TRAIL-induced clonogenic cell death in Type I cells
Having ruled out an essential contribution of Caspase-9 to execution of Type I cells after TRAIL stimulation, we examined the relevance of Smac/DIABLO release. For this purpose, we used a Smac/DIABLO mutant (ΔSmac), which lacks its mitochondrial-targeting sequence and therefore solely resides in the cytosol.14 The MCF-7Casp-3 cells, in which Bid expression was silenced by Bid shRNA1 (Figure 4a), were stably transduced with ΔSmac-encoding vector or the empty control vector. Cells transduced with empty vectors served as control (EV1+EV2). Subcellular fractionation, followed by immunoblotting confirmed that ΔSmac was expressed in the cytosol, whereas the endogenous molecule exclusively resided in the mitochondrial fraction of unstimulated cells (Figure 4b). By expressing ΔSmac in cells that had their mitochondrial pathway blocked by Bid RNAi, we created a situation in which only Smac/DIABLO and no other mitochondrial effectors could be operational in the cytosol.

Smac/DIABLO is essential for clonogenic elimination of Type I cells by TRAIL. (a–d) ΔSmac/DIABLO partially overrules clonogenic resistance of Type I cells to TRAIL treatment. MCF-7Casp-3 cells were stably transduced with an empty RNAi vector (EV1) or the same vector encoding Bid shRNA1, together with an empty expression vector (EV2) or the same vector encoding a cytosolic Smac/DIABLO variant (ΔSmac). (a) Immunoblotting on total cell lysates for Bid and Actin as loading control on the cell lines transduced (Td) as indicated. (b) Cells were separated into cytosol and mitochondria-enriched fractions that were immunoblotted for the presence of Smac/DIABLO. Immunoblotting for Actin served as loading control and validation of the subcellular fractionation. (c,d) MCF-7Casp-3 cells that had been transduced as indicated were plated at 50 000 or 10 000 cells per 10-cm dish for colony visualization (c) and counting (d) purposes, respectively, and left untreated (−) or treated with 50 ng/ml IZ-TRAIL for 15 days. Resistant colonies were visualized with Coomassie blue fixing solution and counted. Colony numbers in (d) are mean+S.D. values of four independent experiments and plates in (c) are representative of these. Statistically significant differences are indicated for ***P<0.001. (e–g) Smac/DIABLO mimetic overrules clonogenic resistance of Type I cells to TRAIL treatment. MCF-7Casp-3 were stably transduced with an empty RNAi vector (EV) or Bid shRNA2 to create a control cell line and a second Bid RNAi cell line. (e) Immunoblotting on total cell lysates for Bid and Actin as loading control on the cell lines transduced (Td) as indicated. (f,g) MCF-7Casp-3 cells stably expressing EV or Bid shRNA2 were plated at 50 000 or 10 000 cells per dish for visualization (f) and counting (g) purposes, respectively, and treated with 50 ng/ml IZ-TRAIL for 15 days, in presence or absence of 10 μM or 20 μM Smac/DIABLO mimetic LBW242. Resistant colonies were visualized with Coomassie blue fixing solution and were counted. Colony numbers in (g) are mean+S.D. values of two independent experiments and plates in (f) are representative of these with only the result of 20 μM LBW242 treatment shown
The EV cells and Bid RNAi cells expressing ΔSmac or control vector (both encoding green fluorescent protein, GFP) were treated with 50 ng/ml TRAIL for 15 days and assayed for clonogenicity (Figure 4c,d). Bid RNAi mediated significant clonogenic resistance to TRAIL, whereas expression of ΔSmac in Bid RNAi cells significantly decreased the number of resistant colonies (Figure 4d). This finding indicates that Smac/DIABLO has an important role in the clonogenic elimination of Type I cells by TRAIL.
We considered that stable expression of ΔSmac/DIABLO in Bid RNAi cells might lead to a selection of cells adapting to its presence. Therefore, we also treated Bid RNAi MCF-7Casp-3 cells acutely with the cell-permeable synthetic Smac/DIABLO mimetic LBW242. This mimetic, LBW242, is a chemical compound that competes with Smac/DIABLO for binding to the BIR3 pocket of XIAP.24 Bid RNAi was performed using a second shRNA (Bid shRNA2), to exclude any off-target effects of Bid shRNA1. This Bid shRNA2 also efficiently silenced Bid expression (Figure 4e). After 15 days of treatment with IZ-TRAIL, in absence or presence of 10 μM or 20 μM LBW242, resistant colonies were visualized and quantified (Figure 4f,g). Bid RNAi with the shRNA2 mediated significant clonogenic resistance of MCF-7Casp-3 cells to TRAIL (Figure 4g). At both doses, LBW242 reduced the clonogenicity of Bid RNAi cells, with the 20 μM concentration bringing it back to the level of EV-transduced cells (Figure 4g). Together, these observations indicate that the release of Smac/DIABLO from mitochondria is essential to limit clonogenicity of Type I cells after TRAIL treatment.
Effector caspase activity is hampered in Bid RNAi cells but restored by Smac/DIABLO mimetic
Our data suggested that the mitochondrial pathway contributes to clonogenic death of Type I cells by releasing effector caspases from XIAP inhibition after their initial activation by Caspase-8/10. To validate this, we compared the proteolytic processing of effector caspases and their substrates in control and Bid RNAi MCF-7Casp-3 cells after TRAIL stimulation.
In repeated assays, we consistently revealed a reduced efficiency in processing of the 36-kDa Caspase-7 proform to its 32-kDa cleavage product (p32) in Bid RNAi cells as compared with EV control cells (Figure 5). This was best observed at the low dose of 25 ng/ml IZ-TRAIL, but also visible at the 50-ng/ml dose used in the clonogenic assay (data not shown). Caspase-7 can remove the pro-domain from its own proform to generate p32,25 and therefore this may reflect reduced Caspase-7 activity. Moreover, the p20 form of Caspase-7, which results from autoprocessing,25 was less efficiently generated in Bid RNAi cells (Figure 5). For Caspase-3, we observed that in Bid RNAi cells, the conversion of the 20-kDa cleavage form (p20) to its fully processed 17-kDa form (p17) was less efficient (Figure 5). This indicated a reduced activity of effector caspases in Bid RNAi cells, as p20 is generated by Caspase-8, which cleaves pro-Caspase-3 at Asp175 between the large and the small subunits, whereas p17 is generated by removal of the pro-domain by (autocatalytic) effector caspase activity.19, 26 This difference in Caspase-3 processing was not revealed in the flow cytometric assay as statistically significant, although there was a modest reduction in case of Bid RNAi cells (Figure 1c). Reduced effector caspase activity in Bid RNAi cells was reflected by a reduced efficiency in proteolytic processing of their substrate PARP (Figure 5).

Effector caspase activity is reduced in Bid-deficient Type I cells, but can be restored by treatment with Smac/DIABLO mimetic LBW242. MCF-7Casp-3 cells stably transduced with an empty RNAi vector (EV) or with Bid shRNA2 were exposed to 25 ng/ml IZ-TRAIL in presence or absence of 20 μM LBW242 compound for the indicated periods of time in h. Total cell lysates were prepared and probed by immunoblotting for Caspase-7, -3 and PARP. Actin served as loading control. Film exposures for the detection of the cleavage products of Caspase-3 and -7 were longer than those applied for detection of the proforms. For Caspase-3 detection, an antibody was used that detects full-length Caspase-3 and its p20, p19 and p17 cleavage fragments
Importantly, treatment with the Smac mimetic, LBW242, restored effector caspase processing and PARP cleavage to the extent observed in EV control cells (Figure 5). These data indicate that the mitochondrial release of Smac/DIABLO allows for full effector caspase activation in Type I cells.
XIAP inhibition is important for TRAIL-induced clonogenic cell death in Type I cells
To study the importance of XIAP inhibition by the mitochondrial pathway for TRAIL-induced clonogenic death in Type I cells, we silenced its expression by RNAi. The Bcl-2-overexpressing MCF-7Casp-3 cells were stably transduced with EV or a vector encoding an XIAP-targeting shRNA that effectively silenced XIAP protein expression (Figure 6a). Clonogenicity of these cells relative to control was examined after treatment with 50 ng/ml TRAIL for 15 days (Figure 6b). The overexpression of Bcl-2 conferred significant clonogenic resistance to TRAIL in MCF-7Casp-3 cells (Figure 6c). The RNAi-mediated downregulation of XIAP in turn significantly reduced clonogenicity of the Bcl-2-overexpressing MCF-7Casp-3 cells after TRAIL treatment (Figure 6c). This indicates that XIAP levels limit the potential of the TRAIL receptor(s) to clonogenically execute Type I tumor cells through the direct, mitochondrion-independent pathway. Release of IAP inhibitors by the mitochondria facilitates this execution, as follows from the potential of ΔSmac/DIABLO and Smac/DIABLO mimetic LBW242 to increment sensitivity to TRAIL in Bid RNAi MCF-7Casp-3 cells (Figure 4d,g). The XIAP RNAi did not restore clonogenic death to control levels, suggesting that the mitochondrial IAP antagonists have additional targets, besides XIAP, through which they enhance cellular execution after TRAIL treatment.

XIAP inhibition is important for clonogenic elimination of Type I cells by TRAIL. MCF-7Casp-3 cells were stably transduced with an empty vector (EV1) or with the same vector encoding Bcl-2. Cells transduced with Bcl-2 were co-transduced with an empty RNAi vector (EV2) or XIAP shRNA. (a) Immunoblotting for Bcl-2, XIAP and Actin as a loading control on total cell lysates of cells transduced (Td). (b, c) EV1- and Bcl-2-overexpressing cells with EV2 or XIAP shRNA were plated at 50 000 or 10 000 cells per 10-cm dish for colony visualization (b) and counting (c) purposes, respectively, and left untreated (−) or treated with 50 ng/ml IZ-TRAIL for 15 days. Resistant colonies were visualized with Coomassie blue fixing solution and counted. Colony numbers in (c) are mean+S.D. values of three plates per treatment and representative of three independent experiments. Plates in (b) are representative of these. Statistically significant differences are indicated for ***P<0.001
Low Caspase-3 expression favors clonogenic survival of Type I cells upon TRAIL treatment
A recent report showed that cell-to-cell variability in protein levels or states can significantly influence the apoptotic response of Type I cells to TRAIL treatment.27 In this regard, decreased levels of pro-apoptotic proteins and/or increased levels of anti-apoptotic proteins could cause clonogenic resistance to TRAIL. To examine this, we selected Bcl-2-overexpressing MCF-7Casp-3 cells and SKW6.4 cells for 2 weeks with TRAIL. Next, by immunoblotting we compared the levels of Caspase-8, -6, -7, -3, XIAP, cIAP-1 and cIAP-2 expression in these cells with those in unselected control cells. In repeated experiments, no significant differences in Caspase-8, -6, cIAP-1 and cIAP-2 expression were observed in TRAIL-selected cells as compared with control cells (Figure 7a,b). In both cell lines, however, Caspase-3 levels were strongly reduced and Caspase-7 levels were somewhat reduced. In MCF-7Casp-3 cells, XIAP levels were unaltered and in SKW6.4 cells they were slightly reduced, but clearly in both cases, cells that resisted TRAIL-induced apoptosis were hallmarked by a higher XIAP-to-effector caspase ratio. This finding underlines that clonogenic execution of Type I tumor cells by TRAIL through the mitochondrion-independent pathway can be limited by the availability of active effector caspases.

Caspase-3 expression levels limit clonogenic execution through the mitochondrion-independent death receptor pathway. Bcl-2-overexpressing MCF-7Casp-3 or SKW6.4 cells were left untreated or were selected for 15 days with 50 and 200 ng/ml IZ-TRAIL, respectively. (a, b) Immunoblotting for Caspase-8, Caspase-6, Caspase-7, Caspase-3, XIAP, cIAP1 and cIAP2 on total cell lysates of unselected (Bcl-2) and TRAIL-selected (Bcl-2 TRAIL) MCF-7Casp-3 (a) or SKW6.4 (b) cells. Actin served as a loading control. Data are representative of three independent experiments
Discussion
Prompted by the unbiased recovery of Bid-targeting shRNA from a RNAi screen in Type I MCF-7Casp-3 cells, we have re-examined the relevance of the mitochondrial pathway for the execution of Type I cells by death receptors. Using Type I MCF-7Casp-3 and T47D breast carcinoma cells, and SKW6.4 B lymphoma cells in clonogenic assays, we have demonstrated that the mitochondrial pathway limits the clonogenic outgrowth of Type I tumor cells after TRAIL treatment.
Type I cells are generally assumed not to require mitochondria for death receptor-induced apoptosis.10 The Type I/Type II distinction was originally made for tumor cells4 and was later proved valid for primary cell types as well. Among these, thymocytes and hepatocytes are the prototype Type I and Type II cells, respectively. The Type II nature of hepatocytes followed from their response to in vivo stimulation with CD95/Fas agonists, which killed wild-type mice by hepatic injury, whereas Bcl-2 transgenic, Bid−/− or Bax;Bak−/− mice proved to be resistant.5, 9, 28 In contrast, genetic deficiency for Bid5 or Bax and Bak29 did not overtly affect the cell death response of thymocytes to CD95/Fas stimulation.
Our study points out that the mitochondrial contribution to death receptor-mediated execution of Type I cells is subtle, but important, particularly when one considers the implications for tumor therapy by death receptor agonists. We demonstrate that the mitochondrial contribution to tumor cell execution by TRAIL is revealed when clonogenic outgrowth of the treated population is considered. In our clonogenic assays, Bid deficiency or Bcl-2 overexpression rescued about 3.5–5% of the MCF-7Casp-3 cells from execution by 50 ng/ml IZ-TRAIL. In case of SKW6.4 B lymphoma cells also about 5% of cells in the population were rescued from execution by 200 ng/ml IZ-TRAIL on Bcl-2 overexpression. This effect, which will become increasingly prominent on lowering the TRAIL dose, is relevant in vivo where death receptors control the damage done by chronically stimulated or autoreactive T lymphocytes (CD95)30 and the metastatic spread of solid tumors (TRAIL receptor).31, 32 Moreover, TRAIL receptor agonists are currently in Phase I/II clinical trials for cancer therapy.33 Blockade of the intrinsic apoptosis pathway is a frequent occurrence in cancer and is expected to impact on the effectiveness of TRAIL therapy, also in case of tumors with a ‘Type I’ classification.
The intrinsic pathway enables effector caspase activity by two mechanisms: (1) Caspase-9 can cleave the effector caspase zymogens, and thereby activate them; (2) Smac/DIABLO and HtrA2/Omi alleviate cleaved effector caspases from inhibition by XIAP. Although Caspase-9 is absolutely required as inducer caspase for cell death responses that operate through the intrinsic pathway,23, 34 this is different for cellular execution by death receptors. We show here that TRAIL kills Type I MCF-7Casp-3 breast carcinoma cells equally well at the clonogenic level whether the cells have Caspase-9 activity or not. Different from Bid−/− thymocytes,8 Caspase-9−/−23, 34 or Apaf-1−/−35 thymocytes responded equally well in caspase activation assays on CD95/Fas treatment as wild-type thymocytes. This agrees with our findings and indicates that the apoptosome and Caspase-9 do not make a significant contribution to effector caspase activation by death receptors in Type I cells. Presumably, Caspase-8/10 activity suffices for the initial activation of the effector caspases. Recent data indicate that in Type II cells also, Caspase-9 activation by the mitochondrial route is not essential for apoptotic execution after death receptor stimulation: in Type II Jurkat cells, the apoptosome did not contribute to CD95/Fas-induced apoptosis in short-term assays.36 Moreover, in hepatocytes, Bid deficiency did not affect CD95/Fas-induced processing of pro-Caspase-3 to the p20 form.37
Importantly, this study by Li et al.37 defined effector caspase inhibition by (X)IAP proteins as the bottleneck for CD95/Fas-mediated execution in Bid−/− hepatocytes: the p20 form of Caspase-3 was not further processed to its p17 form, whereas this processing did occur in wild-type hepatocytes by virtue of Smac/DIABLO release from the mitochondria. This research forms the important precursor of the work by Jost et al.,8 who recently reported that genetic ablation of XIAP sufficed to convert Type II hepatocytes to a Type I response profile on CD95/Fas stimulation. Together, these two studies indicate that for death induction by CD95/Fas in hepatocytes, XIAP inhibition rather than effector caspase activation by Caspase-9 is the critical factor contributed by the intrinsic pathway. A recent study in Jurkat cells provides compelling evidence for the same conclusion.36 In colon carcinoma cells, XIAP downregulation similarly allowed a conversion from a Type II to a Type I response to TRAIL in short-term apoptosis assays.38 Hence, although it was generally assumed that Type II cells require Caspase-9 activity to increment effector caspase activation by the extrinsic pathway, there is no abundant experimental evidence for this.
At the biochemical level, most of the abovementioned studies did not reveal effects of the mitochondrial pathway on CD95/Fas-induced Caspase-3 cleavage or effector caspase activity. However, Yin et al.5 reported a delay in Caspase-3 cleavage and effector caspase activity in Bid−/− thymocytes. Jost et al.8 similarly found reduced or delayed effector caspase activity in CD95/Fas-stimulated Bid−/− thymocytes (Type II), which was partially rescued by XIAP deficiency. We detected a reduced efficiency in effector caspase processing and activity on mitochondrial inhibition by immunoblotting. Although differences were not statistically significant in the flow cytometric assay (Figure 1c), a modest reduction in the percentage of cells with cleaved Caspase-3 was also observed. These data are consistent with the concept that XIAP restrains effector caspase activity.
Using the cytosolic ΔSmac variant, we show that Smac/DIABLO release from mitochondria is essential for clonogenic elimination of Type I tumor cells by TRAIL. Use of the LBW242 Smac/DIABLO mimetic corroborated this, although its mechanism of action involves more than XIAP elimination.39, 40 The IAP family is characterized by BIR domains and contains multiple proteins of diverse function.13, 39 Among these, XIAP, cIAP-1 and cIAP-2 are implicated in death-receptor signaling and all are targeted by Smac/DIABLO mimetics. Among IAPs, XIAP is seen as the only bona fide effector caspase inhibitor, as it alone can shield the catalytic site of the effector caspases. It indeed has a much greater inhibitory activity than cIAP-1 or cIAP-2, whose prominent role lies in the regulation of NF-κB activation.13 The downregulation of XIAP by RNAi consistently reduced clonogenicity of TRAIL-treated MCF-7Casp-3 cells, indicating that Smac/DIABLO release from mitochondria promoted clonogenic death by overruling XIAP activity. However, ΔSmac/DIABLO and in particular LBW242 reduced clonogenicity to a greater extent than XIAP RNAi, suggesting that they in addition promoted cellular execution by another mechanism, most likely by targeting other IAPs.
Elegant studies have recently addressed control points in TRAIL-induced apoptosis at the single cell level.12 Within a Type II tumor cell population (HeLa), individual cells exhibited distinct delays in MOMP after inducer caspase activation. In this period, XIAP – that might be expressed at different levels between cells – restrained effector caspase activity. Accordingly, Spencer et al.27 reported cell-to-cell variability in the timing and probability of cellular execution by TRAIL. Clonogenic assays are apt to reveal such variability and in the two Type I tumor cell lines tested in our study, certain cells within the population were dependent on the mitochondria for clonogenic execution because of low Caspase-3 levels. The downregulation of XIAP by RNAi restored sensitivity in a significant proportion of these cells. We conclude that the difference in TRAIL receptor signaling between Type II and Type I cells is not qualitative, but rather quantitative with ranges of differential responsiveness within a population.
Materials and Methods
Cells and stimulation
Breast carcinoma MCF-7 cells stably expressing human Caspase-3, Bcl-2, the dominant-negative Caspase-9 active-site mutant C287A (dnCaspase-9), the Δ55 Smac/DIABLO truncation mutant, or the Caspase-8- or Bid-targeting shRNAs were generated by retroviral transduction. The SKW6.4 B-cell lymphoma cell line was similarly transduced to express Bcl-2. Simultaneously generated empty vector-transduced cell lines acted as controls in all instances. The MCF-7Casp-3 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) and SKW6.4 cells in Iscove's Modified Dulbecco's Medium (IMDM), supplemented with 8% fetal bovine serum, 2 mM L-glutamine and antibiotics. Isoleucine-zippered TRAIL (IZ-TRAIL)21 was kindly provided by Dr. H. Walczak (Imperial College London, London, UK). Smac/DIABLO-mimetic LBW24240 was a kind gift from Dr. L. Zawel (Novartis Institute for Biomedical Research, Cambridge, MA, USA). LBW242 was dissolved in DMSO. For apoptosis assays and clonogenic assays, cells were stimulated with the indicated dosages of IZ-TRAIL, LBW242 and UV irradiation for the indicated time periods and incubated at 37 °C with 5% CO2. Selection of Bcl-2-expressing MCF-7Casp-3 and SKW6.4 cells was performed for 15 days with 50 and 200 ng/ml TRAIL, respectively.
Constructs
Caspase-3 cDNA was generated by PCR from a pcDNA3 vector and cloned into the retroviral LZRS/pBR vector, with an IRES–Zeocin resistance cassette. The LZRS vectors are derivatives of the LZRS-pBMN-LacZ vector, which was originally provided by Dr. G.P. Nolan (Stanford University School of Medicine, Stanford, CA, USA). The RNAi for Caspase-8, Bid and XIAP was performed using shRNA cloned into the retroviral vector pRETRO-SUPER, with a puromycin resistance cassette (see the study by Tait et al.20). The complementary sense and antisense oligonucleotides used had the following targeting sequences: Caspase-8 shRNA: 5′-GTGCCCAAACTTCACAGCA-3′; Bid shRNA1: 5′-CCAGAACCTACGCACCTAC-3′; Bid shRNA2: 5′-GAAGACATCATCCGGAATA-3′; XIAP shRNA: 5′-GAGGAGAAGCTTTGCAAAA-3′. Bcl-2 cDNA was cloned into the retroviral vector pMX with a Blasticidin resistance cassette for transduction of MCF-7Casp-3 cells and in into the retroviral vector LZRS/pBR in an IRES–GFP configuration for transduction of SKW6.4 cells. DnCaspase-9 cDNA was also cloned in LZRS/pBR in an IRES–GFP configuration. Plasmid pOTB7-DIABLO with full-length human Smac/DIABLO was obtained from the Mammalian Gene Collection (http://mgc.nci.nih.gov). The Δ55 Smac/DIABLO mutant (ΔSmac/DIABLO14) encoding the cytosolic Smac/DIABLO variant was generated by PCR and cloned into the retroviral pMX vector in an IRES–GFP configuration. All constructs were verified by dideoxynucleotide sequencing.
Retroviral gene transduction
To produce retrovirus, LZRS, pRETRO-SUPER and pMX constructs were transfected into the HEK293T cell-derived packaging cell line, Phoenix-Ampho, using FuGENE 6 transfection reagent, according to the manufacturer's instructions (Roche Molecular Biochemicals, Mannheim, Germany). After 48 h, virus-containing medium was collected. For transduction, cells were cultured in this virus-containing medium for 8 h and, after refreshing of the same medium, overnight. The next day, the supernatant was removed and cell culture was continued in normal medium. At day 3 after transduction, cells were selected with 200 μg/ml Zeocin (Invitrogen, Carlsbad, CA, USA) in case of transduction with LZRS/pBR constructs; with 1 μg/ml Puromycin (Sigma-Aldrich, St. Louis, MO, USA) in case of pRETRO-SUPER-Caspase-8 and Bid shRNA constructs; and with 10 μg/ml Blasticidin (Sigma-Aldrich) in case of the pMX-Bcl-2 construct. Cells transduced with the LZRS-IRES-GFP and pMX-IRES-GFP constructs were sorted for GFP expression using a MoFlo high speed cell sorter (Cytomation, Fort Collins, CO, USA).
Cell death assays
Assessment of cleaved Caspase-3 protein levels by FACS was performed as follows: after stimulation, all cells were collected, either directly from the medium or via trypsinization, and fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS). Subsequently, cells were washed twice with 1% bovine serum albumin (BSA) in PBS and once with permeabilizing buffer (PBS with 0.1% saponin and 0.5% BSA). Next, cells were incubated for 20 min with permeabilizing buffer and stained for 1 h with rabbit anti-active Caspase-3 antibody (1 : 50, clone C92-605, BD Biosciences, Erembodegem, Belgium), which recognizes all Caspase-3 cleavage products. Hereafter, cells were washed three times with permeabilizing buffer and stained for 1 h with AlexaFluor 647-conjugated goat anti-rabbit immunoglobulin (Ig; 1 : 100; Molecular Probes, Leiden, the Netherlands). After three more washes with permeabilizing buffer, cells were analyzed by flow cytometry with a FACSCalibur (BD Biosciences, Franklin Lakes, NJ, USA) and FCS Express software (De Novo Software, Thornhill, Canada). Cellular debris was excluded from analysis. Cells were stained with FITC Annexin V according to manufacturer's instructions (BD Biosciences, Erembodegem, Belgium) in combination with propidium iodide (PI) and analyzed by flow cytometry as indicated above.
Clonogenic survival assay
For clonogenic survival assays with MCF-7Casp-3 cells, 10 000 (for counting) and 50 000 (for visualization) were plated in 10-cm polystyrene cell culture dishes (BD Biosciences). Once attached, cells were exposed to 50 ng/ml IZ-TRAIL and cultured for 15 days. Next, surviving colonies were fixed with 75% MeOH/25% acetic acid and stained with 50% MeOH/10% acetic acid/0.2% Coomassie blue solution. The number of colonies was counted by visual inspection. Plating efficiency of non-treated cells was 100%. For clonogenic survival assays with SKW6.4 cells, cells were plated in BD Falcon polystyrene 96-well round-bottom plates (BD Biosciences) in medium containing 50 ng/ml TRAIL at a 1-cell per well concentration. Colonies were counted after 15 days and percentages of colony-forming cells were determined.
Immunoblotting
Of total cell lysates, samples were prepared containing 30 μg total cellular protein, as determined by BioRad protein assay (Biorad, Munchen, Germany). Proteins were separated on 4–12% NuPage Bis–Tris gradient gels (Invitrogen, Carlsbad, CA, USA) in MES buffer, according to manufacturer's instructions. Subsequent immunoblotting was performed as described.20 Proteins were detected with the following antibodies: rabbit anti-Caspase-3 pAb (a kind gift from Dr. Gerald M. Cohen, MRC Toxicology Unit, University of Leicester, Leicester, UK), mouse anti-Caspase-6 mAb 556581 (1 : 250; BD Biosciences), mouse anti-Caspase-7 mAb 551239 (1 : 1000, BD Biosciences), mouse anti-Caspase-8 mAb C15 (1 : 1000; Alexis Biochemicals, Lausen, Switzerland), mouse anti-Caspase-9 mAb 9508 (1 : 1000; Cell Signaling Technology, Danvers, MA, USA), rabbit anti-Bid pAb (1 : 250; rabbit serum, homemade, but available from BD Biosciences), mouse anti-Bcl-2 mAb (1 : 1000; clone Bcl-2-100, mouse ascites fluid; Sigma-Aldrich), mouse anti-Smac/DIABLO mAb 78–1–118 (1 : 700; Upstate, Charlottesville, VA, USA), rabbit anti-PARP pAb 9542 (1 : 2000; Cell Signaling Technology), mouse anti-XIAP mAb 610716 (1 : 4000; BD Biosciences), rabbit anti-cIAP1 pAb 210–885 (1 : 1000; Alexis Biochemicals), rabbit anti-cIAP2 pAb AF817 (1 : 500; R&D Systems, Abingdon, UK) and mouse anti-Actin mAb C4 (1 : 10 000, Chemicon International, Temecula, CA, USA). Second-step antibodies were horseradish peroxidase (HRP)-conjugated swine anti-rabbit Ig (1 : 7500) and rabbit anti-mouse Ig (1 : 7500, both from Dako A/S, Glostrup, Denmark). The enhanced chemiluminescence (ECL) kit was from Pierce Biotechnology (Rockford, IL, USA).
Abbreviations
- BIR:
-
Baculovirus inhibitor repeat
- DISC:
-
Death-inducing signaling complex
- dn:
-
dominant negative
- EV:
-
empty vector
- GFP:
-
green fluorescent protein
- IAP:
-
Inhibitor of apoptosis protein
- IBM:
-
IAP binding motif
- IZ-TRAIL:
-
isoleucine-zippered TRAIL
- RNAi:
-
RNA interference
- MOMP:
-
mitochondrial outer membrane permeability
- shRNA:
-
short hairpin RNA
- RNAi:
-
RNA interference
References
Peter ME, Krammer PH . The CD95(APO-1/Fas) DISC and beyond. Cell Death Differ 2003; 10: 26–35.
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X . Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors. Cell 1998; 94: 481–490.
Wang X . The expanding role of mitochondria in apoptosis. Genes Dev 2001; 15: 2922–2933.
Scaffidi C, Fulda S, Srinivasan A, Friesen C, Li F, Tomaselli K et al. Two CD95 (APO-1/Fas) signaling pathways. EMBO J 1998; 17: 1675–1687.
Yin X-M, Wang K, Gross A, Zhao Y, Zinkel S, Klocke B et al. Bid-deficient mice are resistant to Fas-induced hepatocellular apoptosis. Nature 1999; 400: 886–891.
Strasser A, Harris AW, Huang DCS, Krammer PH, Cory S . Bcl-2 and fas/APO-1 regulate distinct pathways to lymphocyte apoptosis. EMBO J 1995; 14: 6136–6147.
Huang DCS, Hahne M, Schroeter M, Frei K, Fontana A, Villunger A et al. Activation of Fas by FasL induces apoptosis by a mechanism that cannot be blocked by Bcl-2 or Bcl-xL . Proc Natl Acad Sci USA 1999; 96: 14871–14876.
Jost PJ, Grabow S, Gray D, McKenzie MD, Nachbur U, Huang DCS et al. XIAP discriminates between type I and type II FAS-induced apoptosis. Nature 2009; 460: 1035–1039.
Lacronique V, Mignon A, Fabre M, Violet B, Rouquet N, Molina T et al. Bcl-2 protects from lethal hepatic apoptosis induced by an anti-Fas antibody in mice. Nat Med 1996; 2: 80–86.
Barnhart BC, Alappat EC, Peter ME . The CD95 Type I/Type II model. Sem Immunol 2003; 15: 185–193.
Algeciras-Schimnich A, Pietras EM, Barnhart BC, Legembre P, Vijayan S, Holbeck SL et al. Two CD95 tumor classes with different sensitivities to anti-tumor drugs. Proc Natl Acad Sci USA 2003; 100: 11445–11450.
Albeck JG, Burke JM, Aldridge BB, Zhang M, Luaffneburger DA, Sorger PK . Quantitative analysis of pathways controlling extrinsic apoptosis in single cells. Mol Cell 2008; 30: 11–25.
Eckelmann BP, Salvesen GS, Scott FL . Human inhibitor of apoptosis proteins: why XIAP is the black sheep of the family. EMBO Rep 2006; 7: 988–994.
Du C, Fang M, Li Y, Li L, Wang X . Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell 2000; 102: 33–42.
Suzuki Y, Imai Y, Nakayama H, Takahashi K, Takio K, Takahasi R . A serine protease, HtrA2 is released from the mitochondria and interacts with XIAP, inducing cell death. Mol Cell 2001; 8: 613–621.
Ekert PG, Silke J, Hawkins CJ, Verhagen AM, Vaux DL . DIABLO promotes apoptosis by removing MIHA/XIAP from processed caspase-9. J Cell Biol 2001; 152: 483–490.
Srinivasula SM, Hegde R, Saleh A, Datta P, Shiozaki E, Chai J et al. A conserved XIAP interaction motif in caspase-9 and Smac/DIABLO regulates caspase activity and apoptosis. Nature 2001; 410: 112–116.
Deveraux QL, Roy N, Stennicke HR, Van Arsdale T, Zhou Q, Srinivasula SM et al. IAPs block apoptotic events induced by caspase-8 and cytochrome c by direct inhibition of distinct caspases. EMBO J 1998; 17: 2215–2223.
Sun X-M, Bratton SB, Butterworth M, MacFarlane M, Cohen G . Bcl-2 and Bcl-xL inhibit CD95-mediated apoptosis by preventing mitochondrial release of Smac/DIABLO and subsequent inactivation of X-linked inhibitor-of-apoptosis protein. J Biol Chem 2002; 277: 11345–11351.
Tait SWG, de Vries E, Maas C, Keller A, D'Santos C, Borst J . Apoptosis induction by Bid requires unconventional ubiquitination and degradation of its N-terminal fragment. J Cell Biol 2007; 179: 1453–1466.
Ganten TM, Haas TL, Sykora J, Stahl H, Sprick MR, Fas SC et al. Enhanced caspase-8 recruitment to and activation at the DISC is critical for sensitisation of human hepatocellular carcinoma cells to TRAIL-induced apoptosis by chemotherapeutic drugs. Cell Death Differ 2004; 11: S86–S96.
Berns K, Hijmans ME, Mullenders J, Brummelkamp TR, Velds A, Heimerikx M et al. A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 2004; 428: 431–437.
Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS et al. Differential requirement for Caspase 9 in apoptotic pathways in vivo. Cell 1998; 94: 339–352.
Chauhan D, Neri P, Velankar M, Podar K, Hideshima T, Fulciniti M et al. Targeting mitochondrial factor Smac/DIABLO as therapy for multiple myeloma (MM). Blood 2007; 109: 1220–1227.
Van de Craen M, Declercq W, Van den Brande I, Fiers W, Vandenabeele P . The proteolytic procaspase activation network: an in vitro analysis. Cell Death Differ 1999; 6: 1117–1124.
Han Z, Hendrickson EA, Bremner TA, Wyche JH . A sequential two-step mechanism for the production of the mature p17:p12 form of caspase-3 in vitro. J Biol Chem 1997; 272: 13432–13436.
Spencer S, Gaudet S, Albeck JG, Burke JM, Sorger PK . Non-genetic origins of cell-to-cell variability in TRAIL-induced apoptosis. Nature 2009; 459: 428–433.
Wei MC, Zong W-X, Cheng E, Lindsten T, Panoutsakopoulou V, Ross AJ et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292: 727–730.
Lindsten T, Ross AJ, King A, Zong W-X, Rathmell JC, Shiels HA et al. The combined functions of proapoptotic Bcl-2 family members Bak and Bax are essential for normal development of multiple tissues. Mol Cell 2000; 6: 1389–1399.
Weant AE, Michalek RD, Khan IU, Holbrook BC, Willingham MC, Grayson JM . Apoptosis regulators Bim and Fas function concurrently to control autoimmunity and CD8+ T cell contraction. Immunity 2008; 28: 218–230.
Cretney E, Takeda K, Yagita H, Galccum M, Peschon JJ, Smyth MK . Increased susceptibility to tumor initiation and metastasis in TNF-related apoptosis-inducing ligand-deficient mice. J Immunol 2002; 168: 1356–1361.
Grosse-Wilde A, Voloshanenko O, Bailey SL, Longton GM, Schaefer U, Csernok AI et al. TRAIL-R deficiency in mice enhances lymph node metastasis without affecting primary tumor development. J Clin Invest 2008; 118: 100–110.
Ashkenazi A, Holland P, Eckhardt G . Ligand-based targeting of apoptosis in cancer: the potential of recombinant human apoptosis ligand 2/tumor necrosis factor-related apoptosis-inducing ligand (rhApo2L/TRAIL). J Clin Oncol 2008; 26: 3621–3630.
Kuida K, Haydar TF, Kuna C-Y, Gu Y, Taya C, Karasuyama H et al. Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking Caspase-9. Cell 1998; 94: 325–337.
Yoshida H, Kong Y-Y, Yoshida R, Elia AJ, Hakem A, Hakem R et al. Apaf1 is required for mitochondrial pathways of apoptosis and brain development. Cell 1998; 94: 739–750.
Shawgo ME, Shelton SN, Robertson JD . Caspase-9 activation is not required for Fas-mediated apoptosis in Type II Jurkat cells. J Biol Chem 2009; 284: 33447–33455.
Li S, Zhao Y, He X, Kim T-H, Kuharsky DK, Rabinowich H et al. Relief of extrinsic pathway of inhibition by the Bid-dependent mitochondrial release of Smac in Fas-mediated hepatocyte apoptosis. J Biol Chem 2002; 277: 26912–26920.
Ndozangue-Touriguine O, Sebbagh M, Merino D, Micheau O, Bertoglio J, Breard J . A mitochondrial block and expression of XIAP lead to resistance to TRAIL-induced apoptosis during progression to metastasis of a colon carcinoma. Oncogene 2008; 27: 6012–6022.
Varfolomeev E, Vucic D . Unexpected roles of c-IAPs in apoptotic and NF-κB signaling pathways. Cell Cycle 2008; 7: 1511–1521.
Gaither A, Porter D, Yao Y, Borawski J, Yang G, Donovan J et al. A Smac mimetic rescue screen reveals roles for inhibitor of apoptosis proteins in tumor necrosis factor−α signaling. Cancer Res 2007; 67: 11493–11498.
Acknowledgements
We thank Dr. Henning Walczak (Division of Medicine, Imperial College London, London, UK) for IZ-TRAIL, Dr. Leigh Zawel (Oncology, Novartis Institute for Biomedical Research, Cambridge, MA, USA) for LBW242 compound, Dr. Gerald M. Cohen (MRC Toxicology Unit, University of Leicester, Leicester, UK) for anti-Caspase-3 antiserum, Dr. Roderick Beijersbergen and Rogier Rooswinkel for advise, Dr. Rene Bernards and coworkers for making the retroviral RNAi library available and personnel of the flow cytometry facility of the Netherlands Cancer Institute for experimental assistance. This study was supported by grants from the Dutch Cancer Society.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Edited by J Silke
Supplementary Information accompanies the paper on Cell Death and Differentiation website
Supplementary information
Rights and permissions
About this article
Cite this article
Maas, C., Verbrugge, I., de Vries, E. et al. Smac/DIABLO release from mitochondria and XIAP inhibition are essential to limit clonogenicity of Type I tumor cells after TRAIL receptor stimulation. Cell Death Differ 17, 1613–1623 (2010). https://doi.org/10.1038/cdd.2010.39
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cdd.2010.39
Keywords
This article is cited by
-
A literature review of microRNA and gene signaling pathways involved in the apoptosis pathway of lung cancer
Respiratory Research (2023)
-
TRAIL C1595T Variant Critically Alters the Level of sTRAIL in Terms of Histopathological Parameters in Colorectal Cancer
Indian Journal of Clinical Biochemistry (2023)
-
Targeting anti-apoptotic Bcl-2 by AT-101 to increase radiation efficacy: data from in vitro and clinical pharmacokinetic studies in head and neck cancer
Radiation Oncology (2015)
-
A functional genomics screen identifies PCAF and ADA3 as regulators of human granzyme B-mediated apoptosis and Bid cleavage
Cell Death & Differentiation (2014)
-
Lyapunov exponents and phase diagrams reveal multi‐factorial control over TRAIL‐induced apoptosis
Molecular Systems Biology (2011)
