
Abstract
Carbon-based metal-free catalyst has attracted more and more attention. It is a big challenge to improve catalytic activity of metal-free catalyst for decomposition of H2O2 to produce hydroxyl radical (HO•). Here, we report chloro-benquinone (TCBQ) modified on graphene oxide (GO) as metal-free catalyst for strong promotion of HO•. By the incorporation of GO, the HO• production by H2O2 and TCBQ is significantly promoted. Based on density functional theory, TCBQ modified GO (GO-TCBQ) is more prone to be nucleophilic attacked by H2O2 to yield HO• via electron transfer acceleration. Furthermore, the generated HO• can cut GO nanosheets into uniform ultra-small graphene oxide (USGO) through the cleavage of epoxy and C-C bonds. Interestingly, the damaged GO and in situ formed GO fragments can further enhance decomposition of H2O2 to produce HO•. Different from other catalytic processes, the GO-TCBQ metal-free catalysis process can be enhanced by GO itself, producing more HO•, and uniform USGO also can be generated. Thus, the metal free catalysis will be considered a fabrication method for uniform USGO, and may be extended to other fields including detoxifying organic pollutants and the application as disinfectants.
Similar content being viewed by others
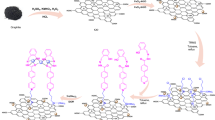
Introduction
Graphene oxide (GO), a highly oxidized form of graphene, has attracted widespread interests due to its unique physical and chemical properties1,2,3. Recently, research on GO-based metal-free catalyst is rapidly increasing4,5,6,7,8. In most cases, metal-based catalysts are hard to remove metal ions from the product, which would have a dominant effect on their further applications9,10. Thus, GO as metal-free catalyst has drawn more and more attention10,11,12.
As known, Fenton reaction is a common method for generating hydroxyl radical (HO•) via decomposition of hydrogen peroxide (H2O2) by metal-based catalysts13,14,15. Interestingly, GO itself can act as catalyst and react with H2O2 due to intrinsic peroxidase catalytic activity16,17,18,19. Sun H. et al. indicated graphene quantum dots (GQDs) could catalyze the decomposition of H2O2, generating HO• to improve the antibacterial performance of H2O2 effectively20. However, peroxidase catalytic activity of GO is usually sensitive to H2O2 with a very low concentration, resulting in the limited catalytic ability during the degradation of organic pollutants15. Therefore, it is a big challenge to improve catalytic activity of GO metal-free catalyst for decomposition of H2O2 to produce HO•.
On the other hand, hydroxyl radical has been reported to oxidize and cut GO into zero dimensional ultra-small graphene oxide (USGO) or GQDs21,22,23,24, which have various potential applications in the fields of optoelectronics25, catalysis26 and biology and medicine27. Now, various methods have been applied for the cleavage of GO into USGO or GQDs. However, most those methods require harsh conditions28,29,30,31 or complicated processes28.
Herein, for the first time, we report chloro-benquinone (TCBQ) modified on GO as metal-free catalyst, with a Janus effect for strong promotion of HO• and formation of USGO with uniform size and shape (Fig. 1). Based on density functional theory (DFT) and metal-free catalysis experiments, GO can effectively accelerate nucleophilic reaction between TCBQ modified GO (GO-TCBQ) and H2O2 to yield HO• due to promotion of electrons transfer from H2O2 to TCBQ. More importantly, the generated HO• can oxidize and cut GO nanosheets into USGO with uniform size and shape. Interestingly, the GO fragments itself can further enhance catalytic decomposition of H2O2 to produce more HO•.

(TCBQ modified on GO as metal-free catalyst is more prone to be nucleophilic attacked by H2O2 to yield HO• via electron transfer acceleration; on the other hand, the generated HO• can cut GO nanosheets into uniform USGO. GO fragments also further enhance decomposition of H2O2 to produce more HO•).
Results and Discussion
Zhu et al.32,33 demonstrated that H2O2 as a nucleophile could nucleophilic attack TCBQ, forming a trichlorohydroperoxyl-1,4-benzoquinone intermediate, which decomposes homolytically to produce HO•. Previous study also indicated that GO possesses intrinsic peroxidase-like activity to catalyze the reduction of H2O216,20. In this study, we used TCBQ modified on the GO surface as metal-free catalyst to enhance H2O2 decomposition, which was named as GO-TCBQ. Some Details about characterization and optimization of GO-TCBQ were shown in Supplementary Material (Fig. S1 and S2).
Figure 2 shows the production of hydroxyl radical in the metal-free catalysis system. GO-TCBQ can significant enhance the decomposition of H2O2 to generate hydroxyl radical. As illustrated in the Fig. 2a, after dosing GO-TCBQ in the H2O2 solution, hydroxyl radical can be detected by electron spin resonance (ESR). As there was no metal catalyst in the system, hydroxyl radical is related to the interaction between GO-TCBQ and H2O2. Figure 2b also shows the quantities of hydroxyl radical in different H2O2 system. It is consistent with previously reported that TCBQ can decompose H2O2 to produce HO•32,34. In compared with TCBQ and GO, the production of hydroxyl radical by GO-TCBQ after 2 h reaction is enhanced 3 times and 13 times higher, respectively. Due to the non-selectivity of hydroxyl radical to electron-rich organic pollutants, metal-free catalyst GO-TCBQ accelerates the removal of organic pollutants with the presence of H2O2 as Fig. 2c indicated. Only GO does little effect on the decomposition of H2O2, so the removal of phenol in the GO-H2O2 system is almost the same as only H2O2 system. Furthermore, the production of hydroxyl radical by GO-TCBQ metal-free catalyst keeps increasing with reaction time. The yield of hydroxyl radical increases fast at the beginning, but the increase slows down after 5 hours. After 24 h reaction, the amount of hydroxyl radical shows 2 times higher than radical produced by 2 h reaction.

(GO = 0.15 mg/mL, TCBQ/GO = 0.3, H2O2 = 2 mM, ultrasonic time = 1 h, pH = 7.0). (a) ESR spectrum of GO-TCBQ with H2O2 after 1 min reaction. (b) Quantitative yield of hydroxyl radicals by LC-MS using salicylic acid method in H2O2 system (H2O2 = 2 mM, 2,3-DHBA as the quantitative product by hydroxyl radical). (c) Removal of phenol with reaction time by metal-free catalysts (original phenol concentration = 10 mg/L). (d) Quantitative yield of hydroxyl radicals with reaction time by GO-TCBQ metal-free catalyst (salicylic acid method).
To explore the promotion of hydroxyl radical by GO-TCBQ, related theoretical calculations based on DFT were analyzed. Highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) determine the way the molecule interacts with other species35. Figure 3 and Table 1 indicate the optimized configurations, and the HOMO/LUMO energy levels of the TCBQ, GO GO-TCBQ obtained by DFT calculations. The LUMO energy level of the GO-TCBQ (−4.13 eV) is lower than that of TCBQ (−3.90 eV) suggests that LUMO level becomes lower via TCBQ modified on GO surface (Fig. 3b). Thus, GO-TCBQ is more prone to obtain electron than TCBQ. It is consistent with a previously suggested that lower LUMO level benefited the injection of electron36. Figure 3c shows the nucleophilic reaction between TCBQ and H2O2. The chemical reactivity of molecular systems is associated closely with the energy difference Eg between HOMO and LUMO37,38, which is calculated for TCBQ and GO-TCBQ system. The Eg between the HOMO level of H2O2 and the LUMO level of TCBQ is 2.28 eV (52.4 kcal/mol), indicating electron transfer from the HOMO of H2O2 to the LUMO of TCBQ. The calculated Eg of GO-TCBQ is 2.05 eV (47.2 kcal/mol), which is considerably lower than that of TCBQ differences of Eg between GO-TCBQ and GO can well explain why the production of hydroxyl radical of H2O2 is enhanced by GO-TCBQ. Due to the promotion of electron transfer, GO-TCBQ is more prone to be attacked by H2O2. The Fig. 3a indicates that the LUMO isosurface of GO-TCBQ is very similar with that of TCBQ. That means electron also transfers from H2O2 to TCBQ in the GO-TCBQ/H2O2 system. Thus, GO promotes nucleophilic reaction through accelerating electron transfer from H2O2 to GO-TCBQ.

(a) The HOMO and LUMO density distribution for optimized configurations. (b) Comparison of energy levels and calculated HOMO-LUMO energy gaps for TCBQ and GO-TCBQ reaction system. (c) Schematic illustrations of nucleophilic reaction between TCBQ (GO-TCBQ) and H2O2.
In the GO-TCBQ metal-free system mentioned above, hydroxyl radical can produce gradually through nucleophilic reaction. However, hydroxyl radical as electrophile can oxidize and decompose GO-TCBQ. As shown in Fig. 3a, the HOMO isosurface of GO-TCBQ is very similar with that of GO, indicating electron transport from GO to hydroxyl radical in this system. Thus, GO is more prone to electrophilic attack by hydroxyl radical. So, the morphology changes of GO during metal-free catalysis were conducted by transmission electron microscopy (TEM) and high-resolution transmission electron microscopy (HRTEM). TEM images in Fig. 4a–e indicate a process of USGO generation in the metal-free system of GO-TCBQ and H2O2. Firstly, a single-layer GO (Fig. 4a) gradually changes after 2h-reaction (Fig. 4b). The surface of GO sheet begins to damage, and some small holes can be found on the GO layer. As mentioned above, H2O2 can attack GO-TCBQ to produce in situ hydroxyl radical via nucleophilic reaction. The hydroxyl radical generated from GO could attack GO directly. Thus, when the solution is filtrated after 2h-reacion of metal-free catalysis (Fig. 4c), the change of GO fragments in the filtrate is significant. Most the structure of GO layer is damaged by the in situ hydroxyl radical. The visible holes or defect sites are generated initially by the attack of hydroxyl radical followed by progressive attraction of more hydroxyl radical to destroy the carbon-carbon bond around initial defect sites39. Thus, it is surprising that USGO with uniform size formed and GO layer disappears with the filtrate of 2h-reaction sample after 12h-reaction (Fig. 4d). This could be attributed to the oxidation of hydroxyl radical generated from GO-TCBQ. Then, GO is attacked and decomposed in situ by hydroxyl radical, where TCBQ is modified. Thus, USGO can be generated uniformly. After a longer time reaction (24–48 h), the USGO particles can be further decomposed into smaller ones (Fig. 4e,f). HRTEM spectra in Fig. 4f,h show the USGO particles are uniform, with a size distribution of 2–4 nm. The average diameter of the USGO is 3.61 nm. As Fig. 4g shown, the marked lattice fringe spacing of the USGO is 0.34 nm, corresponding to (002) crystal phase of graphite21. The size of USGO obtained during GO-TCBQ metal-free catalysis is similar that prepared in other metal catalysis system22,23. The particle sizes of USGO gradually decrease with reaction time (Fig. 4i), which is consistent with the production of hydroxyl radical.

(GO = 0.15 mg/mL, TCBQ/GO = 0.3, H2O2 = 2 mM, ultrasonic time = 1 h, pH = 7.0). (a) GO-TCBQ, (b) GO-TCBQ after 2h-reaction, (c) filtrate of GO-TCBQ after 2h-reaction, (d) filtrate of (c) after 12h-reaction, (e) filtrate of (c) after 24h-reaction, (f) filtrate of (c) after 48h-reaction, (g) fringe patterns of individual USGO, (h) particle size distributions of USGO, (i) particle sizes of USGO with reaction time.
In order to examine the chemical changes of GO during metal-free catalysis, we employed X-ray photoelectron spectroscopy (XPS), fourier transform infrared spectroscopy (FTIR), Raman, X-ray diffraction (XRD) and fluorescence spectroscopy to characterize the GO samples with different reaction time. The XPS 1C spectra in Fig. 5a–d indicate the distribution of functional groups on the surface of GO samples. The major peaks at 284.7, 285.3, 286.8 and 288.8 eV are corresponded to aromatics (C=C/C–C), hydroxyl and epoxy (C–O), carbonyl (C=O), and carboxyl (C(O)O)groups, respectively. Table 2 shows the corresponding functional group contents of C1s spectra in XPS analysis. As the reaction time of metal-free catalysis increased, carbonyl groups (peak at 286.8 eV) largely decrease, from 60.3% to 35.4%. However, carboxyl (288.8 eV), hydroxyl and epoxy (285.3 eV) and aromatics (284.7 eV) groups increase. The C-O group increased from 6.04% to 18.0%, and C(O)O group increased from 0.73% to 3.53%. This indicated carbonyl groups were oxidized into C-O and C(O)O groups during metal-free catalysis. The carbonyl groups on the surface of GO-TCBQ may be easily attacked and oxidized by the in situ hydroxyl radical into the USGO containing carboxyl groups. The increase of hydroxyl groups may be due to the replacement by chlorine atom from GO-TCBQ through nuclephilic substitution of TCBQ and H2O2.

(GO = 0.15 mg/mL, TCBQ/GO = 0.3, H2O2 = 2 mM, ultrasonic time = 1 h, pH = 7.0), (a) GO-TCBQ, (b) GO-TCBQ after 2h-reaction, (c) the filtrate of GO-TCBQ after 2h-reaction, (d) the filtrate of (c) after 24h-reaction.
The functional groups of the GO samples also are determined by the FT-IR spectra shown in Fig. 6a. The peaks at 1630 cm−1 is assigned as C=O stretching of carbonyl groups, 1575 cm−1 represents C=C stretching of aromatic rings, 1380 cm−1 is due to C-H deformation of CH2 and CH3 groups, the absorbance at 1105 cm−1 is attributed to O-H deformation of COOH, and the band at 1040 cm−1 is characteristic of aromatic C-O-C stretching of aryl ethers (epoxy groups). FT-IR results clearly show a reduction in carbonyl (1630 cm−1) responses, and an increase in O-H deformation of COOH (1105 cm−1) response. It is consistent with the XPS results mentioned above. The C=O groups of GO-TCBQ may be easily attacked and oxidized by hydroxyl radical. Furthermore, aromatic C-O-C groups (1040 cm−1) disappear from the GO samples after 2h-reaction of metal-free catalysis. It should be noted that epoxy (aromatic C-O-C) groups can serve as chemically reactive sites for the rupture of the underlying C-C bonds. Thus, in situ hydroxyl radical mainly attacks epoxy groups in the GO samples. On the other hand, CH2/CH3 groups (1380 cm−1) in the Fig. 6a increase after 24h-reaction of metal-free catalysis, indicating further decomposition of GO nanosheets by hydroxyl radical through the cleavage of C-C bonds.

(a) FTIR, (b) XRD, (c) Raman and (d) fluorescence spectra (Ex = 335 nm) of different samples (GO = 0.15 mg/mL, TCBQ/GO = 0.3, H2O2 = 2 mM, ultrasonic time = 1 h, pH = 7.0).
The XRD is used to determine the crystal structure of GO and USGO. The Bragg’s equation is applied to evaluate the distance between graphene layers (nλ = 2dsinθ, where λ is the wavelength, d is the distance between crystal planes, θ is the angle of the diffracted wave, and n is an integer known as the order of the diffracted beam). As shown in Fig. 6b, the original GO-TCBQ has a diffraction peak at 10.76°, and the interlayer spacing is 0.8 nm. The XRD peak shifts to a lower degree after 2h-reaction, and further shifts after filtration. The interlayer distances of GO increase to 0.82 nm and 1.32 nm, respectively. This result could be attributed to the oxygen-containing groups introduced GO during metal-free catalysis, which enhances the interlayer spacing. After 24h-reaction, the XRD peak of USGO shifts to 10.79°, and the interlayer spacing of USGO is 0.78 nm, indicating the decrease of interlayer distance. This may be due to carbonyl groups on the surface of GO attacked by hydroxyl radical, which is according with the XPS and FTIR results mentioned above. Other research indicated oxygen-containing functional groups on the surface of GO affected surface characteristics of GO40,41, especially for interlayer spacing of GO42. In this study, the content of oxygen-containing groups decreased from 67.07% to 56.93% during oxidation process. Thus, the content of oxygen-containing groups was consistent with the decrease of interlayer distance.
The major Raman peaks in GO samples are the D and G band at around 1352 cm−1 and 1600 cm−1, respectively (Fig. 6c). The intensity ratio of disorder D and crystalline G band (ID/IG) reflects the degree of defects in the GO. The GO-TCBQ sample is observed with an intensity ratio ID/IG at 0.88, after 2h-reaction the intensity nearly does not change. But the defect intensity increases to 1.00 after filtration, which is an indication of oxygen-containing groups increasing in the GO fragments. Then ID/IG decreases to 0.76 after 24h-reaction, indicates the USGO with a higher sp2 crystalline structure are gradually formed. Due to oxygen-containing groups of GO attacked and cut by hydroxyl radical, oxygen groups gradually move to the edge of USGO, which is similar with XRD results.
Fluorescence spectra are carried to determine the emission of the GO samples at a fixed excitation of 335 nm. As shown in Fig. 6d, both GO and TCBQ samples show weak peaks with an emission of 380 nm. After 2 h metal-free catalysis, the strong fluorescence peak could be observed at an emission of 426 nm. Compared with original GO, the fluorescence intensity of the GO samples increased 7 times after 2h-reaction. After 24 h, USGO shows a strong peak at an emission wavelength of 438 nm. With a red shift of the emission wavelength, the fluorescence intensity also reaches the maximum value, increasing more than 10 times. The fluorescence properties of USGO should be attributed to the uniform nano-size and the surface state of the sp2 clusters. It is consistent with previous studies that the blue emission of GQDs is due to electron hole recombination or quantum size effect (intrinsic state emission)31,43,44. Chemical functionalities and defects could cause red-shift of the emission peaks45. Thus, the reason of red-shift may be attributed to the increase in C-O and C(O)O groups during GO cutting process (Table 2).
Based on the data and analysis presented above, a mechanism of uniform USGO formation during gentle metal-free catalysis is proposed, as detailed in Fig. 1. There is a Janus effect of process for promotion of hydroxyl radical and USGO generation.
On one hand, the production of hydroxyl radical is significantly enhanced in a gentle metal-free catalysis system. The nucleophilic reaction between TCBQ and H2O2 is promoted via TCBQ modified on the GO surface. GO-TCBQ is more prone to be attacked by H2O2 to yield HO• through the acceleration of electron transfer. On the other hand, the GO nanosheets are further cut into uniform USGO through metal-free catalysis. Firstly, the hydroxyl radical in situ produced from the GO-TCBQ can directly oxidize the surface of the GO. As discussed above, epoxy (aromatic C-O-C) and carbonyl groups as chemically reactive sites on the surface of GO is easily attacked by hydroxyl radical into carboxyl groups. Therefore, the structure of GO layer is damaged by the in situ hydroxyl radical through cleavage of epoxy C-O-C bonds. This is supported by TEM results (Fig. 4b), which clearly shows the small holes and damage on the surface of GO sheet after 2h-reaction of metal-free catalysis. Epoxy groups on the plane of GO were oxidized, resulting in the cutting of GO, which is consistent with previously reported results.
Secondly, GO nanosheets are further cut into extra-small sheets also could be enhanced by GO itself. Due to the structural changes of TCBQ, the promotion of hydroxyl radical by TCBQ and H2O2 nucleophilic reaction would slow down after 5 h (Fig. 2d). However, As Fig. 4 shown, the decomposition of GO was still ongoing after 5 h reaction. Some investigations indicated that carboxyl-modified GO is shown to possess intrinsic peroxidase-like activity16. With smaller size, USGO possess higher peroxidase-like activity17 originates from their ability to catalyze the decomposition of H2O2 to generate hydroxyl radical23. In this study, the in situ formed GO fragments might further enhance the decomposition of H2O2 to produce more hydroxyl radical during 24 h or longer metal-free catalysis. Furthermore, it should be noted that a further cutting of GO nanosheets was by cleavage of C-C bonds. During the initial metal-free catalysis, GO is attacked by hydroxyl radical to form holes and defect sites, around where more hydroxyl radical can be progressive attracted to destroy the carbon-carbon bond18,46.
Therefore, GO-TCBQ, as metal-free catalyst, can produce hydroxyl radical with the presence of H2O2, to enhance degradation of organic pollutants. Besides environmental application in detoxifying pollutants, GO-TCBQ also had other application potentials. It has been found that the GO-TCBQ with ability to catalyze the decomposition of H2O2, generating HO•. Thus, GO-TCBQ can be used for H2O2 detection or wound disinfection.
Conclusion
In summary, a Janus effect of gentle metal-free catalysis facilitated production of hydroxyl radical and the fabrication of the uniform USGO. The production of hydroxyl radical is significantly enhanced by GO-TCBQ metal-free catalyst. GO-TCBQ is more prone to be nucleophilic attacked by H2O2 to yield HO• via electron transfer acceleration. On the other hand, the GO sheets are further cut into uniform USGO by HO•. The damaged GO itself also can decompose of H2O2 to produce HO• to cut GO into USGO. Different from other catalytic processes, the metal-free catalysis can be enhanced by GO itself, producing more hydroxyl radical, and uniform USGO also can be generated. The metal-free catalysis will be considered a fabrication method for uniform USGO, and may be extended to other fields including detoxifying organic pollutants and the application as disinfectants.
Methods
Preparation of GO and GO-TCBQ
Graphite oxide was prepared by a modified Hummers method47,48. The obtained graphite oxide powder was then redispersed in water to yield a yellow-brown suspension. 1 mg/mL GO aqueous suspension was dropwise added into TCBQ solution and then sonicated under ambient condition (300 W) for 60 min. The obtained TCBQ modified GO was used to the preparation of USGO. The basic system consisted of 0.2 mM TCBQ, 0.15 mg/mL GO, 2 mM H2O2, in 0.1 M phosphate buffer (pH 7.4) and was magnetic stirred at 25 °C in water bath. After 2 hours, the solution was filtrated by 0.22 μm membrane, and the filtrate reacted continuously until 48 hours. The colloidal solution was further dialyzed in a dialysis bag (retained molecular weight: 3500 Da) for 2 days and USGO were obtained.
Metal-free catalyzed process of GO-TCBQ and H2O2
Metal-free catalyzed process was carried out in a dark reactor. Salicylic acid method was used to quantify the hydroxyl radical generated by hydrogen peroxide with TCBQ modified GO by ESI-TOF-MS methods. Typically, the basic system initially consisted of TCBQ modified GO and salicylic acid. The pH was adjusted to 7.4 by using PBS solution before reaction. After H2O2 was added, the system was magnetic stirred at 25 °C in water bath. The product was filtrated by 0.22 μm membrane filter immediately after the reaction for 2 h. LC-MS spectra were analyzed by UPLC H-Class/xevo G2-S TOF (Waters, USA) with a mass range of m/z 50–10000 and resolution ≥40. The area of the hydroxylation product (DHBA, m/z 153.02) was used to represent the hydroxyl radical yield.
The degradation experiment was carried out at 25 °C in water bath reactor at optimum conditions, containing 100 mL solution with 1 mM of phenol. The samples were taken at certain intervals and analyzed with high performance liquid chromatography (HPLC, Agilent Technologies Series 1200), using a mixture of methanol and water (30/70%, v/v) containing 10 mM/L H3PO4 as the mobile phase. The separation was performed using ZORBAX SB-C18 (3.5 μm, 2.1 × 150 mm) reversed-phase column at 35 °C with a flow rate of 0.25 mL/min. Sample was analyzed by a UV detector with a wavelength of 210 nm.
Analysis method and characterization
In ESR study, the basic system consisted of halogenated quinones dissolved in acetonitrile (5%), H2O2, and the spin-trapping agent 5,5-Dimethyl-1-Pyrroline-N-Oxide (DMPO) (100 mM), in 100 mM phosphate buffer (pH 7.4) at room temperature. ESR spectra were recorded 1 min after the interactions at room temperature under normal room-lighting conditions on a Bruker (Billerica, MA) AE500 spectrometer operating at 9.8 GHz and a cavity equipped with a Bruker Aquax liquid sample cell. Typical spectrometer parameters were scan range, 100 G; field set, 3470 G; time constant, 200 ms; scan time, 100 s.
The morphology structure of the obtained material was characterized by using TEM (JEOLJEM-100CXII, Japan) and HRTEM (JEM-2100F, Japan) at an accelerating voltage of 300 kV. The FTIR spectra of the samples were recorded on a Spectra GX spectrometer (PerkinElmer, USA) operating under the transmittance mode. The FTIR spectra were acquired in the wavenumber range of 400–4000 cm−1 at the resolution of 1 cm−1. The XRD pattern was recorded on PANalytical X-ray diffraction system (Empyrean, Netherlands). XPS data was obtained on an electron spectrometer (ESCALab 250Xi, VG Scientific, Britain) using Al Kα radiation at the power of 300 W. Survey and multi-region spectra were recorded at C1s and O1s photoelectron peaks. The Raman spectra were obtained on a LabRAM HR800 Raman spectrometer (Horiba Jobin Yvon, France) with an excitation wavelength of 514 nm. The fluorescence spectra were recorded using a Hitachi F-4600 fluorescence spectrometer (Hitachi High-Technologies, Tokyo, Japan).
Theoretical calculations details
The calculations were performed on a Intel Xeon E3–1225v5 server with 8 G memory using Gaussian 03 W49 program package. The input geometry of the GO, TCBQ and GO-TCBQ in the ground state was fully optimized at DFT/B3LYP using 6–311 G basis set (Details are shown in SI). All the calculations, HOMO and LUMO energy levels were performed by using GaussView 4.0 molecular visualization program Package50.
Additional Information
How to cite this article: Zhao, H. et al. Chloro-benquinone Modified on Graphene Oxide as Metal-free Catalyst: Strong Promotion of Hydroxyl Radical and Generation of Ultra-Small Graphene Oxide. Sci. Rep. 7, 42643; doi: 10.1038/srep42643 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Dreyer, D. R., Park, S., Bielawski, C. W. & Ruoff, R. S. The chemistry of graphene oxide. Chem. Soc. Rev. 39, 228–240 (2009).
Chen, D., Feng, H. & Li, J. Graphene oxide: preparation, functionalization, and electrochemical applications. Chem. Rev. 112, 6027–6053 (2012).
Luo, B., Liu, S. & Zhi, L. Chemical approaches toward graphene-based nanomaterials and their applications in energy-related areas. Small 8, 630–646 (2012).
Indrawirawan, S., Sun, H., Duan, X. & Wang, S. Low temperature combustion synthesis of nitrogen-doped graphene for metal-free catalytic oxidation. J. Mater. Chem. A 3, 3432–3440 (2015).
Duan, X., O’Donnell, K., Sun, H., Wang, Y. & Wang, S. Sulfur and nitrogen co-doped graphene for metal-free catalytic oxidation reactions. Small 11, 3036–3044 (2015).
Duan, X. et al. Nitrogen-doped graphene for generation and evolution of reactive radicals by metal-free catalysis. ACS Appl. Mater. Inter. 7, 4169–4178 (2015).
Zhan, Y. et al. Iodine/nitrogen co-doped graphene as metal free catalyst for oxygen reduction reaction. Carbon 95, 930–939 (2015).
Lan, D. H., Chen, L., Au, C. T. & Yin, S. F. One-pot synthesized multi-functional graphene oxide as a water-tolerant and efficient metal-free heterogeneous catalyst for cycloaddition reaction. Carbon 93, 22–31 (2015).
Wei, W. et al. Nitrogen-doped carbon nanosheets with size-defined mesopores as highly efficient metal-free catalyst for the oxygen reduction reaction. Angew. Chem. Int. Edit. 53, 1570–1574 (2014).
Sun, H., Zhou, G., Wang, Y., Suvorova, A. & Wang, S. A new metal-free carbon hybrid for enhanced photocatalysis. ACS Appl. Mater. Inter. 6, 16745–16754 (2014).
Su, Y. et al. Low-temperature synthesis of nitrogen/sulfur co-doped three-dimensional graphene frameworks as efficient metal-free electrocatalyst for oxygen reduction reaction. Carbon 62, 296–301 (2013).
Sun, H. et al. Facile synthesis of nitrogen doped reduced graphene oxide as a superior metal-free catalyst for oxidation. Chem. Commun. 49, 9914–9916 (2013).
Pignatello, J. J., Oliveros, E. & MacKay, A. Advanced oxidation processes for organic contaminant destruction based on the Fenton reaction and related chemistry. Crit. Rev. Env. Sci. Tec. 36, 1–84 (2006).
Fenton, H. LXXIII.—Oxidation of tartaric acid in presence of iron. J. Chem. Soc. 65, 899–910 (1894).
Voitko, K. V. et al. Morphological and chemical features of nano and macroscale carbons affecting hydrogen peroxide decomposition in aqueous media. J. Colloid Interf. Sci. 361, 129–136 (2011).
Song, Y., Qu, K., Zhao, C., Ren, J. & Qu, X. Graphene oxide: intrinsic peroxidase catalytic activity and its application to glucose detection. Adv. Mater. 22, 2206–2210 (2010).
Zheng, A. et al. Highly-efficient peroxidase-like catalytic activity of graphene dots for biosensing. Biosens. Bioelectron. 49, 519–524 (2013).
Liu, G. et al. Quinone-mediated microbial synthesis of reduced graphene oxide with peroxidase-like activity. Bioresource Technol. 149, 503–508 (2013).
Yang, L. et al. π-conjugated carbon radicals at graphene oxide to initiate ultrastrong chemiluminescence. Angew. Chem. Int. Edit. 53, 10109–10113 (2014).
Sun, H., Gao, N., Dong, K., Ren, J. & Qu, X. Graphene quantum dots-band-aids used for wound disinfection. ACS Nano 8, 6202–6210 (2014).
Qu, D. et al. Formation mechanism and optimization of highly luminescent N-doped graphene quantum dots. Sci. Rep. 4, 5294, (2014).
Zhou, X. et al. Photo-Fenton reaction of graphene oxide: a new strategy to prepare graphene quantum dots for DNA cleavage. ACS Nano 6, 6592–6599 (2012).
Bai, H. et al. Insight into the mechanism of graphene oxide degradation via the photo-Fenton reaction. J. Phys. Chem. C 118, 10519–10529 (2014).
Lu, J. et al. One-pot synthesis of fluorescent carbon nanoribbons, nanoparticles, and graphene by the exfoliation of graphite in ionic liquids. ACS Nano 3, 2367–2375 (2009).
Li, X., Rui, M., Song, J., Shen, Z. & Zeng, H. Carbon and graphene quantum dots for optoelectronic and energy devices: a review. Adv. Funct. Mater. 25, 4929–4947 (2015).
Shen, J., Zhu, Y., Yang, X. & Li, C. Graphene quantum dots: emergent nanolights for bioimaging, sensors, catalysis and photovoltaic devices. Chem. Commun. 48, 3686–3699 (2012).
Yang, K., Feng, L., Shi, X. & Liu, Z. Nano-graphene in biomedicine: theranostic applications. Chem. Soc. Rev. 42, 530–547 (2013).
Bacon, M., Bradley, S. J. & Nann, T. Graphene quantum dots. Part. Part. Syst. Char. 31, 415–428 (2014).
Pan, D., Zhang, J., Li, Z. & Wu, M. Hydrothermal route for cutting graphene sheets into blue-luminescent graphene quantum dots. Adv. Mater. 22, 734–738 (2010).
Wu, M. et al. Preparation of functionalized water-soluble photoluminescent carbon quantum dots from petroleum coke. Carbon 78, 480–489 (2014).
Jin, S. H., Kim, D. H., Jun, G. H., Hong, S. H. & Jeon, S. Tuning the photoluminescence of graphene quantum dots through the charge transfer effect of functional groups. ACS Nano 7, 1239–1245 (2013).
Zhu, B., Kalyanaraman, B. & Jiang, G. Molecular mechanism for metal-independent production of hydroxyl radicals by hydrogen peroxide and halogenated quinones. Proc. Natl. Acad. Sci. USA 104, 17575–17578 (2007).
Zhu, B. et al. Mechanism of metal-independent decomposition of organic hydroperoxides and formation of alkoxyl radicals by halogenated quinones. Proc. Natl. Acad. Sci. USA 104, 3698–3702 (2007).
Zhu, B. et al. Unprecedented hydroxyl radical-dependent two-step chemiluminescence production by polyhalogenated quinoid carcinogens and H2O2 . Proc. Natl. Acad. Sci. USA 109, 16046–16051 (2012).
Houk, K. N. Frontier molecular orbital theory of cycloaddition reactions. Accounts Chem. Res. 8, 361–369 (1975).
Salomon, A. et al. Comparison of electronic transport measurements on organic molecules. Adv. Mater. 15, 1881–1890 (2003).
Nitzan, A. & Ratner, M. A. Electron transport in molecular wire junctions. Science 300, 1384–1389 (2003).
Fukui, K. Role of frontier orbitals in chemical reactions. Science 218, 747–754 (1982).
Xing, W., Lalwani, G., Rusakova, I. & Sitharaman, B. Degradation of graphene by hydrogen peroxide. Part. Part. Syst. Char. 31, 745–750 (2014).
Xie, B. et al. Hydrothermal synthesis of layered molybdenum sulfide/N-doped graphene hybrid with enhanced supercapacitor performance. Carbon 99, 35–42 (2016).
Mungse, H. P., Sharma, O. P., Sugimura, H. & Khatri, O. P. Hydrothermal deoxygenation of graphene oxide in sub- and supercritical water. RSC Adv. 4, 22589–22595 (2014).
Xiong, D. et al. Controllable oxygenic functional groups of metal-free cathodes for high performance lithium ion batteries. J. Mater. Chem. A 3, 11376–11386 (2015).
Zhu, S. et al. Surface chemistry routes to modulate the photoluminescence of graphene quantum dots: from fluorescence mechanism to up-conversion bioimaging applications. Adv. Funct. Mater. 22, 4732–4740 (2012).
Peng, J. et al. Graphene quantum dots derived from carbon fibers. Nano Lett. 12, 844–849 (2012).
Sk, M. A., Ananthanarayanan, A., Huang, L., Lim, K. H. & Chen, P. Revealing the tunable photoluminescence properties of graphene quantum dots. J. Mater. Chem. C 2, 6954–6960 (2014).
Lu, H., Zhang, H., Wang, J., Zhou, J. & Zhou, Y. A novel quinone/reduced graphene oxide composite as a solid-phase redox mediator for chemical and biological Acid Yellow 36 reduction. RSC Adv. 4, 47297–47303 (2014).
Li, M. et al. The evolution of surface charge on graphene oxide during the reduction and its application in electroanalysis. Carbon 66, 302–311 (2014).
William, S., Hummers, J. & Offeman, R. E. Preparation of graphitic oxide. J. Am. Chem. Soc. 80, 1339 (1958).
El-Mansy, M., El-Nahass, M., Khusayfan, N. & El-Menyawy, E. DFT approach for FT-IR spectra and HOMO-LUMO energy gap for N-(p-dimethylaminobenzylidene)-p-nitroaniline (DBN). Spectrochim. Acta A 111, 217–222 (2013).
Frisch, A., Nielsen, A. & Holder, A. GAUSSVIEW Users Manual. (Gaussian Inc., Pittsburgh, PA, 2000).
Acknowledgements
We gratefully acknowledge the funding by National Natural Science Foundation of China (No. 51378487 and No. 51425405) and Youth Innovation Promotion Association, CAS (2014037).
Author information
Authors and Affiliations
Contributions
H.Z., Z.F. and H.C. designed the study. D.Z., Q.D. and J.W. performed the experiments. H.Z., D.Z., P.D. and Q.H. analyzed the data. H.Z., D.Z. and J.W. wrote the paper. H.Z., D.Z., C.L., Y.X., Z.F. Y. Z. and J.W. revised the paper. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Zhao, H., Wang, J., Zhang, D. et al. Chloro-benquinone Modified on Graphene Oxide as Metal-free Catalyst: Strong Promotion of Hydroxyl Radical and Generation of Ultra-Small Graphene Oxide. Sci Rep 7, 42643 (2017). https://doi.org/10.1038/srep42643
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep42643
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
