
Abstract
Intraneural accumulation of misfolded proteins is a common feature of several neurodegenerative pathologies including Alzheimer’s and Parkinson’s diseases, and Familial Encephalopathy with Neuroserpin Inclusion Bodies (FENIB). FENIB is a rare disease due to a point mutation in neuroserpin which accelerates protein aggregation in the endoplasmic reticulum (ER). Here we show that cholesterol depletion induced either by prolonged exposure to statins or by inhibiting the sterol reg-ulatory binding-element protein (SREBP) pathway also enhances aggregation of neuroserpin proteins. These findings can be explained considering a computational model of protein aggregation under non-equilibrium conditions, where a decrease in the rate of protein clearance improves aggregation. Decreasing cholesterol in cell membranes affects their biophysical properties, including their ability to form the vesicles needed for protein clearance, as we illustrate by a simple mathematical model. Taken together, these results suggest that cholesterol reduction induces neuroserpin aggregation, even in absence of specific neuroserpin mutations. The new mechanism we uncover could be relevant also for other neurodegenerative diseases associated with protein aggregation.
Similar content being viewed by others


Introduction
Conformational diseases, such as Alzheimer’s and Parkinson’s diseases, spongiform encephalopathies and serpinopathies, are an increasingly common class of neurological disorders characterized by the aggregation of aberrant conformations of proteins. Familial Encephalopathy with Neuroserpin Inclusion Bodies (FENIB)1,2 is an autosomal dominant inclusion body dementia characterized by protein aggregation within the endoplasmic reticulum (ER)1,2. FENIB is associated with mutations in neuroserpin3, a protein belonging to the superfamily of serpins that plays an important role in brain development, neuronal survival, and synaptic plasticity4. Indeed, mutants of neuroserpin in FENIB patients show accelerated rates of polymerization compared with wild type proteins, both at the protein level in vitro5,6 and in cell models1,2. Moreover, neuroserpin is the most important inhibitor of the tissue plasminogen activator (tPA) in the brain which is known to be increased in Alzheimer’s disease7,8,9.
Lipid metabolism has been shown to play a role in FENIB, but also in other neurodegenerative diseases, from Alzheimer’s to Parkinson’s disease10,11,12,13. In FENIB the inhibition of hydroxymethyl glutaryl-CoA reductase (HMGCR) has a critical role in the clearance of mutant neuroserpin from the ER13. Numerous studies have also reported that the modification of cholesterol content can affect amyloid precursor protein (APP) processing, which is needed for neuronal activity11. In this connection, it has been shown that the E693D (Osaka) mutation in APP promotes intracellular accumulation of Amyloidβ (Aβ), thus disturbing amyloid-mediated cholesterol efflux from the cell14. Cholesterol has also been shown to influence APP processing and Aβ generation by modulating Aβ- and Amyloid-γ-secretase activities15,16,17,18. In turn, APP cleavage products regulate cholesterol homeostasis19.
In a recent paper, our group has shown by 3D numerical simulations and mean-field calculations that protein aggregation undergoes a non-equilibrium phase-transition controlled by the rates of protein synthesis and degradation, suggesting a crucial role of intracellular trafficking, particularly from the ER20. According to this study, a decrease in the rate of protein clearance from the ER can lead to rapid and irreversible protein aggregation in the ER itself20, leading to the onset of neurodegenerative diseases.
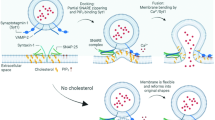
Cholesterol plays a crucial role in regulating the properties of phospholipid membranes, affecting their fluidity and rigidity21,22,23, the function and dynamics of membrane proteins24,25, and thus vesicular trafficking within the cell26,27,28. Hence, by regulating the biosynthesis of cholesterol it is possible to affect the form and function of all the membranes within the cell, including the ER. In particular, experiments show that lowering the level of cholesterol delays ER-to-Golgi transport28,29.
Cholesterol homeostasis is controlled by a family of transcription factors, known as sterol regulatory element binding proteins (SREBPs)30. In cells with sufficient cholesterol supply, SREBPs are transmembrane proteins retained in the ER, associated with SREBP-cleavage-activating protein (SCAP), a cholesterol sensor31. Upon cellular cholesterol depletion, SREBP leaves the ER to reach the Golgi, where cleavage by site-1 protease (S1P) releases the amino-terminal half of SREBP, which can be further cleaved within its membrane-spanning helix by site-2 metalloproteinase (S2P)32. The mature processed form of SREBP is released in the cytosol and can translocate into the nucleus where it modulates the expression of several genes controlling cholesterol and fatty acid homeostasis33, including HMGCR, HMG-CoA synthase (HMGCS), low density lipoprotein receptor (LDLR) and SREBP1/2 itself. SREBP1 activates fatty acid synthesis and SREBP2 cholesterol synthesis and uptake34.
In this paper, we combine experiments and computational analysis to show that lowering the level of cholesterol either by using statins or by inhibiting SREBP1 and SREBP2 pathways with the small molecule betulin has a significant impact on the aggregation of non-mutated neuroserpin within the cell. Statins are competitive inhibitors of HMGCR, a rate limiting enzyme for cholesterol levels. They are widely used as plasma cholesterol lowering drugs in dyslipidemic patients. In spite of the clear effect of statins in lowering morbidity and mortality, in particular in cardiovascular events, many recent studies suggest that prolonged use of statins in Alzheimer’s disease patients leads to mixed outcomes and could contribute to statin-adverse effects35,36,37,38,39. Whereas current guidelines encourage an aggressive use of statins to achieve long-term cholesterol lowering in order to decrease cardiovascular events, the resulting effect on cell membranes is unknown. Here, we focus our experimental analysis on non-mutated (wild type) neuroserpin and directly observe a dramatic increase in aggregates when cells are chonically exposed to statins. To confirm the direct involvement in aggregation of lower cholesterol levels, we use betulin, a pentacyclic triterpene commonly isolated from the birch bark, that specifically inhibits the maturation of SREBPs by enhancing interaction between SCAP and INSIG thus promoting SREBPs retention in the ER40. Also in this case, we observe lower cholesterol and more neuroserpin aggregation. Our findings are consistent with a cholesterol induced decrease in the protein clearance rate, leading to faster aggregation in our model20. To elucidate the biophysical mechanism underlying this observation, we study a mathematical model of coated vesicle release from lipid membranes, illustrating the crucial role of cholesterol for vesicle formation and therefore the impact of its unbalance on intracellular trafficking. Taken together, our results suggest that long-term treatment with statins may affect intracellular trafficking in a way to enhance protein aggregation.
Results
Prolonged treatment with statins enhances neuroserpin aggregation in HeLa cells
Simvastatin (SIM), pravastatin (PRA) and rosuvastatin are all known to be inhibitors of cholesterol synthesis by acting on HMGCR. PRA is, however, reported to be less effective both in patients and in vitro due to its hydrophylicity41. We test the effect of long-term (i.e. up to 8 days) treatment with statins (5 μM SIM or 10 μM PRA or 0.1 μM rosuvastatin or 1 μM rosuvastatin) on the capability of neuroserpin to aggregate in HeLa cells. Figure 1a shows that, in native gel, the treatment with SIM is able to induce multiple bands of neuroserpin, suggesting the presence of aggregates. We obtain similar results for the more powerful drug rosuvastatin, as reported in Fig. 1b 42. In contrast, the exposure to PRA does not affect neuroserpin aggregation, as shown in Fig. 1a. To directly confirm the presence of neuroserpin aggregates after prolonged treatment with statins, we use the Duolink in Situ staining. Using this technique allows the detection of a fluorescent spot if at least two monomers of neuroserpin protein are in close proximity, such as when they form a dimer. We mix (1:1) HeLa with HeLa-GFP cells to confirm that possible aggregation is independent from the specific cell line used. Moreover, in HeLa—GFP cells the cytoplasm is green. We then treat the cells with statins for up to 8 days. Long-time treatment (8 days) with 5 μM SIM or 0.1 μM rosuvastatin induces neuroserpin aggregation while shorter incubation times with 5 μM SIM or 8 days of treatment with 10 μM PRA do not induce any effect, as shown in Fig. 2. Moreover, 8 days of treatment with 0.1 μM rosuvastatin gives a stronger effect with respect to 5 μM SIM Figs 2 and 3.

(a) 100 μg protein of HeLa cells or treated with 10 μM PRA or 5 μM SIM or 0.1 μM rosuvastatin or 1 μM rosuvastatin or 20 μM betulin were loaded on 10% native gel and transferred on PVDF membrane. The sheet was incubated with anti-neuroserpin (ab32901, Abcam, 1:500) overnight at 4 °C and then with the secondary antibody anti goat-HRP (1:5000, ECL Blotting reagents (GE Healthcare RPN2109)/SuperSignal West Femto Maximum Sensitivity Substrate, Thermo scientific) for 1 h at room temperature. Ponceau S solution (P7170, Sigma) was used to verify the correct loading of the samples. (b) Effect of statins or betulin treatments on the level of expression of neuroserpin or HMGCR. 10 μg total protein were loaded on 10% polyacrilamide gel, transferred on PVDF and incubated with anti-neuroserpin (ab32901, AbCAM, 1:500) or with anti-HMGCR (1:1000, MAB5374, Millipore) overnight at 4 °C). Vinculin (1:10000, Sigma) was used as housekeeping.

(a) Hela-GFP and Hela cells were mixed 1:1 and treated with 5 μM SIM or 10 μM PRA up to 8 days, fixed in 3.7% paraformaldeide and incubated with the primary antibody anti-neuroserpin (Ab32901, Abcam) coniugated PLA probes (1:50), overnight at 4 °C. The presence of aggregates are visualized using Duolink in Situ staining according to the manufacturer’s instructions. Nuclei are stained with DAPI. Images are acquired by Leica SP2 laser scanning confocal microscope. Each red spot represents a neuroserpin aggregate. (b) The number of spots per cell for each condition reported in panel a is quantified as described in the methods section.

(a) Hela-GFP and Hela cells were mixed 1:1, treated with 0.1 μM rosuvastatin or 20 μM betulin for 8days, fixed in 3.7% paraformaldeide and incubated with the primary antibody anti-neuroserpin (Ab32901, Abcam) coniugated PLA probes (1:50), overnight at 4 °C. The presence of aggregates are visualized using Duolink in Situ staining according to the manufacturer’s instructions. Nuclei are stained with DAPI. Images are acquired by Leica SP2 laser scanning confocal microscope. Each red spot represents a neuroserpin aggregate. The number of spots per cell for each condition in the images showed in this panel is quantified as described in the methods section. (b) Untreated or treated cells with 0.1 μM rosuvastatin or 20 μM betulin (8 days) were fixed with 3.7% paraformaldeide and then incubated with TNM-AMCA (1 μM) to stain cholesterol for 1 h at room temperature. Images were acquired by Leica SP2 laser scanning confocal microscope.
Effect of the treatment with SIM, rosuvastatin or PRA on cholesterol and HMGCR levels in HeLa cells
Filipin is commonly used to detect cholesterol, but it is not very stable, rapidly photobleaches and results in acute toxicity for living cells43. To avoid these problems, we use a fluorescently labeled theonellamide (TNM-AMCA). TNM-AMCA was originally isolated from a marine sponge, and has been reported to bind in a specific manner to the 3β-hydroxyl group of cholesterol without acute toxicity44,45,46. 5 μM SIM, 0.1 μM rosuvastatin or 20 μM betulin affect the level of cholesterol after 8 days of treatment (Figs 3 and 4), while PRA is less effective and does not change significantly the level of cholesterol41 (Fig. 4). Finally we investigate by western blot whether the treatment with statins affects the expression level of the key enzyme of cholesterol synthesis, HMGCR. As shown in Fig. 1c, no significant changes in the level of expression of this enzyme occurs after long-time treatment either with SIM or PRA.

Untreated or 5 μM SIM or 10 μM PRA treated HeLa cells for the time reported in the figure, were fixed with 3.7% paraformaldeide and then incubated with TNM-AMCA (1 μM) to stain cholesterol for 1 h at room temperature. Images were acquired by Leica SP2 laser scanning confocal microscope.
Effect of the treatment with SIM, rosuvastatin or PRA on the level of expression of SREBP1 and SREBP2
SREBPs are transmembrane proteins expressed at the level of ER, associated with SCAP which is a cholesterol sensor31. Intracellular trafficking of SREBPs is sensitive to cholesterol depletion: when the level of cholesterol is reduced, SREBP leaves the ER to reach the Golgi where it is cleaved. Then, the mature processed form of SREBP is released in the cytosol and translocates into the nucleus where it modulates the expression of several genes controlling cholesterol and fatty acid homeostasis33 and SREBP1/2 itself. Furthermore, it is known that SREBP1 regulates fatty acid synthesis and SREBP2 cholesterol synthesis and uptake34. We thus investigate by immunofluorescence the effect of statins on the level of expression of SREBP1 and SREBP2. Figure 5 shows that SREBP1 is not affected by the either SIM or PRA treatments and SREBP2 increases significantly after 8 days of treatment with SIM. Rosuvastatin, in contrast, affects mainly SREBP1 (Fig. 6a).

Subconfluent cells are fixed with 3.7% paraformaldeide after 8 days treatment with statins and incubated with anti-SREBP1 (1:100, ab28481) or anti SREBP-2 (1 μg/ml ab30682, Abcam) overnight at 4 °C. Next, the cells are incubated with anti-rabbit Alexa Fluo488 for 1 h at room temperature. Actin is stained with 1 μg/ml Actistain-555 phalloidin for 1 h at room temperature. Nuclei are stained with DAPI. Untreated cells are also shown for reference. Images are acquired by Leica SP2 laser scanning confocal microscope.

(a) Level of expression of SREBP1 and SREBP2 after rosuvastatin or betulin treatment in Hela cells. Subconfluent cells a fixed with 3.7% paraforldeide after 8 days of rosovastatin (0.1 μM) or betulin (20 μM) treatment and incubated with anti-SREBP1 (1:100, ab28481) or anti SREBP-2 (1 μg/ml ab30682, Abcam) overnight at 4 °C. Then, the cells are incubated with anti-rabbit AlexaFluo488 for 1 h at room temperature. Nuclei are stained with DAPI. Untreated cells are also shown for reference. Images are acquired by Leica SP2 laser scanning confocal microscope. (b) Western blot of SREBP1 and SREBP2 in Hela cells untreated and after 8 days of treatment with (20 μM betulin. 10 μg total protein were loaded on 10% polyacrilamide gel, transferred on PVDF and incubated or anti-SREPB1 (1:500, ab28481, AbCam) or anti-SREBP2 (1:100, ab30682, AbCam) were used overnight at 4 °C. Mouse anti-alpha Tubulin antibody (1:5000, Sigma) for 1 h at room temperature was used as housekeeping. A secondary antibody anti goat-HRP (1:5000, ECL Blotting reagents (GE Healthcare RPN2109)/SuperSignalTM West Femto Maximum Sensitivity Substrate, Thermo scientific) was used for 1 h at room temperature to detect chemiluminiscence. The bands reported are the precursor of SREBP1 and SREBP2, pre-SREBP.
Effect of the treatment with betulin on the level of expression of cholesterol, neuroserpin aggregation, SREBP1 and SREBP2
An alternative route to interfere with the cholesterol level and study its effect on neuroserpin aggregation is to inhibit SREBPs activity using betulin, a small molecule that is known to inhibit the maturation of SREBP by inducing interaction of SCAP and Insig40,47. Betulin induces a decrease of cholesterol after 8 days of treatment (Fig. 3)40 and an accumulation of the precursor of SREBPs (pre-SREBP), in agreement with refs 48,49 (Fig. 6b). Under these conditions, we show that the treatment for 8 days with 20 μM betulin induces neuroserpin aggregation (Figs 1 and 3a).
Computational simulations reveal aggregation induced by the impairment of intracellular trafficking
In a previous paper our group developed a model three dimensional to explain the critical role for protein aggregation of the non-equilibrium conditions in the ER20. Here we implement the model in order to understand how the impairment of intracellular trafficking might induce protein aggregation. The model performs coarse—grained simulations of diffusing monomers and polymers in a closed system, representing the ER, under a constant rate of protein production kin. Proteins exit the ER with a rate kout when they are close to the boundaries simulating vesicular trafficking. Hence, the parameter kout encapsulates the efficiency of intracellular trafficking which can be affected by the level of cholesterol in the cell. Figure 7a shows that reducing kout, which should simulate an impairment in the intracellular trafficking leads to an increase in protein aggregation, thus recapitulating in silico the experimental observations.

(a) The main figure shows the average number of aggregates observed at a fixed time as a function of kout/kin, the ratio between rate of exit from the ER by means of vesicular transport and the rate of protein production. A reduced value of kout leads to the formation of aggregates. The inset shows the time dependence of the average number of aggregates for different values of kout. (b) The phase diagram of the vesicle formation model. The stiffness ratio between the membrane and the external scaffold Km/Ks as a function of the intensity of the mutual interaction with respect to the scaffold rigidity εLJ/Ks. For soft membranes there is almost always vesicle formation (yellow zone) or the breaking of the coat-membrane interface, if binding is not strong enough (white zone). Increasing the membrane stiffness leads to a new phase appears where the coat-membrane interface is stable but vesicle can not form (grey zone). Typical configurations in the different phases are reported on the right-hand side of the panel.
Mathematical model illustrates mechanical regulation of vesicle formation
The mechanism by which the level of cholesterol determines the value of kout is related to its role in affecting the biophysical properties of membranes21,22,23,24,25 and thus their ability to form the vesicles needed for intracellular trafficking26,27,28. Vesicle formation in the ER is assisted by the Coat protein complex II (COPII) that is recruited on the surface of the ER membrane50. The COPII complex self-assembles into a cage structure that encapsulate the lipid membrane forming a vesicle51. The success of the process crucially depends on the binding strength of the COPII to the membrane and on its bending stiffness, as we can illustrate by a simple mathematical model. We considers an elastic membrane coupled to a curvature induced coat, representing the action of a COPII cage in the ER. The model, described in details in the methods section includes two key dimensionless parameters: (i) the ratio between the bending stiffness of the membrane Km and the one of the coating scaffold Ks, and (ii) the ratio between the scaffold bending energy and its interaction energy with the membrane εLJ. Simulations show that vesicle can only form is Km/Ks is small and εLJ/Ks is sufficiently large. For larger values of Km/Ks, the coating scaffold is not able to bend the membrane into a vesicle while if εLJ/Ks is small the coat detaches from the membrane. This behaviour is summarized in the phase diagram reported in Fig. 7b. A reduction in the level of cholesterol in the ER membrane affects both Km21,22,23 and εLJ29, thus displacing the system in different regions of the phase diagram. In particular, experiments show that cholesterol depletion leads to a reduced recruitment of the Sec23p component of the COPII complex on the ER membrane, delaying ER-to-Golgi transport29.
Discussion
Long-term lowering serum low density lipoprotein cholesterol with statin drugs is used extensively and has proven very effective to reduce the incidence of cardiovascular events. De-novo cholesterol which is the target of statin therapy is found in all membranes and lipid based bodies, contributing to vesicle formation and migration, as well as other membrane functions52. We can therefore expect that regulating the biosynthesis of cholesterol could change the form and function of every membrane within the cell. For this reason, statin therapies could also cause potential harm53. The role of ER in the aggregation of misfolded proteins, such as neuroserpin, and therefore as a crucial determinant of cellular toxicity, is important in many neurodegenerative diseases. Our group recently demonstrated the presence of a phase transition towards rapid protein aggregation due to a breakdown of homeostasis in intracellular organelles, such as the ER, controlled by the rates of protein synthesis and clearance, kin and kout, respectively20. The open question stated in the conclusions of our earlier paper was related to the identification of possible biological processes and factors that would tune these key parameters into an aggregation-prone pathological phase20. In the present paper, we combine experiments and computational models to investigate this critical issue.
We focus our attention on cholesterol depletion by statins as a potential inhibitor protein clearance from the ER. We investigate three different statins in a chonic treatement (8 days): SIM, rosuvastatin and PRA. Rosuvastatin has been shown to be more effective than SIM42, while PRA is less effective when compared with SIM41. Here we show indeed that long-term treatment with SIM or rosuvastatin decreases the level of cholesterol, increases the level of expression of SREBP, compensating for cholesterol depletion, and finally enhances neuroserpin aggregation even in the absence of specific mutations. In contrast, PRA is less effective than the others two statins and therefore does not affect neither the level of cholesterol nor neuroserpin aggregation41.
To ensure that the aggregation we observe for neuroserpin is really due to the lowering of cholesterol in the cell rather to a direct interaction between statins and neuroserpin, we interfere with the cholesterol level indirectly by targeting SREBP through betulin, whose action ultimately leads to cholesterol depletion40. We found that betulin decreases the level of cholesterol and accumulates pre-SREBPs40,48,49. Also in this case, we find an enhancement of neuroserpin aggregation after long term treatment (8 days).
It is well known that N-glycosylation plays determinant roles in protein folding and trafficking, and N-glycosylated proteins are especially important in regulating extracellular activities. Recent papers show that aberrant N-glycosilation happens frequently in relation to human diseases, including Alzheimer’s54 and FENIB, as pointed out in a recent paper55. Furthermore, a systematic and quantitative analysis of surface proteins was carried out in HepG2 liver cells treated with statin showing that many glycosilation sites are downregulated compared to untreated cells56. On the other hand, statins inhibit the pathway of dilichol, which plays a critical role in protein N-glycosilation and acts as a membrane anchor for the formation of a precursor oligosaccharide57. Therefore, the long-treatment with statins could lead to important impairment of intracellular trafficking, effectively decreasing the clearance rate kout20. This is shown directly by numerical simulations of a protein aggregation model where reducing kout leads to faster aggregation.
The role of cholesterol on the biophysical properties of lipid membranes has been the subject of extensive experimental investigation. It is known that the level of cholesterol affects the membrane bending stiffness Km21,22,23 the dynamics of membrane proteins24,25 and has thus an effect on vesicle formation and trafficking26,27,28. In particular, experiments show that the attachment of coating proteins, such as COPII, to membranes is dependendent on cholesterol29. Here, we develop simple mathematical model that can incorporate into a single framework all this experimental observations and help predict the effect of statins on the intracellular trafficking. While the model is too simple to provide a quantitative explanation of vesicle formation in dependence on the cholesterol level, the phase diagram illustrates the possibility that small changes of key membrane biophysical parameters such as the bending stiffness or the binding affinity of coat proteins, all known to be affected by cholesterol, result in a net impairment of vesicle formation.
Recent results in the literature show an increased level of neuroserpin in Alzheimer’s disease patients, the critical role of tPA and the association between neuroserpin and Aβ plaques in Alzheimer’s brain tissues58. Our paper shows that neurosperpin aggregation can be modulated by environmental factors affecting intracellular trafficking, even in the absence of deleterious mutations that are known to induce aggregation1,2,5,6. The aggregation mechanism we propose is fairly general, depending only on the modification of kinetic rates for protein clearance and it could therefore be relevant also for other neurodenerative pathologies. For instance, it would be extremely interesting to clarify the possible role of statins in Aβ aggregation and more generally on the development of Alzheimer’s disease.
Methods
Cell lines and treatments
Human Hela cells (ATCC-CRL-2) or HeLa-GFP cells (AKR-213 Cell Biolabs Inc.) were cultured in 80%DMEM, 10% FBS with the addition of 1% antibiotics and 1% L-glutamine at 37 °C/5% CO2. SIM (S-6196, Sigma) needs to be activated by opening of the lactone ring before use in cell culture. We used the protocol described by59. Briefly, 40 mg/ml of SIM were dissolved in 100% ethanol with subsequent addition of 0.1 N NaOH59. The solution was heated at 50 °C for 2 h and then neutralized with HCl to pH 7.2. The resulting solution was bought to a final volume (1 ml) with distilled water and aliquots were stored at −80 °C for no more then 3 months. Rosuvastatin (SML1264, Sigma) was dissolved in DMSO stock solution (20 mM) and stored in aliquots at −20 °C. Pravastatin (P4498 Sigma) was dissolved in distilled water and stored in aliquots at −80 °C. Betulin (B9757, Sigma) was dissolved in 100% etanol (500 μM) and stored in aliquots at −20 °C. The medium has been replaced every 4 days with fresh medium containing statins or betulin or fresh medium for untreated cells.
Immunofluorescence
Subconfluent cells were fixed with 3.7% paraformaldeide, incubated with 0.1%Triton X-100 in PBS for 15 min at room temperature, then with 3%BSA in PBS for 30 min at room temperature and finally with anti-SREBP-1 (1:100, ab28481 Abcam. 3.7% PFA fixed cells were incubated with 1% BSA/10% goat serum/0.3 M glycine/0.1% tween in PBS for 1 h at room temperature and incubated with anti-rabbit (1:250, Alexa Fluor 488, ab 150077) for 1 h at room temperature. 1 μg/ml Actinstain-555 phalloidin (Cytoskeleton Inc) is used to stain actin for 1 h at room temperature. The nuclei were counterstained with DAPI and the slides mounted with Pro-long anti fade reagent (Life technologies). The images were acquired with a Leica TCS NT confocal microscope.
Duolink assay
Subconfluent cells were fixed with 4% paraformaldeide and incubated with 0.1%Triton X-100 in PBS for 15 min at room temperature, then with 5%BSA in PBS for 1 h at room temperature and finally incubated with anti-neuroserpin (ab32901 AbCam) coniugated PLA probes (1:50) overnight at 4 °C. We detected neuroserpin aggregates with Duolink In Situ staining. The Duolink In Situ staining are based on in situ PLA, which is a proximity ligation assay technology. A pair of oligonucleotide labeled secondary antibodies (PLA probes) generates a signal only when the two PLA probes have bound in close proximity (at a distance of less than 40 nm), either to the same primary antibody or to two primary antibodies bound to the sample in close proximity. The signal from each detected pair of PLA probes is visualized as an individual fluorescent spot. Duolink In Situ Probemaker PLUS enables quick and convenient conjugation of the PLA PLUS oligonucleotide arm directly to the primary antibody (DUO92009 and DUO92010 for PLUS and MINUS probe). The cross reacted proteins were detected according to the manufacturer’s instruction (sigma.com/duolin, section 7.3 Detection protocol). The nuclei were mounted with Duolink in situ mounting medium containing DAPI (DUO82040, Sigma). The images were acquired with a Leica TCS NT confocal microscope. These PLA signals were quantified as described in the section spot detection.
Cholesterol staining
Cells were fixed with 4% paraformaldehyde (PFA) in PB 0.1 M for 10 minutes at room temperature (RT). Then, they were incubated with 50 μg/ml Digitonin for 5 minutes and then for 1 hour with 4% Bovine Serum Albumin (BSA) in PBS. The cells were then incubated with TNM-AMCA (1 μM, gently gifted by Shinichi Nishimura and Minoru Yoshida, RIKEN Center for Sustainable Resource Science, Saitama, Japan) for 1 hour. All the specimens were mounted with Fluoromount Aqueous Mounting Medium without DAPI (Sigma-F4680) and the images were acquired with Leica SP2 laser scanning confocal microscope.
Western Blot
Confluent cells were lysed by boiling in a modified Laemmli sample buffer (2% SDS, 20% glycerol, and 125 mM Tris-HCl, pH6.8). The protein concentration was measured by BCA Protein Assay Kit (Thermo Scientific). Equal amount of proteins were loaded on gel and separated by SDS-PAGE and transferred to a PVDF membrane (Whatman). After blocking, primary and HRP-linked secondary antibodies, specific bindings were detected by chemiluminescence system (GE Healthcare). Goat polyclonal antibody anti-neuroserpin (1:500, ab32901, AbCAM) or rabbit anti-HMGCR (1:1000, MAB5374, Millipore) or anti-SREPB1 (1:500, ab28481, AbCam) or anti-SREBP2 (1:100, ab30682, AbCam) were used overnight at 4 °C. Mouse anti-vinculin (1:10000, V9264, Sigma) or anti-alpha Tubulin antibody (1:5000, Sigma) for 1 h at room temperature was used as housekeeping. A secondary antibody anti goat-HRP (1:5000, ECL Blotting reagents (GE Healthcare RPN2109)/SuperSignalTM West Femto Maximum Sensitivity Substrate, Thermo scientific) was used for 1 h at room temperature to detect chemiluminiscence.
For non denaturating gels, cells were resuspended in 50 mM Tris-HCl, pH7.4 containing 5 mM EDTA, 1% Triton-X100 and Protease inhibitors cocktail (1 ml/100 ml lysate, P8340, Sigma), passed for 4 times through an insulin syringe and maintained in ice for 20 min. After a brief centrifugation, the protein concentration was measured by BCA Protein Assay Kit (Thermo Scientific). 100 μg protein were loaded on 10% non denaturating gel, transferred on PVDF membrane as described above. The PVDF membrane was reversible stained with Ponceau S solution (P7170, Sigma) to verify the correct loading of the samples.
Spot detection
Image analysis of Duolink In Situ staining was performed using the bioimage informatics platform Icy60. To detect spots, we first use the Thresholder plugin to eliminate the random noise in the images. We used the automated mode so that the algorithm automatically calculates the optimal threshold value. We then applied the Spot Detector plugin61 that is based on an UnDecimated Wavelet Transform tool designed to detect spots. Then, we used the default values, enabling the scale = 2 setup with a threshold equal to 100 and a size filter to accept only spots with a size bigger than three pixels. We also extracted the number of cell nuclei and compute the ratio between the number of spots and the number of cells.
Statistical Analysis
Statistical significance analysis is performed using the Kolmogorov-Smirnov test.
Polymer aggregation model
Simulations are performed using the protein aggregation model described in ref. 20 where individual protein molecules sit on a three dimensional cubic lattice. Monomers diffuse with rate kD and attach to neighboring monomers or polymer endpoints with rate kH. Polymers move collectively by reptation with a length-dependent rate kR/i2, where i is the number of monomers in the polymer, and locally by end rotations, with rate kE and kink motions with rate kK. A polymer can attach to another polymer with rate kH if their endpoints meet, and can fragment by breaking an internal bond with rate kf. Inspired by experimental results on neuroserpin polymerization62, we allow for polymerization after at least one of the monomers has been activated with rate kA. Active monomers can also become latent with rate kL and after that they do not aggregate. We consider a system of size (L×L×H) with L = 100L0 and H = 25L0, where L0 is the linear size of a monomer, with periodic boundary conditions along x and y and closed boundary conditions along z. Monomers enter the system from both closed boundaries with rate kin and monomers and polymers can exit from the same boundaries with rate kout/i3. Numerical simulations are performed using Gillespie Montecarlo algorithm63. We measure time in units of 1/kin and set  , kL = 0,
, kL = 0,  ,
,  and vary kout.
and vary kout.
Vesicle formation model
We consider a simple model with three basic interaction energies: two bending energies related, respectively, to the elasticity of the membrane and external cage, and a potential representing their mutual interaction. The membrane is modeled by a polygon with Nm nodes connected by inextensible bonds and resisting bending through an angular spring of energy

where the parameter θ0 = θm = 0 is the equilibrium value of the angle between two bonds, and K = Km is the bending stiffness of the membrane. The coating scaffold is modeled, instead, as an open polygon with Ns nodes, rigid bonds and a bending energy given by eq. 1 with K = Ks and θ0 = θs ≥ 0. In order to simulate a realistic structure, we chose Nm/Ns = 10, corresponding to the ratio between the typical lipid distance in a biological membrane and the characteristic length of the COPII proteins in a cage51. For the membrane-scaffold interaction we employ a Lennard-Jones potential where the depth of the potential well is equal to εLJ. Starting from a pseudo-circular equilibrium configuration, where the scaffold does not perturb the membrane, we simulate the vesicle formation by changing gradually the equilibrium angle of the scaffold θs. The simulations are performed using the LAMMPS molecular dynamics simulator package64 with a constant NVE integration and the addition of a viscous damping force to reduce oscillations.
Additional Information
How to cite this article: Giampietro, C. et al. Cholesterol impairment contributes to neuroserpin aggregation. Sci. Rep. 7, 43669; doi: 10.1038/srep43669 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Miranda, E., Römisch, K. & Lomas, D. A. Mutants of neuroserpin that cause dementia accumulate as polymers within the endoplasmic reticulum. J Biol Chem 279, 28283–91 (2004).
Miranda, E. et al. The intracellular accumulation of polymeric neuroserpin explains the severity of the dementia fenib. Hum Mol Genet 17, 1527–39 (2008).
Roussel, B. D. et al. Unravelling the twists and turns of the serpinopathies. FEBS J 278, 3859–67 (2011).
Yepes, M. et al. Neuroserpin reduces cerebral infarct volume and protects neurons from ischemia-induced apoptosis. Blood 96, 569–76 (2000).
Belorgey, D., Crowther, D. C., Mahadeva, R. & Lomas, D. A. Mutant neuroserpin (S49P) that causes familial encephalopathy with neuroserpin inclusion bodies is a poor proteinase inhibitor and readily forms polymers in vitro . J Biol Chem 277, 17367–73 (2002).
Belorgey, D. et al. Neuroserpin portland (SER52ARG) is trapped as an inactive intermediate that rapidly forms polymers: implications for the epilepsy seen in the dementia FENIB. Eur J Biochem 271, 3360–7 (2004).
Fabbro, S., Schaller, K. & Seeds, N. W. Amyloid-beta levels are significantly reduced and spatial memory defects are rescued in a novel neuroserpin-deficient Alzheimer’s disease transgenic mouse model. Journal of neurochemistry 118, 928–938 (2011).
Subhadra, B., Schaller, K. & Seeds, N. W. Neuroserpin up-regulation in the Alzheimer’s disease brain is associated with elevated thyroid hormone receptor-β1 and hud expression. Neurochemistry international 63, 476–481 (2013).
Lee, T. W., Tsang, V. W. & Birch, N. P. Physiological and pathological roles of tissue plasminogen activator and its inhibitor neuroserpin in the nervous system. Frontiers in cellular neuroscience 9 (2015).
Jones, L., Harold, D. & Williams, J. Genetic evidence for the involvement of lipid metabolism in Alzheimer’s disease. Biochim Biophys Acta 1801, 754–61 (2010).
Pierrot, N. et al. Amyloid precursor protein controls cholesterol turnover needed for neuronal activity. EMBO Mol Med 5, 608–25 (2013).
Montag, J. et al. A genome-wide survey for prion-regulated miRNAs associated with cholesterol homeostasis. BMC Genomics 13, 486 (2012).
Roussel, B. D. et al. Sterol metabolism regulates neuroserpin polymer degradation in the absence of the unfolded protein response in the dementia FENIB. Hum Mol Genet 22, 4616–26 (2013).
Nomura, S., Umeda, T., Tomiyama, T. & Mori, H. The E693δ (Osaka) mutation in amyloid precursor protein potentiates cholesterol-mediated intracellular amyloid β toxicity via its impaired cholesterol efflux. J Neurosci Res 91, 1541–50 (2013).
Fassbender, K. et al. Simvastatin strongly reduces levels of Alzheimer’s disease beta -amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo . Proc Natl Acad Sci USA 98, 5856–61 (2001).
Grimm, M. O. W. et al. Independent inhibition of Alzheimer disease beta- and gamma-secretase cleavage by lowered cholesterol levels. J Biol Chem 283, 11302–11 (2008).
Refolo, L. M. et al. A cholesterol-lowering drug reduces beta-amyloid pathology in a transgenic mouse model of Alzheimer’s disease. Neurobiol Dis 8, 890–9 (2001).
Yao, Z.-X. & Papadopoulos, V. Function of beta-amyloid in cholesterol transport: a lead to neurotoxicity. FASEB J 16, 1677–9 (2002).
Green, R. L., Lagacé, R. E., Oldroyd, N. J., Hennessy, L. K. & Mulero, J. J. Developmental validation of the AMPSTR NGM select PCR amplification kit: A next-generation str multiplex with the SE33 locus. Forensic Sci Int Genet 7, 41–51 (2013).
Budrikis, Z., Costantini, G., La Porta, C. A. M. & Zapperi, S. Protein accumulation in the endoplasmic reticulum as a non-equilibrium phase transition. Nat Commun 5, 3620 (2014).
Song, J. & Waugh, R. E. Bending rigidity of sopc membranes containing cholesterol. Biophysical journal 64, 1967 (1993).
Needham, D. & Nunn, R. S. Elastic deformation and failure of lipid bilayer membranes containing cholesterol. Biophysical journal 58, 997 (1990).
Dimova, R. Recent developments in the field of bending rigidity measurements on membranes. Advances in colloid and interface science 208, 225–234 (2014).
Espenshade, P. J., Li, W.-P. & Yabe, D. Sterols block binding of COPII proteins to SCAP, thereby controlling SCAP sorting in ER. Proceedings of the National Academy of Sciences 99, 11694–11699 (2002).
Armstrong, C. L. et al. Effect of cholesterol on the lateral nanoscale dynamics of fluid membranes. European Biophysics Journal 41, 901–913 (2012).
Wang, Y., Thiele, C. & Huttner, W. B. Cholesterol is required for the formation of regulated and constitutive secretory vesicles from the trans-golgi network. Traffic 1, 952–962 (2000).
Zhang, J., Xue, R., Ong, W.-Y. & Chen, P. Roles of cholesterol in vesicle fusion and motion. Biophys J 97, 1371–80 (2009).
Ridsdale, A. et al. Cholesterol is required for efficient endoplasmic reticulum-to-Golgi transport of secretory membrane proteins. Molecular biology of the cell 17, 1593–1605 (2006).
Runz, H., Miura, K., Weiss, M. & Pepperkok, R. Sterols regulate ER-export dynamics of secretory cargo protein TS-O45-G. EMBO J 25, 2953–65 (2006).
Bengoechea-Alonso, M. T. & Ericsson, J. SREBP in signal transduction: cholesterol metabolism and beyond. Curr Opin Cell Biol 19, 215–22 (2007).
Feramisco, J. D. et al. Intramembrane aspartic acid in SCAP protein governs cholesterol-induced conformational change. Proc Natl Acad Sci USA 102, 3242–7 (2005).
Brown, M. S. & Goldstein, J. L. A proteolytic pathway that controls the cholesterol content of membranes, cells, and blood. Proc Natl Acad Sci USA 96, 11041–8 (1999).
Horton, J. D., Goldstein, J. L. & Brown, M. S. SREBPs: transcriptional mediators of lipid homeostasis. Cold Spring Harb Symp Quant Biol 67, 491–8 (2002).
Goldstein, J. L., DeBose-Boyd, R. A. & Brown, M. S. Protein sensors for membrane sterols. Cell 124, 35–46 (2006).
Friedhoff, L. T., Cullen, E. I., Geoghagen, N. S. & Buxbaum, J. D. Treatment with controlled-release lovastatin decreases serum concentrations of human beta-amyloid (Abeta) peptide. Int J Neuropsychopharmacol 4, 127–30 (2001).
Fassbender, K. et al. Effects of statins on human cerebral cholesterol metabolism and secretion of Alzheimer amyloid peptide. Neurology 59, 1257–8 (2002).
Simons, M. et al. Treatment with simvastatin in normocholesterolemic patients with Alzheimer’s disease: A 26-week randomized, placebo-controlled, double-blind trial. Ann Neurol 52, 346–50 (2002).
Vega, G. L. et al. Reduction in levels of 24s-hydroxycholesterol by statin treatment in patients with Alzheimer disease. Arch Neurol 60, 510–5 (2003).
Doraiswamy, P. M., Steffens, D. C. & McQuoid, D. R. Statin use and hippocampal volumes in elderly subjects at risk for Alzheimer’s disease: a pilot observational study. Am J Alzheimers Dis Other Demen 19, 275–8 (2004).
Tang, J.-J. et al. Inhibition of SREBP by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques. Cell Metab 13, 44–56 (2011).
Contermans, J., Smit, J. W., Bär, P. R. & Erkelens, D. W. A comparison of the effects of simvastatin and pravastatin monotherapy on muscle histology and permeability in hypercholesterolaemic patients. Br J Clin Pharmacol 39, 135–41 (1995).
McKenney, J. M. Pharmacologic characteristics of statins. Clinical cardiology 26, 32–38 (2003).
Kotler-Brajtburg, J., Price, H. D., Medoff, G., Schlessinger, D. & Kobayashi, G. S. Molecular basis for the selective toxicity of amphotericin b for yeast and filipin for animal cells. Antimicrob Agents Chemother 5, 377–82 (1974).
Nishimura, S. et al. Marine antifungal theonellamides target 3beta-hydroxysterol to activate RHO1 signaling. Nat Chem Biol 6, 519–26 (2010).
Nishimura, S. et al. Visualization of sterol-rich membrane domains with fluorescently-labeled theonellamides. PLoS One 8, e83716 (2013).
Arita, Y. et al. Targeting cholesterol in a liquid-disordered environment by theonellamides modulates cell membrane order and cell shape. Chem Biol 22, 604–10 (2015).
Soyal, S. M., Nofziger, C., Dossena, S., Paulmichl, M. & Patsch, W. Targeting SREBPs for treatment of the metabolic syndrome. Trends Pharmacol Sci 36, 406–16 (2015).
Krycer, J. R., Phan, L. & Brown, A. J. A key regulator of cholesterol homoeostasis, SREBP-2, can be targeted in prostate cancer cells with natural products. Biochem J 446, 191–201 (2012).
Quan, H. Y. et al. Betulinic acid alleviates non-alcoholic fatty liver by inhibiting SREBP1 activity via the AMPK-mTOR-SREBP signaling pathway. Biochem Pharmacol 85, 1330–40 (2013).
Kuehn, M. J., Herrmann, J. M. & Schekman, R. COPII-cargo interactions direct protein sorting into ER-derived transport vesicles. Nature 391, 187–90 (1998).
Zanetti, G. et al. The structure of the COPII transport-vesicle coat assembled on membranes. eLife 2, e00951 URL https://dx.doi.org/10.7554/eLife.00951 (2013).
Ikonen, E. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol 9, 125–38 (2008).
Kiortsis, D. N., Filippatos, T. D., Mikhailidis, D. P., Elisaf, M. S. & Liberopoulos, E. N. Statin-associated adverse effects beyond muscle and liver toxicity. Atherosclerosis 195, 7–16 (2007).
Wang, J. Z., Grundke-Iqbal, I. & Iqbal, K. Glycosylation of microtubule-associated protein tau: an abnormal posttranslational modification in alzheimer’s disease. Nat Med 2, 871–5 (1996).
Moriconi, C. et al. Interactions between n-linked glycosylation and polymerisation of neuroserpin within the endoplasmic reticulum. FEBS J 282, 4565–79 (2015).
Xiao, H., Tang, G. X. & Wu, R. Site-specific quantification of surface n-glycoproteins in statin-treated liver cells. Anal Chem 88, 3324–32 (2016).
Burda, P. & Aebi, M. The dolichol pathway of n-linked glycosylation. Biochim Biophys Acta 1426, 239–57 (1999).
Fabbro, S. & Seeds, N. W. Plasminogen activator activity is inhibited while neuroserpin is up-regulated in the Alzheimer disease brain. Journal of neurochemistry 109, 303–315 (2009).
Dong, W., Vuletic, S. & Albers, J. J. Differential effects of simvastatin and pravastatin on expression of Alzheimer’s disease-related genes in human astrocytes and neuronal cells. J Lipid Res 50, 2095–102 (2009).
de Chaumont, F. et al. Icy: an open bioimage informatics platform for extended reproducible research. Nat Methods 9, 690–6 (2012).
Olivo-Marin, J.-C. Extraction of spots in biological images using multiscale products. Pattern recognition 35, 1989–1996 (2002).
Noto, R. et al. The tempered polymerization of human neuroserpin. PLoS One 7, e32444 URL http://dx.doi.org/10.1371 (2012).
Gillespie, D. T. A general method for numerically simulating the stochastic time evolution of coupled chemical reactions. Journal of Computational Physics 22, 403–434 URL http://www.sciencedirect.com/science/article/pii/0021999176900413 (1976).
Plimpton, S. Fast parallel algorithms for short-range molecular dynamics. Journal of Computational Physics 117, 1–19 URL http://www.sciencedirect.com/science/article/pii/S002199918571039X (1995).
Acknowledgements
We thank S. Nishimura and M. Yoshida, RIKEN Center for Sustainable Resource Science, Saitama, Japan who kindly provided the cholesterol probe TNM-AMCA; GC and SZ are supported by the ERC advanced grant SIZEFFECTS. SZ acknowledges support from the Academy of Finland FiDiPro progam, project 13282993. CAMLP thanks the visiting fellow program of Aalto Science Institute.
Author information
Authors and Affiliations
Contributions
C.G. carried out the native elecrophoresis, M.C.L. and F.M. carried out immunofluorescences and western blots experiments, G.C. performed numerical simulations and image analysis, S.Z. designed the model and assisted in the writing of the paper. C.A.M.L.P. designed the project, performed and supervised experiments, and wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Giampietro, C., Lionetti, M., Costantini, G. et al. Cholesterol impairment contributes to neuroserpin aggregation. Sci Rep 7, 43669 (2017). https://doi.org/10.1038/srep43669
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep43669
This article is cited by
-
The probable role of tissue plasminogen activator/neuroserpin axis in Alzheimer’s disease: a new perspective
Acta Neurologica Belgica (2024)
-
Neuroserpin, a crucial regulator for axogenesis, synaptic modelling and cell–cell interactions in the pathophysiology of neurological disease
Cellular and Molecular Life Sciences (2022)
-
Marine natural products targeting the eukaryotic cell membrane
The Journal of Antibiotics (2021)
-
Neuroserpin: structure, function, physiology and pathology
Cellular and Molecular Life Sciences (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
