
Abstract
Bacteriophages display remarkable genetic diversity and host specificity. In this study, we explore phages infecting bacterial strains of the Enterobacteriaceae family because of their ability to infect related but distinct hosts. We isolated and characterized two novel virulent phages, SH6 and SH7, using a strain of Shigella flexneri as host bacterium. Morphological and genomic analyses revealed that phage SH6 belongs to the T1virus genus of the Siphoviridae family. Conversely, phage SH7 was classified in the T4virus genus of the Myoviridae family. Phage SH6 had a short latent period of 16 min and a burst size of 103 ± 16 PFU/infected cell while the phage SH7 latent period was 23 min with a much lower burst size of 26 ± 5 PFU/infected cell. Moreover, phage SH6 was sensitive to acidic conditions (pH < 5) while phage SH7 was stable from pH 3 to 11 for 1 hour. Of the 35 bacterial strains tested, SH6 infected its S. flexneri host strain and 8 strains of E. coli. Phage SH7 lysed additionally strains of E. coli O157:H7, Salmonella Paratyphi, and Shigella dysenteriae. The broader host ranges of these two phages as well as their microbiological properties suggest that they may be useful for controlling bacterial populations.
Similar content being viewed by others
Introduction
Bacteriophages, the viruses of bacteria, are characterized by their nanometer size as well as their obligatory parasitism. They are present in the same ecological niche as their hosts and contribute to bacterial ecology and evolution1,2. The vast majority of phages also possess a proteinaceous tail (Caudovirales order) that enables the specific recognition and subsequent adsorption to a receptor at the surface of the host bacterium. The Caudovirales are further divided into three families: Myoviridae (long contractile tail), Siphoviridae (long noncontractile tail) and Podoviridae (short tail). In the past decade, comparative genomic and proteomic analyses have been essential in revealing the diversity and evolutionary relationships between phages as well as the levels of interaction with their bacterial hosts. A better understanding of these bacterial viruses has led to several prospective biotechnological applications. For example, virulent phages have potential for preventing or treating bacterial disease3,4, detecting bacteria5, and decontaminating surfaces6,7.
Phages are usually species-specific and even strain-specific8, however, some polyvalence is observed, predominantly among phages of Enterobacteria9,10 and staphylococci11. These polyvalent phages are able to infect strains from either different genera or species12.
Members of the Enterobacteriaceae family are also closely related and are sometimes taxonomically difficult to distinguish13,14. The ability of these polyvalent lytic phages to infect various host species makes host range an imperfect indicator of phage relatedness. Therefore, phages of the same morphological family are grouped in genera/species based on similarities in genome sequences15.
Phages with broad host ranges are most often isolated from natural microbial communities that promote genetic exchanges16,17. Under selective pressure, tail fiber genes appear to evolve faster than other phage genes, presumably because it seems advantageous to gain new specificities to infect different hosts as well as to enter other ecological niches1,18. The T4virus genus is one of the best-characterized phage groups for which tail fibers have been extensively studied and shown relative rapid adaptation. They have extended host ranges, mostly due to their unique ability to recognize various host receptors (outer membrane protein or lipopolysaccharide (LPS)) using the same tail fiber protein (gp37T4)19. Members of this genus have high genetic identity and the modest differences observed are related to their adaptation to novel host constraints20,21. For example, coliphages T4 and AR1 share a highly conserved core genome but have distinct host ranges22. Phage T4 infects E. coli B and K-12 but not E. coli O157:H7, while AR1 is very specific to strains of E. coli O157:H723. Another example was observed in the T1virus genus of the Siphoviridae family. Coliphage Rtp is closely related to coliphage T1 but acquired a unique tail tip that allows it to recognize a different host receptor. Phage T1 uses the FhuA outer membrane protein as a receptor and requires TonB for infection while phage Rtp uses LPS24.
Because of the renewed interest in lytic phages as biocontrol agents, new phages are sought after as they offer the possibility of increasing bacterial strain coverage in the design and development of phage cocktails11,25. In an effort to isolate new phages effective against several strains, we investigated phages infecting the Enterobacteriaceae family. From sewage samples in Tunisia, we isolated two virulent phages that belong to the Myoviridae and Siphoviridae families. Their genomic, proteomic and microbiological characteristics provided an overview of their evolutionary relationships with other previously characterized phages and suggest that they may have potential to control bacterial populations.
Results
Isolation of bacteria and phages
A Gram-negative bacterial isolate, SF1, was recovered from a wastewater sample in Tunisia. The 16S rRNA sequencing and MLST analysis revealed that this strain belongs to the Shigella flexneri species with the sequence type ST216. This strain was used as the host organism to search for virulent phages. Phages were isolated from water samples of the wadi of Chotrana in Tunisia. Two plaques with distinct morphology were picked at random and single-plaque purified at least three times. These phage isolates were named SH6 and SH7. Phage SH6 formed clear plaques of 2 mm in diameter after only 3 hours of incubation at 37 °C. Phage SH7 formed very small plaques of around 0.2 mm in diameter after overnight incubation at 37 °C.
Host range and microbiological properties
The host range of phages SH6 and SH7 was then tested on 35 bacterial strains by spot test of diluted phage lysate (Table 1). Among the 35 strains tested, phage SH7 was able to infect 27 bacterial strains while phage SH6 infected 9 strains. Both isolated phages infected their host strain S. flexneri, the host strain of the T-odd and T-even coliphages, E. coli B11303, and 7 other E. coli K-12 derived strains. Phage SH7 also infected 16 strains of the Shiga toxin-producing E. coli O157:H7, one strain of Salmonella Paratyphi, and one strain of Shigella dysenteriae. For phage SH6, a lysis zone was observed at 100 and 10−1 with the 16 strains of E. coli O157:H7 but they were likely the result of lysis from without (at high multiplicity of infection) as no clear plaques could be observed or propagated.
A one-step growth curve experiment using their host strain S. flexneri SF1 revealed that phage SH6 has a short latent period of only 16 min and a burst size of 103 ± 16 PFU/infected cell. The latent period of phage SH7 was 23 min with a burst size of 26 ± 5 PFU/infected cell. The pH sensitivity of the two phages was determined by exposing them for one hour at 37 °C to pH ranging from 2 to 11. Phage SH6 was sensitive to acidic conditions as it was inactivated when exposed to pH 2 and pH 3. It also had a 2-Log reduction at pH 4 but was stable from pH 5 to pH 11. Phage SH7 was stable from pH 3 to 11 and inactivated only at pH 2.
Morphological analysis
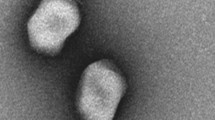
Electron microscopy observations of purified phage SH6 revealed an icosahedral capsid of 62 ± 2 nm in diameter with a long noncontractile flexible tail of 161 ± 2 nm in length by 13 ± 1 nm in width, morphology indicative of the Siphoviridae family (Fig. 1A). Phage SH7 had an elongated capsid of 112 ± 5 nm in length by 89 ± 3 nm in width and a long contractile tail of 116 ± 4 nm in length by 22 ± 2 nm in width. SH7 belongs to the Myoviridae family (Fig. 1B).

Electron micrographs of phage SH6 (A) and phage SH7 (B).
Proteomic analysis
The purified phages SH6 and SH7 were analyzed by liquid chromatography/tandem mass spectrometry (LC-MS/MS). The LC-MS/MS analysis detected 11 structural proteins in phage SH6 with a sequence coverage ranging from 14 to 55% (Table 2). These proteins included the portal protein, capsid protein, tail proteins, tail tape measure protein, tail fiber proteins and four other proteins of unknown function. Analysis of phage SH7 by LC-MS/MS detected 13 structural proteins with a sequence coverage ranging from 4 to 31% including proteins of the capsid, tail, tail fibers, neck and procapsid core. Other than the phage SH7 structural proteins, we also identified one ADP-ribosylase protein with the lowest coverage (3%, 76 kDa), which corresponds to an enzyme that is usually not present in the phage structure. The target of the phage ADP-ribosylase is the host RNA polymerase. The ADP-ribosylation of RNA polymerase is involved in increasing gene expression after infection but it is not essential for phage development26,27. While it is unclear if this is a contaminant of the phage purification process or a protein encapsidated with the phage genome, a similar protein was also detected by mass spectrometry for phage AR122.
Genomic analysis of phage SH6
The genome of phage SH6 is composed of 50,552 bp of double-stranded DNA with 45.8% G+C content, in the same range as phage T1 (48,836 bp; 45.6% G+C)28. The SH6 genome is predicted to encode 82 ORFs with only ATG as initiation codon and only 27 ORFs (33%) have assigned functions (Table S1). No tRNA was identified. Phage SH6 is novel but has high nucleotide sequence identity to other phages of the T1virus genus including S. flexneri phage pSf-2 (89%) and coliphage T1 (83%). Of note, the T1 genome encodes 77 ORFs and is missing homologous sequence to the 3’ region (from ORF78 to ORF82) of phage SH6 (Fig. 2). At the amino acid level, SH6 shares 57 and 48 ORFs with phages pSf-2 and T1, respectively, with more than 90% identity. Of these, SH6 has 6 ORFs with 100% identity to T1 ORFs (ORF46/Minor tail, ORF47/Tail assembly, ORF65/Holin and ORF22, ORF37, ORF39 of unknown functions) and 2 ORFs with 100% identity to phage pSf-2 ORFs (two putative proteins (ORF12, ORF56) of unknown functions). Despite the overall genome conservation between SH6, pSf-2 and T1, diversity occurred upstream of the DNA packaging module. In addition, phage SH6, like pSf-2, is missing the three homing HNH endonucleases dispersed throughout the genome of phage T1. Additional differences between T1 and SH6/pSf-2 were found in the central regions of their genomes: ORF49SH6 and ORF49pSf-2 share no homology with their T1 counterparts (ORF31 and ORF32) situated in the same genome location but in the opposite orientation. Interestingly, ORF50SH6 has 45% identity to the homolog ORF30corT1, a receptor-blocking lipoprotein excluding FhuA-dependent phages29. This phage-encoded mechanism protect phage progeny from inactivation by cell wall particles when released from lysed cells30.

Each line represents a different phage genome and each arrow represents a putative protein. The blue gradient represents ORFs whose translated products share from 90 to 99.5% amino acid identity. The upper part of SH6 genome shows identity with phage T1 while the lower part shows identity to phages pSf-2. Conserved genomic regions are connected by grey shading. Arrows with thick outlines and bold numbers represent structural proteins detected by LC-MS/MS.
Module analysis of phage SH6
The phage SH6 genome can be divided into at least four functional modules, including DNA packaging, phage structure, DNA recombination/replication, and host lysis. The two subunits of the SH6 terminase (ORF27 and ORF28) share 99% aa identity with the phage T1 terminase subunits, suggesting a common packaging process. The terminase complex of phage T1, comprised one small and one large subunit, recognizes a pac site to catalyze the headful DNA packaging into preassembled empty capsids using its endonuclease and ATPase activities. This headful mechanism results in terminal redundancy and circular permutation15,28.
ORF29SH6 to ORF48SH6 appears to be proteins involved in capsid and tail morphogenesis. In this region, phage SH6 ORFs share more than 95% identity with 79% of T1 ORFs and 68% of pSf-2 ORFs, including the portal protein, the minor and major capsid proteins, the minor and major tail proteins, the tail tape measure protein and the tail assembly protein. This is consistent with the conserved morphology observed between phages of the T1virus genus.
Phage SH6 host recognition proteins include two tail fiber proteins, like T1 phages28. ORF48SH6 is a tail fiber protein of 1,149 amino acids that shares 98% and 92% identity with the homologous ORFs of phages pSf-2 and T1, respectively. Phage SH6 was able to infect the T1 host strain, E. coli B11303. However, ORF54SH6 appears to be a second 559 aa tail fiber protein, which is the most striking difference between phage SH6 and previously characterized t1viruses. There are few identities between the N-terminal domain of ORF54SH6 and the tail fiber proteins of Shigella phages Shfl1 (49%) and pSf-2 (45%) as well as with the one of the coliphage T1 (34%). In fact, the C-terminal portion shares identity with the tail tip proteins of E. coli phages SSL2009a (50%) and JL1, classified within the Hk578virus genus31,32. It contains a conserved 51 aa peptidase_S74 domain with 94% identity to those two tail tip proteins. Peptidase_S74 is a chaperone of endosialidase that acts as a tailspike protein responsible for host polysialic acid capsule recognition, binding and degrading activity33. It seems that ORF54SH6 is a combination of the tail fiber proteins of a T1virus and a Hk578virus, which likely explain the expanded host range of phage SH6 compared to pSf-2, which reportedly could infect only some strains of S. flexneri34.
Three ORFs constitute the recombination module of phage SH6. ORF51SH6, ORF52SH6 and ORF53SH6 share 98%, 95% and 90% aa identity with ORF29recET1 (exodeoxyribonuclease VIII), ORF28erfT1 (recombinase) and ORF27ssbT1 (single stranded DNA binding protein), respectively. Two ORFs are within the replication module of SH6, the DNA primase ORF55 and the ATP-dependent helicase ORF57, with high identity (99%) to ORF24priA and ORF22helA of phage T1.
The genome of SH6 contains three closely linked ORFs coding for a holin, an endolysin and a spanin, three proteins that act successively at the end of the latent period for phages release. First, ORF65SH6 is 100% identical to ORF13holT1, which encodes a small holin protein (71 aa) and creates pores in the cytoplasmic membrane. Second, ORF66SH6 is 99% identical to ORF14LysT1, which encodes the endolysin protein that cleaves β-1,4-linkages in the peptidoglycan of bacterial cell walls28. Finally, ORF67SH6 has 78% identity to spanin protein ORF11T1 which spans the periplasm, making a physical connection between the inner and outer membranes35.
Phylogeny of phage SH6
Phylogenetic trees were constructed to further investigate the relatedness of phage SH6 to the 32 known phages of the T1virus genus and 5 phages of the Hk578virus genus. To associate the newly isolated phage SH6 to one of the previously established clusters36, we examined the common features of the same three proteins, including the large terminase (Fig. 3A), the portal protein (Fig. 3C) and the major capsid (Fig. 3D). The phylogenetic trees of each of these three proteins showed that phage SH6 is in the same branch as phages T1, pSf-2, Shfl1, JMPW1, JMPW2 and ADB-2, indicating that these phages belong to cluster C of the T1virus genus. Interestingly, examination of a fourth protein, the second tail fiber (ORF54SH6), revealed that cluster C was more closely related to phages of the Hk578virus genus and phage pSf-1 than to the other phages of the T1virus genus (Fig. 3B). It seems that an acquisition of tail fiber protein happened during the evolution of this cluster of T1virus genus and the Hk578virus genus.

Evolutionary relationships between SH6, the 32 known phages of the T1virus genus and the 5 known phages of the Hk578virus genus according to four proteins: (A) Large subunit of terminase, (B) Tail fiber, (C) Portal protein and (D) Major capsid.
Genomic analysis of phage SH7
The SH7 genome is composed of 164,870 bp of double stranded DNA with a surprisingly low G+C content of 35.5%, as its Shigella host has about 50–51% G+C37. This low G+C content has been suggested to be advantageous for phage DNA transcription and replication by facilitating its recognition by specific host factors38. Phage SH7 genome encodes for 265 putative ORFs (Table S2). The best matches for most of these ORFs are to phages proteins belonging to the T4 superfamily. Of the matches, 137 (52%) could be annotated with a putative function. Comparative genomic analyses with phage T4 revealed that SH7 genes maintain a conserved gene orientation (Fig. 4).

Each ORF is represented by an arrow. The blue gradient represents ORFs whose translated products share from 90 to 99.5% amino acid identity. Conserved genomic regions are connected by grey shading. Arrows with thick outlines and bold numbers represent structural proteins detected by LC-MS/MS.
In phage T4, 62 ORFs were previously predicted to be essential, including proteins involved in nucleotide metabolism, transcription regulation and virion structure and assembly38. Homologs of all of these essential ORFs were found in the genome of phage SH7 with high identities, including 10 ORFs with 100% identity to phage T4 (ORF74, ORF138, ORF157, ORF158, ORF161, ORF164, ORF180, ORF202, ORF235 and ORF244). A difference was noted in the DNA topoisomerase subunit, which is encoded by a single gene (ORF3) for phage SH7 instead of two genes (gp60T4 and gp39T4) in T4. However, ORF3SH7 shares homology with both T4 genes, suggesting that they have bound or dissociated during the evolutionary process.
Noteworthy, SH7 genome is missing homologs to 14 of the 15 mobile elements encoded by phage T4, including three group I introns (I-Tev (I-III)) and 12 freestanding homing endonucleases genes (7 seg (A-G) and 5 mob (A-E))39. The SH7 genome contains only one homing endonuclease product equivalent to mobET4 (62% aa identity), located between the genes coding for the beta and the alpha subunits of the aerobic ribonucleotide reductase. Previous studies also revealed a limited number of such mobile elements for other t4viruses such as coliphages AR1 and RB4920,22. On the other hand, 11 tRNA genes were identified in the SH7 genome with the following specificities: Arg (UCU), His (GUG), Asn (GUU), Tyr (GUA), Met (CAU), Thr (UGU), Ser (UGA), Pro (UGG), Gly (UCC), Leu (UAA) and Gln (UUG). The number of tRNAs varies considerably among the genomes of phages of the T4virus genus21. The tRNAs are not essential but are presumably required to ensure optimum progeny size40.
Another striking difference between phages T4 and SH7 lies in their tail fiber proteins. Phages of the T4 family recognize their cellular receptor (outer membrane protein C or LPS) using the C-terminus of the distal long tail fiber protein encoded by gp37T4-like protein41,42. The distal long tail fiber protein ORF241SH7differs from gp37T4, sharing only 37% identity, mostly located at the N-terminus of this protein. In phage T4, the N-terminal region of gp37 is involved in the interaction with its neighboring gp36T4 and is highly conserved among the T-even phages43. Interestingly, ORF241SH7 is 95% identical to the gp37-like protein of phage AR1, most likely explaining the ability of phage SH7 to infect strains of E. coli O157:H7 as AR1 is a typing phage for this serotype22.
ORF242SH7 is likely the tail fiber adhesin implicated in the dimerization of the distal long tail fiber but does not share any homology with gp38T4. On the other hand, it is 95% identical to the gp38-like protein of phage wV7, another typing phage of E. coli O157:H741. The short tail fibers are encoded by a single gene, gp12T4, and their role is to extend and bind irreversibly to the core region of the host LPS44. ORF153SH7 shares 64% identity with gp12T4 and is 99% identical to the short tail fiber proteins of phages e11/12 and AR1.
Similarity matrix of phage SH7
Phylogenic analysis of the whole proteome of phage SH7 with the other 54 phages of the T4virus genus available in public databases demonstrated that phages within each cluster shared close relatedness at the amino acid level (Fig. 5). This result at the protein level is consistent with the whole genome nucleotide dot blot of the 37 t4viruses45. According to the ten clusters defined previously, phage SH7 appears in the same cluster as the prototypic phage T4 (cluster A). This cluster also includes phages PST, ECML_134, RB32, RB14, pSs-1, ime09, HY01, Shfl2, slur07, slur02, slur04, wV7, RB51, phiD1, slur14, RB27, AR1, vB_EcoM_ACG_C40 and e11/12.

The heatmap is generated based on the number of proteins shared by phages. Deeper shade of blue indicates a closer relationship.
Discussion
In this study, we isolated and characterized two virulent phages, SH6 and SH7, from sewage in Tunisia using a strain of S. flexneri as host bacterium. Morphological and genomic analyses revealed that phage SH6 belongs to the Siphoviridae family and the T1virus genus, while phage SH7 belongs to the Myoviridae family and the T4virus genus.
Both isolated phages have broad host ranges. Other studies have shown that sequential or simultaneous addition of multiple hosts should be used to isolate phages with broader host range. However with these methods the host range of these phages seems limited to the bacterial species used during their isolation16,46. The current study indicates that it is possible to isolate such phages using a single host. Phage SH6 infected its host strain S. flexneri and 8 strains of E. coli. Phage SH7 lysed the same strains as phage SH6 but also strains of E. coli O157:H7 (16), S. Paratyphi (1), and S. dysenteriae (1).
Phages SH6 and SH7 owe their extended host range to the acquisition of new tail fiber genes. Phage SH6 is closely related to phages pSf-2 and T1 infecting, respectively, S. flexneri and E. coli. The major difference between SH6 and his closely related phages lies in the C-terminal portion of their second tail fiber protein (ORF54SH6) which shares similarity with coliphages SSL-2009a and JL1, classified in the Hk578virus genus31,32. On the other hand, phage SH7 is closely related to coliphage T4 but has a wider host range47. The major differences between phages SH7 and T4 are in their tail fiber proteins (gp37, gp38 and gp12), which have more similarity to coliphages AR1 and wV7. Since phages AR1 and wV7 are typing phages of E. coli O157:H722,41, this could explain the ability of phage SH7 to infect this serotype. However, phage AR1 is very specific to E. coli O157:H7 and cannot lyse strains of E. coli K-1247.
The relative lack of specificity of phage SH7 is interesting from a therapeutic perspective. This phage can be amplified on a nonpathogenic host E. coli K-12 strain instead of the pathogenic strains of E. coli O157:H7, perhaps contributing to an easier approval of a biocontrol product based on this phage48.
It has been shown previously that acquisition of new tail fiber genes could be due to recombination between divergent relatives49, yet these phages are geographically dispersed and from diverse habitats. Phage AR1 was isolated in 1990 from cow stools in the US47, JL1 from sewage in China a few years ago32, pSf-2 from sewage in Korea34, while SH6 and SH7 were isolated from sewage in Tunisia in 2014. These observations also clearly indicate that related phages are spread around the world, while likely ongoing adaptation to local host strains.
It is also valuable to consider the conditions under which phages can control bacterial populations. For this reason, we investigated the rate and time at which a new phage population can increase, as well as its susceptibility to variable pHs. For phage SH6, the latent period was relatively short (16 min) and the burst size about 103 ± 16 PFU/infected cell. These values are consistent with the range observed for phage T1 (13 min, 100 PFU/infected cell50,51) but phage SH6 appears more optimized than phage pSf-2 (30 min, 16 PFU/infected cell34). The phage SH7 latent period and burst size (23 min, 26 ± 5 PFU/infected cell) are similar to those observed for phages AR1 (20–25 min, 34 PFU/infected cell47) and T4 (18–37 min, 12–152 PFU/infected cell52). The relative ease of their isolation and their short lytic cycle suggests that these phages were in an environment where new host cells are constantly provided as otherwise they would deplete local resources and possibly go extinct.
Phage SH6 was sensitive to acidic conditions, like phage T153, but stable from pH 5 to 11. On the other hand, phage SH7 was very stable from pH 3 to 11 while phage T4 is sensitive to acidic conditions under pH 554.
While it remains to be seen if these phages can be used for biosanitation purposes or even to treat infectious diseases, these above properties (broader host range, rapid lytic cycle and stability at various pH) will have to be taken into accounts depending of the applications. For example in the case of phage oral application, stomach acid may negatively affect the infectivity of phage SH6. As such, it may require some protection during his passage through the gastrointestinal tract, perhaps through microencapsulation in alginate-chitosan55. On the other hand, both phages appear to be highly stable in slightly acidic, neutral, as well as basic solutions or environments.
One of the main limitations to the use of lytic phages for biocontrol or therapeutic purposes is related to the selection of strains resistant to phages. Since phages SH6 and SH7 are highly effective and belong to different phage families, their use in combination may reduce the risk of the rapid emergence of phage resistant strains in the environment. Finally, the newly characterized phages SH6 and SH7 expand our knowledge about the diversity and evolution of enterophages in the environment.
Materials and Methods
Bacterial strain, phage isolation and growth conditions
One bacterial isolate was previously obtained by plating a wastewater sample retrieved from the wadi of Megrine in Tunisia on Salmonella-Shigella agar (Biokar) and incubating for 24 h at 37 °C. The bacterial species of the isolate was identified using 16S rRNA sequencing and an API 20 E strip test (BioMérieux). This strain was also genotyped using the multi-locus sequence type (MLST) of seven housekeeping genes (adk, fumC, gyrB, icd, mdh, purA and recA) as described previously56. Its allelic profile and sequence type (ST) were determined using the E. coli MLST database (http://mlst.warwick.ac.uk/mlst/dbs/Ecoli/dbs/Ecoli). This strain was used as a host for plaque purification, propagation and titration of the phage stocks. The two phages described here were isolated from a water sample retrieved from the wadi of Chotrana in Tunisia as described previously57. Phages and hosts are available at the Félix d’Hérelle Reference Center for Bacterial Viruses of the Université Laval (Québec, Canada; www.phage.ulaval.ca) under the following names: phages SH6 (HER521), and SH7 (HER522) as well as S. flexneri SF1 (HER1521).
Microbiological assays
The host range was tested by adding 10 μl of serial dilutions (100 to 10−6) of a phage suspension on a lawn of each bacterial strain mixed with 3 ml of BHI containing 0.75% (w/v) agar. Plaque formation was observed after overnight incubation at 37 °C, indicating a complete lytic cycle on the host. For host range analysis, 35 bacterial strains obtained from the Félix d’Hérelle collection were tested (Table 1). The susceptibility of the two phages to different pHs was determined by incubating them in BHI broth adjusted to pH 2 to 11 for one hour as described previously57. The one-step growth curve assay was carried out in triplicate. Briefly, phages were mixed with 2 ml of a mid-exponential phase culture of Shigella flexneri (optical density at 600 nm of 0.8) with a starting multiplicity of infection (MOI) of 0.05. Phages were allowed to adsorb for 2 min at 37 °C, then the mixture was centrifuged for 30 sec at 16,000x g. The pellet was resuspended in 10 ml of BHI broth. This suspension was incubated at 37 °C and phages were titered every 3 min for a total of 40 min. Plates were incubated for 18–24 h at 37 °C. The burst size was calculated by subtracting the initial titer from the final titer then dividing by the initial titer. The latent phase correspond to the middle of the exponential phase of the curve58.
Electron microscopy
Phages were prepared and observed as described elsewhere59. Capsid sizes and tail lengths were determined by measuring at least 10 phage specimens stained with uranyl acetate (2%).
Structural proteins examination
One liter of phage lysate was concentrated with 10% polyethylene glycol (PEG) 8000 and 29.2 g of sodium chloride (Laboratoire Mat), and purified on a discontinuous CsCl (Fisher Scientific) gradient followed by a continuous CsCl gradient as described previously60,61. Structural phage proteins were detected directly from the purified phage preparation by liquid chromatography/tandem mass spectrometry (LC-MS/MS) at the Plateforme Protéomique, Centre de Génomique de Québec (Université Laval). A custom database was generated using the putative predicted proteins from the sequences of the phage genomes and compared with the peptides identified. These results were analyzed with Scaffold Proteome software version 4.4.5.
Genome sequencing and bioinformatics analyses
Phage DNA was extracted from 500 ml of lysate using a Plasmid maxi kit (Qiagen) with modifications62. Genome sequencing was performed using the Nextera XT DNA library preparation kit (Illumina) according to the manufacturer’s instructions. The libraries were sequenced using a MiSeq reagent kit v2 (Illumina; 500 cycles) on a MiSeq system. De novo assembly was performed with Ray assembler version 2.2.0 using k-mer sizes of 31 and 51 and the mean coverage depths for each single phage contig was 5549 and 1369 for SH6 and SH7, respectively. Coverage was calculated with Samtools (http://samtools.sourceforge.net). The open reading frames (ORFs) were identified using NCBI ORF finder (http://www.ncbi.nlm.nih.gov/gorf/orfig.cgi) 63 and GeneMark.hmm prokaryotic64 (http://exon.gatech.edu/GeneMark/gmhmmp.cgi). ORFs were analyzed using BioEdit 7.2.0 and validated if they contained at least 30 amino acids (aa), had an initiation codon (AUG, UUG or GUG) and were preceded by a Shine-Dalgarno sequence. BLASTp analysis against the NCBI protein database was used to predict the function of the ORFs. Hits were considered when the E-value was lower than 10−3. The percent identity between proteins was calculated by dividing the number of identical residues by the size of the smallest protein. Theoretical molecular weight (MW) and isoelectric point (pI) of the proteins were obtained using the compute pI/MW available on the ExPASy Web page (http://web.expasy.org/compute_pi/). The examination of tRNA genes was conducted with ARAGORN65 and tRNAscan-SE v.1.2166.
Phylogeny of phage SH6
The amino acid sequences of the terminase, the second tail fiber protein (fibBT1), the neck passage structure and the major capsid protein from a set of phages of the T1virus and Hk578virus genus were aligned using MAFFT67. Since the terminase of phage JK06 is truncated, it was not included in the phylogenetic analysis. Similarly, no orthologous protein to the second tail fiber was found in the genome of phage vB_XveM_DIBBI, so it was not included in the analysis. The best amino-acid substitution model was determined using ProtTest3.468 and was implemented in PhyML 3.0 to calculate the best tree. The Shimodaira-Hasegawa-like procedure was used to determine the branch support values69. Finally, Newick utility package70 and ETE3.0.0b3571 were used to render the trees. All trees were midpoint rooted.
Similarity matrix of phage SH7
All protein sequences from selected T4virus genomes were extracted. The proteins were grouped in orthologous clusters using CogSoft72 with an E-value lower than 10−3 requiring that the alignment of orthologous proteins span at least 75% of the protein lengths. The clusters were then parsed with in-house Python scripts to obtain a protein presence/absence binary matrix where each column represents a cluster. The rows were populated with the phage genomes. When the phage had a protein that belonged to cluster x, the value 1 was given to the corresponding column/cluster, otherwise the value 0 was given. The similarity matrix was generated in R73 with the function dist with the binary method to calculate the distance. The heatmap was generated with the gplots package74 in R. The order of the rows and columns was manually adjusted when needed.
Nucleotide sequence accession numbers
The annotated phage genomic sequences were deposited in GenBank under the accession numbers KX828710 (SH6) and KX828711 (SH7).
Additional Information
How to cite this article: Hamdi, S. et al. Characterization of two polyvalent phages infecting Enterobacteriaceae. Sci. Rep. 7, 40349; doi: 10.1038/srep40349 (2017).
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Casjens, S. R. Comparative genomics and evolution of the tailed-bacteriophages. Curr. Opin. Microbiol. 8, 451–458 (2005).
Rohwer, F. Global Phage Diversity. Cell 113, 141 (2003).
Bruttin, A. & Brüssow, H. Human Volunteers Receiving Escherichia coli Phage T4 Orally: a Safety Test of Phage Therapy. Antimicrob. Agents Chemother. 49, 2874–2878 (2005).
Sarker, S. A. et al. Oral Phage Therapy of Acute Bacterial Diarrhea With Two Coliphage Preparations: A Randomized Trial in Children From Bangladesh. EBioMedicine 4, 124–137 (2016).
Cowley, L. A. et al. Analysis of whole genome sequencing for the Escherichia coli O157:H7 typing phages. BMC Genomics 16 (2015).
Hagens, S. & Loessner, M. J. Application of bacteriophages for detection and control of foodborne pathogens. Appl. Microbiol. Biotechnol. 76, 513–519 (2007).
Woolston, J. et al. Bacteriophages lytic for Salmonella rapidly reduce Salmonella contamination on glass and stainless steel surfaces. Bacteriophage 3 (2013).
Ackermann, H. W. et al. Guidelines for bacteriophage characterization. Adv. Virus Res. 23, 1–24 (1978).
Parra, B. & Robeson, J. Selection of polyvalent bacteriophages infecting Salmonella enterica serovar Choleraesuis. Electron. J. Biotechnol. 21, 72–76 (2016).
Park, M. et al. Characterization and Comparative Genomic Analysis of a Novel Bacteriophage, SFP10, Simultaneously Inhibiting both Salmonella enterica and Escherichia coli O157:H7. Appl. Environ. Microbiol. 78, 58–69 (2012).
Haddad, L. E. et al. Improving the Safety of Staphylococcus aureus Polyvalent Phages by Their Production on a Staphylococcus xylosus Strain. PLOS ONE 9, e102600 (2014).
Malki, K. et al. Bacteriophages isolated from Lake Michigan demonstrate broad host-range across several bacterial phyla. Virol. J. 12, 164 (2015).
Delmas, J., Breysse, F., Devulder, G., Flandrois, J.-P. & Chomarat, M. Rapid identification of Enterobacteriaceae by sequencing DNA gyrase subunit B encoding gene. Diagn. Microbiol. Infect. Dis. 55, 263–268 (2006).
Hong Nhung, P. et al. Phylogeny and species identification of the family Enterobacteriaceae based on dnaJ sequences. Diagn. Microbiol. Infect. Dis. 58, 153–161 (2007).
Casjens, S. R. Diversity among the tailed-bacteriophages that infect the Enterobacteriaceae. Res. Microbiol . 159, 340–348 (2008).
Jensen, E. C. et al. Prevalence of Broad-Host-Range Lytic Bacteriophages of Sphaerotilus natans, Escherichia coli, and Pseudomonas aeruginosa . Appl. Environ. Microbiol. 64, 575–580 (1998).
Eisen, J. A. Horizontal gene transfer among microbial genomes: new insights from complete genome analysis. Curr. Opin. Genet. Dev. 10, 606–611 (2000).
Haggård-Ljungquist, E., Halling, C. & Calendar, R. DNA sequences of the tail fiber genes of bacteriophage P2: evidence for horizontal transfer of tail fiber genes among unrelated bacteriophages. J. Bacteriol. 174, 1462–1477 (1992).
Montag, D., Hashemolhosseini, S. & Henning, U. Receptor-recognizing proteins of T-even type bacteriophages: The receptor-recognizing area of proteins 37 of phages T4 TuIa and TuIb. J. Mol. Biol. 216, 327–334 (1990).
Comeau, A. M., Bertrand, C., Letarov, A., Tétart, F. & Krisch, H. M. Modular architecture of the T4 phage superfamily: A conserved core genome and a plastic periphery. Virology 362, 384–396 (2007).
Nolan, J. M., Petrov, V., Bertrand, C., Krisch, H. M. & Karam, J. D. Genetic diversity among five T4-like bacteriophages. Virol. J. 3, 1 (2006).
Liao, W.-C. et al. T4-Like Genome Organization of the Escherichia coli O157:H7 Lytic Phage AR1. J. Virol. 85, 6567–6578 (2011).
Yu, S.-L. et al. Characterization of a phage specific to hemorrhagic Escherichia coli O157:H7 and disclosure of variations in host outer membrane protein OmpC. J. Biomed. Sci. 5, 370–382 (1998).
Wietzorrek, A., Schwarz, H., Herrmann, C. & Braun, V. The Genome of the Novel Phage Rtp, with a Rosette-Like Tail Tip, Is Homologous to the Genome of Phage T1. J. Bacteriol. 188, 1419–1436 (2006).
El Haddad, L. et al. Efficacy of two Staphylococcus aureus phage cocktails in cheese production. Int. J. Food Microbiol. 217, 7–13 (2016).
Wilkens, K., Tiemann, B., Bazan, F. & Rüger, W. ADP-ribosylation and early transcription regulation by bacteriophage T4. Adv. Exp. Med. Biol. 419, 71–82 (1997).
Goff, C. G. & Setzer, J. ADP ribosylation of Escherichia coli RNA polymerase is nonessential for bacteriophage T4 development. J. Virol. 33, 547–549 (1980).
Roberts, M. D., Martin, N. L. & Kropinski, A. M. The genome and proteome of coliphage T1. Virology 318, 245–266 (2004).
Uc-Mass, A. et al. An orthologue of the cor gene is involved in the exclusion of temperate lambdoid phages. Evidence that Cor inactivates FhuA receptor functions. Virology 329, 425–433 (2004).
Decker, K., Krauel, V., Meesmann, A. & Heller, K. J. Lytic conversion of Escherichia coli by bacteriophage T5: blocking of the FhuA receptor protein by a lipoprotein expressed early during infection. Mol. Microbiol. 12, 321–332 (1994).
Li, S. et al. Characterization and Genome Sequencing of a Novel Coliphage Isolated from Engineered Escherichia coli . Intervirology 53, 211–220 (2010).
Pan, F. et al. Complete genome sequence of Escherichia coli O157:H7 lytic phage JL1. Arch. Virol. 158, 2429–2432 (2013).
Stummeyer, K., Dickmanns, A., Mühlenhoff, M., Gerardy-Schahn, R. & Ficner, R. Crystal structure of the polysialic acid–degrading endosialidase of bacteriophage K1F. Nat. Struct. Mol. Biol. 12, 90–96 (2005).
Jun, J. W. et al. Isolation and Comparative Genomic Analysis of T1-Like Shigella Bacteriophage pSf-2. Curr. Microbiol. 72, 235–241 (2015).
Summer, E. J. et al. Rz/Rz1 Lysis Gene Equivalents in Phages of Gram-negative Hosts. J. Mol. Biol. 373, 1098–1112 (2007).
Niu, Y. D., McAllister, T. A., Nash, J. H. E., Kropinski, A. M. & Stanford, K. Four Escherichia coli O157:H7 Phages: A New Bacteriophage Genus and Taxonomic Classification of T1-Like Phages. PLoS ONE 9 (2014).
Nie, H. et al. Complete genome sequence of Shigella flexneri 5b and comparison with Shigella flexneri 2a. BMC Genomics 7, 173 (2006).
Miller, E. S. et al. Bacteriophage T4 Genome. Microbiol. Mol. Biol. Rev. 67, 86–156 (2003).
Sandegren, L., Nord, D. & Sjöberg, B.-M. SegH and Hef: two novel homing endonucleases whose genes replace the mobC and mobE genes in several T4-related phages. Nucleic Acids Res. 33, 6203–6213 (2005).
Wilson, J. H. Function of the bacteriophage T4 transfer RNA’s. J. Mol. Biol. 74, 753–757 (1973).
Kropinski, A. M. et al. Escherichia coli O157:H7 Typing Phage V7 Is a T4-Like Virus. J. Virol. 86, 10246 (2012).
Beckendorf, S. K., Kim, J. S. & Lielausis, I. Structure of bacteriophage T4 genes 37 and 38. J. Mol. Biol. 73, 17–35 (1973).
Riede, I., Drexler, K. & Eschbach, M. L. The nucleotide sequences of the tail fiber gene 36 of bacteriophage T2 and of genes 36 of the T-even type Escherichia coli phages K3 and Ox2. Nucleic Acids Res. 13, 605–616 (1985).
Thomassen, E. et al. The Structure of the Receptor-binding Domain of the Bacteriophage T4 Short Tail Fibre Reveals a Knitted Trimeric Metal-binding Fold. J. Mol. Biol. 331, 361–373 (2003).
Grose, J. H. & Casjens, S. R. Understanding the enormous diversity of bacteriophages: The tailed phages that infect the bacterial family Enterobacteriaceae . Virology 468–470, 421–443 (2014).
Yu, P., Mathieu, J., Li, M., Dai, Z. & Alvarez, P. J. J. Isolation of Polyvalent Bacteriophages by Sequential Multiple-Host Approaches. Appl. Environ. Microbiol. 82, 808–815 (2016).
Ronner, A. B. & Cliver, D. O. Isolation and Characterization of a Coliphage Specific for Escherichia coli 0157:H7. J. Food Prot. 53, 944–947 (1990).
Santos, S. B. et al. Selection and Characterization of a Multivalent Salmonella Phage and Its Production in a Nonpathogenic Escherichia coli Strain. Appl. Environ. Microbiol. 76, 7338–7342 (2010).
Nilsson, A. S. & Haggård-Ljungquist, E. Detection of Homologous Recombination among Bacteriophage P2 Relatives. Mol. Phylogenet. Evol. 21, 259–269 (2001).
Delbrück, M. The Burst Size Distribution in the Growth of Bacterial Viruses (Bacteriophages)1. J. Bacteriol. 50, 131–135 (1945).
Borchert, L. D. & Drexler, H. T1 Genes Which Affect Transduction. J. Virol. 33, 1122–1128 (1980).
Hadas, H., Einav, M., Fishov, I. & Zaritsky, A. Bacteriophage T4 Development Depends on the Physiology of its Host Escherichia Coli . Microbiology 143, 179–185 (1997).
Jończyk, E., Kłak, M., Międzybrodzki, R. & Górski, A. The influence of external factors on bacteriophages—review. Folia Microbiol. (Praha) 56, 191–200 (2011).
Ly-Chatain, M. H. The factors affecting effectiveness of treatment in phages therapy. Front. Microbiol. 5 (2014).
Ma, Y. et al. Microencapsulation of Bacteriophage Felix O1 into Chitosan-Alginate Microspheres for Oral Delivery. Appl. Environ. Microbiol. 74, 4799–4805 (2008).
Wirth, T. et al. Sex and virulence in Escherichia coli: an evolutionary perspective. Mol. Microbiol. 60, 1136–1151 (2006).
Hamdi, S. et al. Characterization of Five Podoviridae Phages Infecting Citrobacter freundii . Antimicrob. Resist. Chemother. 1023, doi: 10.3389/fmicb.2016.01023 (2016).
Duplessis, M., Russell, W. M., Romero, D. A. & Moineau, S. Global gene expression analysis of two Streptococcus thermophilus bacteriophages using DNA microarray. Virology 340, 192–208 (2005).
Fortier, L.-C. & Moineau, S. Morphological and Genetic Diversity of Temperate Phages in Clostridium difficile . Appl. Environ. Microbiol. 73, 7358–7366 (2007).
Sambrook, J. F. & Russel, D. W. Molecular Cloning: A Laboratory Manual, 3rd ed., Vols 1, 2 and 3J.F. Sambrook and D.W. Russell, ed., Cold Spring Harbor Laboratory Press, 2001, 2100 pp., soft cover | Sigma-Aldrich. (2001).
Chibani Azaïez, S. R., Fliss, I., Simard, R. E. & Moineau, S. Monoclonal Antibodies Raised against Native Major Capsid Proteins of Lactococcal c2-Like Bacteriophages. Appl. Environ. Microbiol. 64, 4255–4259 (1998).
Deveau, H., Van Calsteren, M.-R. & Moineau, S. Effect of Exopolysaccharides on Phage-Host Interactions in Lactococcus lactis . Appl. Environ. Microbiol. 68, 4364–4369 (2002).
Rombel, I. T., Sykes, K. F., Rayner, S. & Johnston, S. A. ORF-FINDER: a vector for high-throughput gene identification. Gene 282, 33–41 (2002).
Lukashin, A. V. & Borodovsky, M. GeneMark.hmm: new solutions for gene finding. Nucleic Acids Res. 26, 1107–1115 (1998).
Laslett, D. & Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16 (2004).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Katoh, K. & Standley, D. M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 30, 772–780 (2013).
Darriba, D., Taboada, G. L., Doallo, R. & Posada, D. ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165 (2011).
Shimodaira, H. An Approximately Unbiased Test of Phylogenetic Tree Selection. Syst. Biol. 51, 492–508 (2002).
Junier, T. & Zdobnov, E. M. The Newick utilities: high-throughput phylogenetic tree processing in the Unix shell. Bioinformatics 26, 1669–1670 (2010).
Letunic, I. & Bork, P. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. gkr201, doi: 10.1093/nar/gkr201 (2011).
Kristensen, D. M. et al. A low-polynomial algorithm for assembling clusters of orthologous groups from intergenomic symmetric best matches. Bioinformatics 26, 1481–1487 (2010).
R Development Core Team. R: a language and environment for statistical computing. Vienna, Austria: the R Foundation for Statistical Computing. ISBN: 3-900051-51-07-0. http://www.R-project.org/(2011). Available at: http://www.gbif.org/resource/81287 (Accessed: 12th July 2016).
Warnes, G. et al. gplots: Various R programming tools for plotting data, R package version 2.14.0. Available at: http://www.crantastic.org/packages/gplots/versions/34110 (Accessed: 12th July 2016).
Acknowledgements
We would like to thank Barbara-Ann Conway (Medical Writer & Editor) for editorial assistance. S.M. holds a Tier 1 Canada Research Chair in Bacteriophages.
Author information
Authors and Affiliations
Contributions
S.M., K.S., R.K. conceived and designed the study. S.M. afforded the materials and critically revised the manuscript. S.H. performed experiments, did the bioinformatics analysis and drafted the manuscript. G.R. participated in the data analysis and helped in the coordination of the experiments. D.T. did the genome sequencing and the electron microscopy analysis. S.L. designed the figures and helped in the bioinformatics analysis. All authors read and approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Hamdi, S., Rousseau, G., Labrie, S. et al. Characterization of two polyvalent phages infecting Enterobacteriaceae. Sci Rep 7, 40349 (2017). https://doi.org/10.1038/srep40349
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep40349
This article is cited by
-
Dominance of phage particles carrying antibiotic resistance genes in the viromes of retail food sources
The ISME Journal (2023)
-
Systematic analysis of putative phage-phage interactions on minimum-sized phage cocktails
Scientific Reports (2022)
-
Duck sewage source coliphage P762 can lyse STEC and APEC
Virus Genes (2022)
-
Genomic characterization of three bacteriophages targeting multidrug resistant clinical isolates of Escherichia, Klebsiella and Salmonella
Archives of Microbiology (2022)
-
Isolation, phenotypic characterization and comparative genomic analysis of 2019SD1, a polyvalent enterobacteria phage
Scientific Reports (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
