
Abstract
Biological hydrogen production is based on activity of specific enzymes called hydrogenases. Hydrogenases are oxygen sensitive metalloenzymes containing Ni and/or Fe atoms at the active site, catalyzing reversible reduction of protons. Generally, [Fe-Fe] hydrogenases prefer proton reduction to molecular hydrogen, a potential energy carrier molecule that can be produced by bioprocesses in sustainable manner. Thus, monitoring tools have been developed to study the relationship between [Fe-Fe] hydrogenases and biohydrogen production in bioreactors at DNA and RNA levels. In the present study, novel molecular tools are introduced for quantitative monitoring of clostridial [Fe-Fe] hydrogenases at the protein level. Aerobic and anaerobic biopanning (for inactive and active [Fe-Fe] hydrogenase, respectively) of phage displayed single-chain variable fragment (scFv) antibody libraries aided in isolating nine potential scFvs. The enriched antibodies demonstrated high specificity towards Clostridium spp. [Fe-Fe] hydrogenases allowing detection from pure and mixed cultures. Additionally, the antibodies showed different binding characteristics towards hydrogenase catalytic states, providing a possible means for functional detection of clostridial [Fe-Fe] hydrogenases. From hydrogenase-antibody interaction studies we observed that though antibody binding reduced the enzyme catalytic activity, it facilitated to retain hydrogen evolution from oxygen exposed hydrogenases.
Similar content being viewed by others


Introduction
Depletion of traditional energy reserves, global warming and increased environmental pollution have strongly urged for alternative energy sources. Hydrogen (H2) is considered an alternative energy carrier due to its high energy yield, low heating value and non-polluting emission1. To increase sustainability in energy production, H2 can be produced biologically by dark fermentation using organic wastes as substrates in bioprocesses2.
Hydrogenases are the key enzyme involved in the metabolism of molecular H2. The enzyme is grouped into three classes based on the metal cofactor present at the active site, namely [Fe-Fe], [Ni-Fe] and [Fe] hydrogenases. In general, hydrogenases catalyze the reversible conversion of dihydrogen to protons and electrons through the reaction, H2 ↔ 2H+ + 2e− 3. However, the oxygen (O2) sensitivity of hydrogenase is a limitation for some practical applications4. Studies indicate that O2 enters to the enzyme active site through the gas migration channels and on binding to the [Fe] moiety at the active site, generates reactive O2 species that destroys the enzyme catalytic function5,6,7.
Among the hydrogenase classes, [Fe-Fe] hydrogenases favors in proton reduction to molecular H2 and exhibits the highest catalytic activity4. This makes [Fe-Fe] hydrogenases the natural target for molecular prospections in biohydrogen production systems8. In bioprocess, quantitative monitoring of hydrogenase enzymes is important and can provide more in-sight on the process performance. Several groups have described tools for hydrogenase monitoring at DNA and RNA levels and reported how quantitative analyses of hydrogenases can reveal important clues about the bioprocess performance8,9,10. Previously our group reported the means to quantify Clostridium butyricum [Fe-Fe] hydrogenase at gene and transcript levels in an open bioprocess system10. Later on, the application of quantitative PCR and melting curve analysis of clostridial [Fe-Fe] hydrogenase as monitoring tools in an open bioreactor, assisting in elucidating biohydrogen production and changes in the functional community during the open fermentation process was investigated8. The relationship between H2 production kinetics and gene transcript levels of several Clostridium spp. isolated from continuous stirred tank reactor was investigated by Morra et al.9. The authors reported similar transcriptional profiles, but different H2 evolution kinetics among the isolates indicating a post-transcriptional down regulation of clostridial hydrogenases9. In the present study, to add another level for molecular prospections of hydrogenase enzyme in biological processes, we describe the isolation of specific antibodies for clostridial [Fe-Fe] hydrogenases.
Display of proteins or peptide libraries on the surface of filamentous phages, invented by Smith in 1985, has become a widely applied and powerful tool for screening antibodies for different molecular targets11. With the help of phage display, screening conditions can be adjusted according to specific needs. More specifically, phage displayed libraries have been used in protein engineering12, protein-protein interaction studies13, epitope mapping14, specific screening and quantification15, recognizing function-specific16 and diverse protein conformations17,18 of aerobic enzymes. However, screening of antibodies specific towards O2 labile targets has been rarely conducted. Bruggeman et al.19, have reported successful isolation of recombinant antibodies specific towards O2 sensitive flavin. Here, we report the enrichment of recombinant antibodies with specific binding characteristics towards [Fe-Fe] hydrogenases from Clostridium spp. using aerobic and anaerobic biopanning techniques.
Results
Biopanning for anti-hydrogenase antibodies
Biopanning of phage displayed antibody libraries were conducted to enrich and isolate scFvs specific towards catalytically active and inactive Clostridium acetobutylicum [Fe-Fe] hydrogenases. Streptavidin/avidin paramagnetic beads and streptavidin/neutravidin plates were chosen as the binding platforms for antibody panning of chemically biotinylated inactive and active hydrogenases, respectively. The biopanning surfaces were alternated at each panning round to avoid unspecific enrichment. Prior to biopanning, the purity of His-tag purified C. acetobutylicum hydrogenases were analyzed by SDS-PAGE (see Supplementary Fig. S1).
Under anoxic conditions, [Fe-Fe] hydrogenases are capable of reducing protons to molecular H2. However, upon exposure to O2, the catalytic activities of hydrogenase enzyme are irreversibly inactivated. Consequently, prior to the panning against active hydrogenases, the buffers were purged with nitrogen gas and stored in an anaerobic glove box. The antibody panning was conducted under strict anoxic conditions in anaerobic glove box. In the case for inactive hydrogenases, the biopanning was performed aerobically with O2 saturated buffers. The percentage ratio of output to input phages from biopanning panning rounds indicated clear phage enrichments (see Supplementary Table S1).
Cloning the enriched scFv genes from pEB32X phagemid vector into pAK600 expression vector allowed screening antibodies specific towards the target antigens. Ninety four (from inactive panning, see Supplementary Fig. S2) and ninety two (from active panning, see Supplementary Fig. S3) single clones were randomly selected and tested for antigen binding by alkaline phosphatase assay. Based on the signal level, twelve and eight scFvs that specifically recognized inactive and active hydrogenases, respectively, were selected. In the following text, the selected antibodies will be specified by the clone number and suffixes ‘In’ and ‘Ac’, representing antibodies screened from inactive and active biopanning, respectively. Sequencing revealed that all the 8 antibodies recognizing active hydrogenase (7Ac, 23Ac, 31Ac, 43Ac, 49Ac, 59Ac, 82Ac and 88Ac) were different and among the twelve inactive hydrogenase specific antibodies only two unique clones (7In and 48In) were found. The amino acid sequences of complementary determining regions in the selected scFvs are presented in Supplementary Table S2.
Antibodies recognize hydrogenases from Clostridium spp. and differentiate between enzyme functional forms
The scFv antibody clones were investigated for their binding towards hydrogenases in Clostridium spp. by sandwich immunoassay. The supernatant obtained after centrifuging the lysed cells (lysate) were employed as immunoassay antigens. The scFv genes (48In, 7Ac, 23Ac, 31Ac, 43Ac, 49Ac, 59Ac, 82Ac and 88Ac) (see Supplementary Table S3) and were cloned to pAK400cb vector, which allows to express the scFvs as an N-terminal fusion with biotin carboxyl carrier protein (BCCP) domain in the cytoplasm of Escherichia coli Origami B20. A biotin moiety is enzymatically added to BCCP by the cell, and hereby the scFv-BCCP fusion affords a straightforward way for the production of biotinylated antibodies for efficient immobilization. With the exception of 7In, expression and in vivo biotinylation of scFv-BCCP fusion proteins was successful in the redox modified E. coli strain.
Upon confirmation of hydrogenase activity (see Supplementary Table S4), sandwich immunoassay was performed with C. acetobutylicum lysate, scFv-BCCP (capture antibody) and scFv-phoA (tracer antibody) fusion proteins. The lysates of E. coli XL1 and Origami B carrying empty plasmids were included as negative controls. Clearly detectable absorbance (A405 nm) values were observed for all scFv-BCCP fusions with 49Ac-phoA, 59Ac-phoA and 82Ac-phoA fusions as tracer antibodies. Whereas, negligible or very low signals were observed with 7Ac, 23Ac, 31Ac, 43Ac and 88Ac as tracer antibodies (Fig. 1a). High absorbance values were observed when 48In was used either as capture or tracer antibody. Based on the results, 82Ac and 48In were the best performing tracer antibodies. The order of capture scFv-BCCP fusions in accordance to the signal obtained with 82Ac as tracer was: 48In > 7Ac > 59Ac > 49Ac > 88Ac > 82Ac > 31Ac > 43Ac > 23Ac.

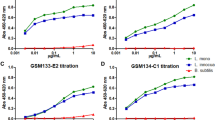
Sandwich and one-step immunoassays of C. acetobutylicum hydrogenases (affinity purified and in crude cell lysates) with the isolated scFvs.
The above graphs depicts (a) the binding specificities of the enriched antibody clones (7Ac, 23Ac, 31Ac, 43Ac, 49Ac, 59Ac, 82Ac, 88Ac and 48In) towards C. acetobutylicum lysate, (b) dose response immunoassay with the 7Ac-BCCP and 82Ac-phoA immunoassay pairs against C. acetobutylicum lysate and purified C. acetobutylicum hydrogenase. The data points from C. acetobutylicum lysate (Empty square), and purified active hydrogenase (filled star) were fitted linearly (The solid line represents the linear regression; R-square values: 0.99, purified active hydrogenase; 0.99, C. acetobutylicum lysate) and (c) one-step immunoassay demonstrating the binding specificities of the enriched scFvs’ towards affinity purified and biotinylated active and inactive hydrogenases. The data represents mean absorbance values (A405 nm) subtracted from the background signals (Sandwich immunoassay, E. coli XL1 and Origami B lysates containing empty pAK600 and pAK400cb plasmids; One-step Immunoassay, E. coli XL1 lysate containing empty pAK600 plasmid). The error bars indicate standard deviation of averaged data (A405) from triplicate microtiter wells.
The assay configuration based on the antibody pair 7Ac (capture) and 82Ac (tracer) was tested against varying concentrations of C. acetobutylicum lysate and recombinant active C. acetobutylicum [Fe-Fe] hydrogenase (Fig. 1b). E. coli XL1 and Origami B lysates containing pAK600 and pAK400cb vectors devoid of scFv genes were included as negative controls. A linear fit was observed with escalated concentrations of purified active hydrogenase (R-square, 0.99) and C. acetobutylicum lysate (R-square, 0.99) concentrations (Fig. 1b).
To confirm the binding specificity of scFvs towards functionally different forms of [Fe-Fe] hydrogenases, the scFv-phoA fusions (7Ac, 23Ac, 31Ac, 43Ac, 49Ac, 59Ac, 82Ac, 88Ac, 7In and 48In) was tested in one-step immunoassay for their specificity towards the biotinylated recombinant active and inactive C. acetobutylicum hydrogenases (see Supplementary Table S4). The antibodies exhibited different binding characteristics towards catalytically active and inactive hydrogenases. From the experimental data presented in Fig. 1c, it can be inferred that antibodies 7Ac, 23Ac, 31Ac, 43Ac, 59Ac, 82Ac and 88Ac exclusively recognized the active hydrogenase, whereas this was not the case for the clones 49Ac, 7In and 48In.
From bioprocess monitoring perspective, it is important to test whether the selected antibodies are capable of recognizing hydrogenases from closely related Clostridium spp. For that reason, C. butyricum, an efficient mesophilic H2 producer, was chosen in this study. The amino acid sequence similarity of [Fe-Fe] hydrogenases from C. acetobutylicum (AAB03723) and C. butyricum (ACD62594.1) by ClustalW2 was identified to be 67% (see Supplementary Fig. S4).
After confirming hydrogenase activity in the lysate (see Supplementary Table S4), the sandwich immunoassay configurations recognizing the C. acetobutylicum [Fe-Fe] hydrogenase were investigated for their capacity to detect [Fe-Fe] hydrogenase from C. butyricum. The assay was performed in an anaerobic glove box with the inclusion of blank E. coli XL1-pAK600 and Origami B-pAK400cb carrying empty plasmids as negative controls. Interestingly, absorbance signals were obtained from all the binder combinations tested (Fig. 2a). Among the tracer antibodies tested, 49Ac-phoA gave the highest signal with most of the capture antibodies tested and the immunoassay pair exhibiting the highest signal was 23Ac-BCCP and 49Ac-phoA. The order of capture antibodies (BCCP fusions) in accordance to the signal obtained with 49Ac tracer is: 23Ac > 7Ac > 43Ac > 48In > 31Ac > 49Ac > 59Ac > 88Ac > 82Ac.

Sandwich immunoassay with scFv-BCCP and scFv-phoA fusion proteins as capture and tracer antibodies, respectively.
The above graphs depict (a) the binding specificities of the enriched anti-hydrogenase antibodies towards C. butyricum lysate and (b) linear regression (R2 = 0.98) data from sandwich immunoassay using 23Ac-BCCP and 49Ac-phoA immunoassay pairs and C. butyricum lysate. The plotted data represents blank (E. coli lysates containing empty pAK400cb and pAK600 plasmids) subtracted mean absorbance values (A405 nm) and standard deviations from triplicate microtiter wells.
Dose-dependent immunoassay with 23Ac-BCCP (capture antibody) and 49Ac-phoA (tracer antibody) was performed on streptavidin coated microtiter wells against increasing concentrations of C. butyricum lysate. Figure 2b depicts the mean absorbance signal obtained from triplicate microtiter well readings with 0–600 mg L−1 of antigen. A linear fit with an R-square value of 0.98 in absorbance response observed during the immunoassay.
scFvs bind selectively to [Fe-Fe] hydrogenases
In order to obtain purified antibodies for the following experiments, the scFv genes were cloned from pAK600 into pLK06H vector and transformed into E. coli XL1 strain. The cells were induced with 100 μM IPTG overnight and lysed. The filtered lysates were purified (His-tag based immobilized metal affinity chromatography) and buffer exchanged with 1X phosphate buffered saline (PBS). The purified proteins were analyzed by SDS-PAGE (see Supplementary Fig. S5).
As the antibodies employed in study showed different expression profiles, scFv-phoA-(6X)His fusions discerned as strong bands in SDS-PAGE (59Ac, 82Ac and 48In) were selected as tracer antibodies for one-step immunoassay with chemically biotinylated antigen lysates [C. acetobutylicum (positive control), enriched activated sludge sample21 and E. coli XL1 (negative control)]. Background phoA activity was checked by including His-tag purified empty E. coli XL1-pAK600 as tracer antibody. The mean alkaline phosphatase signals from triplicate well readings corresponding to antigen-antibody interaction are presented in Fig. 3. Similar to the previous results, the scFvs recognized hydrogenases in the positive control (C. acetobutylicum lysate). Relative to data obtained from XL1-pAK600 lysate, purified 59Ac, 82Ac and 48In antibodies generated 2–2.5 fold higher phoA activities with C. acetobutylicum lysate as assay antigen. Inclusion of an enriched activated sludge sample (predominated with Clostridium spp.)21, enabled to investigate whether the antibodies recognized [Fe-Fe] hydrogenases from mixed microbial consortia. Antibodies 59Ac and 82Ac recognized the antigen in activated sludge sample and on comparison with XL1-pAK600 yielded higher phoA activity. Alkaline phosphatase activity by phoA gene was not recorded for any anti-hydrogenase antibodies with E. coli lysate as immunoassay antigen. These results indicate that the antibodies are capable of recognizing hydrogenases from mixed microbial cultures. Additionally, the experimental data confirms the antibody binding specificity towards [Fe-Fe] hydrogenases.

One-step immunoassay with affinity purified scFv-phoA fusion proteins [59Ac, 82Ac, 48In and XL1-pAK600 (empty vector)] and biotinylated antigens (C. acetobutylicum, activated sludge and E. coli lysates).
The horizontal dashed line indicates alkaline phosphatase activity level from immunoassay tests with the empty vector and respective antigen lysates. The graphical data represents mean absorbance values (A405 nm) and error bars from triplicate microtiter wells.
Antigen-antibody interactions impart changes in enzyme characteristics
The effect of scFv binding on hydrogenase enzyme characteristics was investigated. The changes in enzyme activities imparted by antigen-antibody interactions were studied using recombinant hydrogenases and scFv immobilized on streptavidin beads (7Ac, 59Ac, 82Ac and 48In as fused to BCCP domain). Prior to experiment, the presence of scFv on streptavidin beads was confirmed by sandwich immunoassay with purified hydrogenase antigen and 82Ac-phoA as tracer antibody (see Supplementary Table S5). Lysates from cells harboring empty vectors (Origami-pAK400cb and XL1-pAK600) were used as assay controls. After confirmation, the streptavidin beads coated with scFvs were incubated anaerobically with catalytically active recombinant hydrogenase in rotating mixer [1 hour at room temperature (RT)] in two sets. Until the wash step, both the sets were prepared in similar conditions. The washes were conducted in parallel for both the sets, i.e. in Set 1, antigen-scFv-beads were washed with anaerobic TBT-0.05 amended with 20 mM sodium dithionite and for Set 2 using aerobic TBT-0.05 buffer. As washing removes the unbound hydrogenases, H2 evolution is performed solely by the scFv bound hydrogenases.
In the experiment, two controls were included. Bead control (purified hydrogenases coated to streptavidin beads, i.e. no scFvs’) demonstrates the background activity of antigen coated to antibody-null streptavidin beads. Enzyme control (purified hydrogenases in aerobic and anaerobic 50 mM Tris-HCl) enabled to identify the original enzyme catalytic activity and validate hydrogenase inactivation on exposure to O2 saturated buffer. The mean H2 productions (nmol) and standard deviations from triplicate methyl viologen oxidation experiments (in 2 ml reaction buffer, anoxic 50 mM Tris-HCl supplemented with 5 mM methyl viologen as electron donor) are presented in Table 1. In Bead control, Set 1 produced negligible enzyme activity and headspace H2 content was absent when washed with aerobic TBT-0.05 (Bead control, Set 2). Similar O2 mediated enzyme inactivation was observed in Enzyme control (Set 2). In comparison with Enzyme control, the hydrogenase catalytic activity was greatly affected with scFv binding (see Supplementary Table S6). The interactions with scFvs resulted in 3–5 fold reduced H2 production. Interestingly, upon exposure to O2 the antigen-scFv complex (Set 2) retained methyl viologen oxidation property, producing comparable H2 to the corresponding sample in Set 1. The percentage drop in enzyme activity upon O2 exposure for antigen bound 7Ac, 59Ac, 82Ac and 48In antibodies were 25%, 11%, 54% and 73%, respectively. The t-Test results for the experimental data (n = 3; Table 1) postulates that the drop in enzyme activity observed for hydrogenase bound 82Ac and 48In antibodies is statistically significant (82Ac, p = 0.027; 48In, p = 0.025). Whereas, antigen bound to 7Ac and 59Ac antibodies imparted statistically insignificant (7Ac, p = 0.189; 59Ac, p = 0.649) decrease in hydrogenase catalytic activity.
Discussion
H2 gas has a pivotal role in many biological processes as energy carrier. Since hydrogenases are the key enzymes involved in H2 metabolism in biological systems, they are rational targets for such molecular level explorations8,9,10. The present study aims in employing anti-hydrogenase antibodies as tools for protein level monitoring of hydrogenases.
Enrichment and isolation of antibodies recognizing O2 labile proteins is a challenging task. The target enzyme in this study is irreversibly inactivated in presence of O2, and such process might be reflected as changes in the epitopes potentially recognized the antibodies. Inactivation of [Fe-Fe] hydrogenase on exposure to O2 undergoes several steps. Under aerobic conditions O2 enters into the protein structure and binds to enzyme catalytic site through gas migration channels, generating reactive oxygen species4,5,6,7. This imparts irreversible oxidative damage of electron transfer domains and active site. Thus, aerobic and anaerobic biopanning of phage displayed antibody libraries were conducted against purified inactive and active C. acetobutylicum hydrogenases, respectively. From the results, we observed increased phage enrichment from aerobic biopanning (see Supplementary Table S1) and amino acid sequence similarities among antibodies enriched against inactive hydrogenases: only two unique clones were found. This may indicate the loss of antigenic epitopes as the hydrogenase structural integrity is affected during the inactivation process22.
In order to employ the enriched scFvs’ as monitoring tool in bioprocess systems, we studied their binding characteristics towards hydrogenases from different Clostridium spp. and mixed microbial communities23. Firstly, the scFvs enriched against C. acetobutylicum [Fe-Fe] hydrogenases were capable of recognizing the target antigen from C. butyricum. This binding characteristics apparently reflects the relatively high degree of similarity among clostridial [Fe-Fe] hydrogenases at protein level24. The amino acid sequence involved in the formation of the active site at the C-terminal region of [Fe-Fe] hydrogenases is highly conserved among clostridial type [Fe-Fe] hydrogenases25,26. By selecting optimal antibody pairs, more specific hydrogenase recognition can be obtained: antibody pair (7Ac-BCCP and 82Ac-phoA) showed stronger binding characteristics towards C. acetobutylicum [Fe-Fe] hydrogenase compared with [Fe-Fe] hydrogenase from C. butyricum (Figs 1a and 2a). Secondly, the immunoassay results (Fig. 3) demonstrates that the scFvs clear recognition profile [Fe-Fe] hydrogenases. These interpretations points towards the capacity of scFvs from this study as monitoring tools in hydrogen-fermenting systems.
Previous reports indicate that scFvs interactions with active enzymes may demonstrate reduced or inhibitory effects on the enzyme catalytic property27,28. Similar antibody mediated changes in enzyme properties were observed in this study. On comparison with Enzyme control (see Supplementary Table S6), hydrogenases bound to antibody showed reduced catalytic activities. When exposed to O2, hydrogenases bound to 82Ac and 48In antibodies conferred a drop in H2 production. Very interesting finding was that the hydrogenase bound by 7Ac and 59Ac scFv retained methyl viologen oxidation property in the studied conditions (Table 1). This inflection in enzyme characteristics allows to speculate that the antibodies 7Ac and 59Ac recognized epitopes near to hydrogenase gas migration channels. However, this finding requires more experimental validations and will be scrutinized in future investigations.
This is the first study to report successful generation of recombinant antibodies recognizing anaerobic microbial [Fe-Fe] hydrogenases. The antibodies described here can be useful for the protein level monitoring purposes when applied together with other molecular monitoring tools. Future investigations on epitope mapping, elucidating scFv binding kinetics and antibody interactions with [Fe-Fe] hydrogenases from a phylogenetically distinct source will illuminate immense potentials in biotechnological applications, for example in affinity-purification of [Fe-Fe] hydrogenases for structural studies and attempts to improve the O2 tolerance of [Fe-Fe] hydrogenases by specific molecular interactions.
Materials and Methods
The methods for recombinant expression and purification of C. acetobutylicum hydrogenase, preparation of bacterial cell lysates and methyl viologen oxidation assay are provided in Supplementary Information.
Phage libraries, helper phage and solid surfaces
High-diversity phage-displayed antibody libraries and VCS-M13 helper phage (Stratagene, USA) were used in the biopanning process29. Dynabeads MyOne Streptavidin T1 (Invitrogen, USA) and avidin paramagnetic beads (Gentaur Molecular Products, Belgium) were selected as solid surfaces for aerobic panning (inactive hydrogenase). For anaerobic panning (active hydrogenase) streptavidin (Kaivogen, Finland) and neutravidin (Pierce NeutrAvidin, Thermo Scientific, USA) coated microtiter plates were used.
Bacterial strains, plasmids and culture media
E. coli XL1 (Stratagene, USA) strain was used for phage infection, production of enriched phages and expression of scFv-phoA fusion protein in pAK600 vector. The pAK600 vector assists in periplasmic expression of scFv-bacterial alkaline phosphatase (phoA) fusion protein. Origami B (Novagen, USA) strain harboring pAK400cb vector was used for expressing scFv-BCCP fusion proteins for sandwich immunoassay20. Recombinant antibody expression in E. coli XL1 was conducted by cloning scFv genes into pLK06H vector using SfiI restriction sites. pLK06H is an ampicillin-resistant version of pAK600 vector constructed to introduce hexhistag at the C-terminal end of phoA gene29.
Antibody recognition towards hydrogenases in bacterial lysates was conducted using C. acetobutylicum DSM 792, C. butyricum DSM 2478, activated sludge predominated with Clostridium spp.21 and E. coli XL1 lysates. E. coli BL21 (DE3) ∆iscR pFEGA was used for recombinant expression of C. acetobutylicum hydrogenase30. Low salt Lysogeny agar (LA) plates and Super broth (SB) medium were prepared as described31. The biopanning reagents such as Polyethylene Glycol 8000-Sodium Chloride [PEG8000-NaCl], Tris Base-Sodium Chloride [TBS; pH 7.5], TBS-Bovine Serum Albumin-Tween 20 (0.5%) [TBT-0.5; pH 7.5], TBT-0.05 (pH 7.5) and TBS-Sodium azide (0.02% w/v)-Tween 20 (0.05%) [TSAT-0.05; pH 7.5] solutions were prepared as described previously32.
Biopanning
His-tag purified hydrogenases and E. coli BL21 (DE3) lysate were chemically biotinylated (EZ-Link Sulfo-NHS-Biotin, Thermo Scientific, USA) and buffer exchanged twice by NAPTM column (SephadexTM-G-25 DNA grade, GE healthcare, UK) with 1X PBS. Biotinylation and buffer exchange of purified active hydrogenases was performed in an anaerobic glove box (Don Whitley Scientific, UK) and stored anaerobically at 4 °C (Anaerocult, Merk, Germany).
Mixed libraries (phage diversity, 5 × 1012 c.f.u.) were used for the first round of phage display. Streptavidin beads and streptavidin wells were chosen as biopanning platforms for inactive and active hydrogenases, respectively. Subtractive panning was performed by incubating the phage stock with selection platforms and biotinylated blank BL21 (DE3) lysate (70 mg L−1). For the blank lysate, E. coli BL21 (DE3) was grown and lysed by lysozyme (1 g L−1) and freeze-thaw treatments in the presence of 1X protease inhibitor cocktail mix (cOmplete EDTA-free, Roche, Germany). The protein concentrations were measured by QuickstartTM Bradford assay kit (Bio-rad, USA). In anaerobic panning, the input phage library was prepared by incubating the phage stock individually with empty streptavidin wells and BL21 (DE3) lysate for 15 minutes followed with phage precipitation using PEG8000-NaCl solution. For aerobic panning, the subtractive panning was performed by incubating phage stock with BL21 (DE3) lysate coated on streptavidin beads for 5 minutes and thereafter collected. The following biopanning steps were performed as schematically presented in Supplementary Fig. S6.
In the second round, neutravidin coated microtiter plate wells and avidin coated beads were used as selection platforms with reduced phage library (5 × 1011 c.f.u.) and protein concentrations (10 mg L−1). For the final round, streptavidin coated microtiter wells and beads were used with more stringent panning conditions (5 × 1010 c.f.u and 5 mg L−1).
Identification of anti-hydrogenase antibodies
The enriched scFv genes (from 3rd biopanning round) were sub-cloned ‘en-masse’ into pAK600 vector using SfiI restriction sites and transformed to E. coli XL1 cells. Individual clones were cultured in SB medium and scFv expression was induced with 250 μM IPTG (OD600 nm 0.6–0.7) and grown for 16–20 hours (23 °C and 300 rpm). The cells were lysed as previously described and lysate containing scFv-phoA fusion protein was used in the following assays.
First in the screening, chemically biotinylated recombinant C. acetobutylicum hydrogenases (70 mg L−1) were added into pre-washed (TBT-0.05) streptavidin wells and incubated under aerobic (for inactive hydrogenase) or anaerobic (for active hydrogenase) conditions at RT for 1 hour (intermittent shaking). The wells were washed thrice with TBT-0.05 and E. coli XL1 lysates containing scFv-phoA fusion proteins were incubated with immobilized hydrogenases. To determine any unspecific binding, scFv-phoA fusions were incubated separately with empty streptavidin wells and biotinylated BL21 (DE3) lysate. E. coli XL1-pAK600 lysates lacking antibody gene was also included as control to determine background phoA activity. Following the incubation, the wells were washed as described previously and alkaline phosphatase substrate (p-nitrophenyl phosphate, Sigma) was added. The plates were incubated at 37 °C with slow shaking for 1–2 hours. The scFv-phoA fusions with binding towards hydrogenase (measured as alkaline phosphatase activity at A405 nm) were selected and sequenced (Forward primer M13R-pUC(−40), Macrogen).
Screening for antibodies specific towards active hydrogenase was performed in an anaerobic glove box. The buffers for anaerobic immunoassay screening were pre-sparged with N2 for 2 hours and stored in the anaerobic chamber.
Antibody purification
The scFv genes cloned from pAK600 into pLK06H vector were transformed to E. coli XL1 cells. Single clones cultured in SB medium were induced with 100 μM IPTG (16–20 hours at 23 °C/300 rpm). His-tag purification of scFv-phoA fusion proteins was conducted as described by the manufacturer (Novagen, USA).
For antigen-antibody interaction studies, scFv-BCCP fusion proteins (7Ac, 59Ac, 82Ac and 48In) were purified using streptavidin beads. One milliliter of E.coli lysate containing scFv-BCCP fusions were mixed with 20 μl of pre-washed beads (PBS) at RT for 3 hours in a rotating mixer. The beads were washed four times with 1 ml TBT-0.05 buffer and stored in 200 μl TSAT-0.05 buffer at 4 °C. Presence of antibody on streptavidin beads were confirmed by sandwich immunoassay with purified hydrogenase, scFv-BCCP coated beads and 82Ac-phoA as antigen, capture and tracer antibodies respectively. Empty Origami B-pAK400cb and XL1-pAK600 strains were included as immunoassay negative controls.
Immunoassay
In order to obtain biotinylated capture antibodies for sandwich immunoassay, scFv genes [one from aerobic panning (48In) and eight from anaerobic panning (7Ac, 23Ac, 31Ac, 43Ac, 49Ac, 59Ac, 82Ac and 88Ac)] were amplified using rm13_1 and rm13_2 primers (see Supplementary Table S3). The amplified product was sub-cloned into pAK400cb vector using NdeI and EcoRI restriction sites and transformed into Origami B.
E. coli XL1-scFv-phoA (tracer antibody expression) and Origami B-scFv-BCCP (capture antibody expression) strains were grown in SB medium supplemented with 0.1% glucose, 10 mg L−1 tetracycline, 25 mg L−1 chloramphenicol and 15 mg L−1 kanamycin (Origami B). Overexpression of scFv-BCCP and scFv-phoA fusion proteins were initiated at OD600 nm 0.6–0.7 with 250 μM IPTG (23 °C/300 rpm) for 24 and 48 hours, respectively. Following cell lysis, the immunoassay experiments were conducted in anaerobic glove box as described previously. The capture antibodies (scFv-BCCP fusions), antigens (C. acetobutylicum and C. butyricum lysates) and tracer antibodies (scFv-phoA fusions) were added to streptavidin wells in the order. Empty E. coli XL1-pAK600 and Origami B-pAK400cb were included as negative control.
One-step immunoassay was conducted to observe antibody specificities towards [Fe-Fe] hydrogenases in environmental samples. Briefly, the immunoassay was conducted using streptavidin well individually immobilized with 70 mg L−1 biotinylated C. acetobutylicum (positive control), enriched activated sludge sample and E. coli XL1 lysates (negative control) and 10 mg L−1 His-tag purified scFv-phoA fusions (59Ac, 82Ac and 48In). His-tag purified E. coli XL1 lysate was included as the negative control. The alkaline phosphatase activities were calculated as mean signals (A405nm) obtained from triplicate well readings.
The dose dependent immunoassay was conducted under anaerobic conditions using the antibody pairs that exhibited maximal signal for respective target antigens (7Ac-BCCP:82Ac-phoA, C. acetobutylicum lysate and purified active hydrogenase; 23Ac-BCCP:49Ac-phoA, C. butyricum lysate). Empty E. coli XL1-pAK600 and Origami B-pAK400cb lysates were included as negative controls.
Antigen-antibody interactions on enzyme characteristics
Antibodies expressing scFv-BCCP fusions (7Ac, 59Ac, 82Ac and 48In) were grown, induced, lysed and coated to 20 μl streptavidin beads in two Sets. The beads were transferred to anaerobic glove box. Each scFv bound beads were incubated with 40 ng of purified hydrogenase (1 hour/RT). The unbound hydrogenases were removed by washing the beads (4X) in parallel - Set 1 with TBT-0.05 containing 20 mM sodium dithionite and Set 2 with aerobic TBT-0.05. Care was taken to prevent O2 escape from wash buffer by maintaining O2-saturated buffer outside the glove box, keeping the container lid tightly sealed and checking for O2 presence with anoxic water containing 0.002 gL−1 resazurin. After the wash steps, the beads were resuspended in anoxic 1X PBS and the hydrogenase activities were analyzed by methyl viologen oxidation assay. The assay included two controls: Bead control (streptavidin beads coated with purified hydrogenases) and Enzyme control [recombinant hydrogenase (40 ng) prepared in aerobic and anaerobic 50 mM Tris-HCl (pH 8.0)].
Statistical analysis
The data from antigen-antibody interaction studies (individual sample in Set 1 to the corresponding sample in Set 2) were statistically analyzed by Two-Sample t-Test Assuming Unequal Variances using Data Analysis function in Microsoft Excel 2007. The p value [P(T <= t) two-tail] < 0.05 indicates statistical significance.
Additional Information
How to cite this article: Mangayil, R. et al. Recombinant antibodies for specific detection of clostridial [Fe-Fe] hydrogenases. Sci. Rep. 6, 36034; doi: 10.1038/srep36034 (2016).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
Balat, H. & Kirtay, E. Hydrogen from biomass - Present scenario and future prospects. International Journal of Hydrogen Energy 35, 7416–7426 (2010).
Elsharnouby, O., Hafez, H., Nakhla, G. & El Naggar, M. H. A critical literature review on biohydrogen production by pure cultures. International Journal of Hydrogen Energy 38, 4945–4966 (2013).
Stephenson, M. & Stickland, L. H. Hydrogenase: a bacterial enzyme activating molecular hydrogen: The properties of the enzyme. Biochem. J. 25, 205–214 (1931).
Bingham, A. S., Smith, P. R. & Swartz, J. R. Evolution of an [FeFe] hydrogenase with decreased oxygen sensitivity. In International Journal of Hydrogen Energy 37, 2965–2976 (2012).
Cohen, J. et al. Molecular dynamics and experimental investigation of H2 and O2 diffusion in [Fe]-hydrogenase. Biochem. Soc. Trans. 33, 80–82 (2005).
Stripp, S. T. et al. How oxygen attacks [FeFe] hydrogenases from photosynthetic organisms. Proc. Natl. Acad. Sci. USA 106, 17331–17336 (2009).
Swanson, K. D. et al. [FeFe]-hydrogenase oxygen inactivation is initiated at the H cluster 2Fe subcluster. J. Am. Chem. Soc. 137, 1809–1816 (2015).
Tolvanen, K. E. S., Santala, V. P. & Karp, M. T. [FeFe]-hydrogenase gene quantification and melting curve analysis from hydrogen-fermenting bioreactor samples. Int. J. Hydrogen Energy 35, 3433–3439 (2010).
Morra, S. et al. Expression of different types of [FeFe]-hydrogenase genes in bacteria isolated from a population of a bio-hydrogen pilot-scale plant. Int. J. Hydrogen Energy 39, 9018–9027 (2014).
Tolvanen, K. E. S. et al. Profiling the hydA gene and hydA gene transcript levels of Clostridium butyricum during continuous, mixed-culture hydrogen fermentation. Int. J. Hydrogen Energy 33, 5416–5421 (2008).
Smith, G. P. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science 228, 1315–1317 (1985).
Dwyer, M. A., Lu, W., Dwyer, J. J. & Kossiakoff, A. A. Biosynthetic phage display: a novel protein engineering tool combining chemical and genetic diversity. Chem. Biol. 7, 263–274 (2000).
Hertveldt, K., Beliën, T. & Volckaert, G. General M13 phage display: M13 phage display in identification and characterization of protein-protein interactions. Methods Mol. Biol. 502, 321–339 (2009).
Fack, F. et al. Epitope mapping by phage display: Random versus gene-fragment libraries. J. Immunol. Methods 206, 43–52 (1997).
Schirrmann, T., Meyer, T., Schütte, M., Frenzel, A. & Hust, M. Phage display for the generation of antibodies for proteome research, diagnostics and therapy. Molecules 16, 412–426 (2011).
Eisenhardt, S. U., Schwarz, M., Bassler, N. & Peter, K. Subtractive single-chain antibody (scFv) phage-display: tailoring phage-display for high specificity against function-specific conformations of cell membrane molecules. Nat. Protoc. 2, 3063–3073 (2007).
Chung, W. Y. et al. Phage-display derived single-chain fragment variable (scFv) antibodies recognizing conformational epitopes of Escherichia coli heat-labile enterotoxin B-subunit. J. Immunol. Methods 339, 115–123 (2008).
Haque, A. & Tonks, N. K. The use of phage display to generate conformation-sensor recombinant antibodies. Nat. Protoc. 7, 2127–2143 (2012).
Bruggeman, Y. E. et al. Phage antibodies against an unstable hapten: Oxygen sensitive reduced flavin. FEBS Lett. 388, 242–244 (1996).
Santala, V. & Lamminmäki, U. Production of a biotinylated single-chain antibody fragment in the cytoplasm of Escherichia coli. J. Immunol. Methods 284, 165–175 (2004).
Mangayil, R., Karp, M. & Santala, V. Bioconversion of crude glycerol from biodiesel production to hydrogen. Int. J. Hydrogen Energy 37, 12198–12204 (2012).
Buhrke, T., Lenz, O., Krauss, N. & Friedrich, B. Oxygen tolerance of the H2-sensing [NiFe] hydrogenase from Ralstonia eutropha H16 is based on limited access of oxygen to the active site. J. Biol. Chem. 280, 23791–23796 (2005).
Sinha, P., Roy, S. & Das, D. Genomic and proteomic approaches for dark fermentative biohydrogen production. Renew. Sustain. Energy Rev. 56, 1308–1321 (2016).
Gorwa, M. F., Croux, C. & Soucaille, P. Molecular characterization and transcriptional analysis of the putative hydrogenase gene of Clostridium acetobutylicum ATCC 824. J. Bacteriol. 178, 2668–2675 (1996).
Girbal, L. et al. Homologous and heterologous overexpression in Clostridium acetobutylicum and characterization of purified clostridial and algal Fe-only hydrogenases with high specific activities. Appl. Environ. Microbiol. 71, 2777–2781 (2005).
Calusinska, M., Joris, B. & Wilmotte, A. Genetic diversity and amplification of different clostridial [FeFe] hydrogenases by group-specific degenerate primers. Lett. Appl. Microbiol. 53, 473–480 (2011).
Zhou, N., Paemen, L., Opdenakker, G. & Froyen, G. Cloning and expression in Escherichia coli of a human gelatinase B-inhibitory single-chain immunoglobulin variable fragment (scFv). FEBS Lett. 414, 562–566 (1997).
Saini, D., Kala, M., Jain, V. & Sinha, S. Targeting the active site of the placental isozyme of alkaline phosphatase by phage-displayed scFv antibodies selected by a specific uncompetitive inhibitor. BMC Biotechnol. 5, 1–13 (2005).
Huovinen, T. et al. Two ScFv antibody libraries derived from identical VL-VH framework with different binding site des. Protein Eng. Des. Sel. 26, 683–693 (2013).
Akhtar, M. K. & Jones, P. R. Deletion of iscR stimulates recombinant clostridial Fe–Fe hydrogenase activity and H2-accumulation in Escherichia coli BL21(DE3). Appl. Microbiol. Biotechnol. 78, 853–862 (2008).
Sambrook, J. & W Russell, D. Molecular Cloning: A Laboratory Manual. Cold Spring Harb. Lab. Press. Cold Spring Harb. NY 999 (2001).
Brockmann, E. C., Lamminmäki, U. & Saviranta, P. Engineering dihydropteroate synthase (DHPS) for efficient expression on M13 phage. Biochim. Biophys. Acta - Gen. Subj. 1724, 146–154 (2005).
Acknowledgements
The research was funded by the Maj and Tor Nessling Foundation (Project no. 2012356) and The Academy of Finland (Project nos 126974 and 139830). M.K. acknowledges the sabbatical year financial support from The Finnish Cultural Foundation (Suomen Kulttuuri rahasto). The authors wish to thank Hiroshi Takami and Wataru Motsuchi from Fujirebio, Inc. and Dr. Patrik Jones for kindly providing E. coli BL21 (DE3) ∆iscR pFEGA strain.
Author information
Authors and Affiliations
Contributions
U.L. and V.S. designed and conceptualized the project; R.M., M.K., U.L. and V.S. designed the experiments; R.M., performed the experiments; R.M., U.L. and V.S. analyzed the data; U.L. provided the phage displayed antibody libraries, VCSM13 helper phages and supervised the biopanning experiments; M.K. and V.S. supervised the study; R.M. wrote the manuscript and M.K., U.L. and V.S. contributed extensively in revising the manuscript. All authors have read and approved the final manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Mangayil, R., Karp, M., Lamminmäki, U. et al. Recombinant antibodies for specific detection of clostridial [Fe-Fe] hydrogenases. Sci Rep 6, 36034 (2016). https://doi.org/10.1038/srep36034
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep36034
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
