
Abstract
Alpha subunits of heterotrimeric G proteins (Gα) are involved in a variety of cellular functions. Here we report an optogenetic strategy to spatially and temporally manipulate Gα in living cells. More specifically, we applied the blue light-induced dimerization system, known as the Magnet system, and an alternative red light-induced dimerization system consisting of Arabidopsis thaliana phytochrome B (PhyB) and phytochrome-interacting factor 6 (PIF6) to optically control the activation of two different classes of Gα (Gαq and Gαs). By utilizing this strategy, we demonstrate successful regulation of Ca2+ and cAMP using light in mammalian cells. The present strategy is generally applicable to different kinds of Gα and could contribute to expanding possibilities of spatiotemporal regulation of Gα in mammalian cells.
Similar content being viewed by others

Introduction
Heterotrimeric guanine nucleotide-binding proteins (heterotrimeric G proteins) are made up of alpha (Gα), beta (Gβ) and gamma (Gγ) subunits. They act as molecular switches inside cells, and their main function is signal transduction working together with G protein-coupled receptors (GPCR). Gα dissociates from the Gβγ dimer when responds to a ligand-induced conformation change of GPCR, and then activates effector proteins in particular signal transduction pathways, evoking downstream signaling cascades1.
To achieve precise manipulation of Gα, chemically-inducible dimerization (CID) system2 has been applied previously. Putyrski and Schultz developed a rapamycin-based system to bypass the GPCR by direct activation of Gα, which induces downstream signaling cascades. They demonstrated that the plasma membrane recruitment of constitutively active form of Gαq (herein after referred to as Gαq) and Gαs results in the activation of their effectors3,4. Despite the achievement gained through the robust protein-protein interaction using CID system, it hardly reaches spatiotemporal control because of the irreversibility and the diffusiveness of the chemical dimerization. On the other hand, optogenetic tools like optoXRs5, which allow achieving high spatiotemporal precision, have been developed for manipulation of Gα. OptoXRs are composed of an extracellular component derived from light sensitive rhodopsin and an intracellular component derived from GPCR. A limitation of optoXRs is that rhodopsin gets broad absorption band. Practically, fluorescent tools used as biomarkers or biosensors are commonly required to evaluate the effect of the optogenetic perturbation on the cell. However, optoXRs are often incompatible with these tools because the excitation wavelength of the fluorescent tools may potentially perturb the function of optoXRs having broad absorption band.
In this study, we certify a strategy that Gα achieves activation via light-controlled translocation of Gα from the cytoplasm to the plasma membrane using photoswitchable dimerization systems. In contrast to optoXRs, this strategy affords unique advantage in selectable usage of photoswitchable dimerization systems having narrow absorption spectrums, thereby allowing the feasibility of combinational application with other fluorescent tools. Additionally, this strategy is generally applicable to different classes of Gα. To be more specific, we report individual approaches to manipulate Gαq and Gαs based on the Magnet system and the PhyB/PIF6 system, and we show the light-dependent regulation of two second messengers: Ca2+ and cAMP.
Results
Design scheme
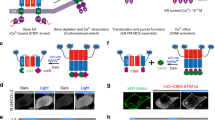
Magnet system consisting of nMagHigh1 and pMagFast1 is a recently developed dimerization system6, which is based on a photoreceptor VIVID derived from Neurospora crassa7,8,9. Heterodimerization occurs between nMagHigh1 and pMagFast1 upon blue light illumination (Fig. 1A). The Magnet system shows high interaction affinity and fast switch-off kinetic, absorbs a narrow spectrum of blue light (Supplementary Fig. S1) and its heterodimerization can be induced with low light power. These facilitative properties of the Magnet system show the feasibility to gain precise spatiotemporal control of Gα. In utilization of the photoswitchable Magnet system, we designed a strategy that blue light induces Gαq to move from the cytoplasm toward the plasma membrane, where Gαq activates the effector phospholipase C-beta (PLCβ) and triggers the cytosolic Ca2+ release (Fig. 1A).

The present approach for manipulating Ca2+ via Gαq using the Magnet system.
(A) Light-inducible dimerization of the Magnet system allows Gαq to translocate to the plasma membrane and trigger cytosolic Ca2+ release through the phospholipase C-beta (PLCβ)-inositol triphosphate receptor (IP3R) pathway. (B) Constructs for nMagHigh1-mKikGR-CAAX and pMagFast1-Gαq.
In this translocation-based approach, pMagFast1 linked with Gαq at the C-termini is localized in the cytoplasm, while nMagHigh1 tagged with mKikGR is anchored to the plasma membrane using a membrane localization sequence (CAAX motif)10. The fluorescent protein mKikGR can be used to monitor the expression level of the probe and mark the cells of interest. After blue light stimulation, heterodimerization occurs between nMagHigh1 and pMagFast1, and thereby induces the translocation of pMagFast1-linked Gαq from the cytoplasm to the plasma membrane. The translocation of Gαq to the plasma membrane activates the downstream effector PLCβ, which produces inositol 1,4,5-triphosphate (IP3) and diacylglycerol (DAG). Subsequently, IP3 binds to IP3 receptor (IP3R) on endoplasmic reticulum (ER) and leads to the release of Ca2+ from the intracellular store sites to the cytosol3,11,12.
Blue light-induced membrane recruitment of Gαq using the Magnet system
Translocation to the plasma membrane induced by the dimerization of nMagHigh1 and pMagFast1 is a prerequisite for the activation of Gαq. To definitively ascertain the interaction between nMagHigh1 and pMagFast1 upon blue light stimulation, we performed the cell-based translocation assay using a total internal reflection fluorescence (TIRF) microscope. nMagHigh1-mKikGR-CAAX and DsRedEx2-pMagFast1-Gαq were coexpressed in COS-7 cells. nMagHigh1 was targeted to the plasma membrane while pMagFast1 was targeted to the cytoplasm. We traced the fluorescence intensity of DsRedEx2 in the plasma membrane and observed a remarkable increase of TIRF signal directly after 488 nm laser illumination (Supplementary Fig. S2A). In order to verify the light dependency of the fluorescence change, we introduced C71S8 mutation to nMagHigh1 and pMagFast1, which impairs the photoswitching dimerization of these two proteins. As expected, the C71S substitutions generated scarcely TIRF signal change upon 488 nm light illumination (Supplementary Fig. S2B). These results demonstrate that the dimerization of nMagHigh1 and pMagFast1 is switched on upon blue light illumination, and this blue light-dependent dimerization can induce the recruitment of cytosolic DsRedEx2-pMagFast1-Gαq to the nMagHigh1-decorated plasma membrane.
Blue light-induced Ca2+ release
nMagHigh1-mKikGR-CAAX and pMagFast1-Gαq were transfected to the human embryonic kidney 293 (HEK 293) cells. As confirmed by the fluorescence of mKikGR, nMagHigh1 domain was anchored to the plasma membrane (Fig. 2A). To quantify the Ca2+ level induced by activated Gαq, we transfected a Ca2+ indicator R-GECO113, which was excited at 559 nm spectrally distinct from the absorption spectrum of the Magnet system (Supplementary Fig. S1). Ca2+ responses were transiently evoked in the cells immediately after nMagHigh1 and pMagFast1 were activated upon blue light illumination at 473 nm (Fig. 2B). This result demonstrates that blue light-dependent membrane recruitment of pMagFast1-Gαq induces the Ca2+ release in the cells. In contrary, cells in the absence of the light stimulation failed to detect any Ca2+ response, implying the excitation light (559 nm) for R-GECO1 did not perturb the interaction of the Magnet system as expected from its absorption spectrum (Fig. 2C and Supplementary Fig. S1). There was not any blue light-dependent response observed in the negative control that cells only expressing R-GECO1 (Supplementary Fig. S3A,B). We also did not detect any Ca2+ signal under the blue light illumination when the C71S substitutions were employed to this approach, which well explained the light dependence of this approach (Supplementary Fig. S3C,D).

Control of Ca2+ release with blue light and comparison with opto-α1AR.
(A) Subcellular localization of nMagHigh1-mKikGR-CAAX at the plasma membrane (upper panel) and its line profile (lower panel). (B–E) A comparison between the present approach based on the Magnet system and conventional opto-α1AR. For Ca2+ imaging, the cells were expressed with R-GECO1 and continuously exposed to excitation light at 559 nm. In the Magnet-based approach, the response of R-GECO1 was observed when the cells were stimulated with activation light at 473 nm (B) but not observed in the absence of the activation light (C); In opto-α1AR, the response of R-GECO1 was observed with (D) or without (E) blue light illumination at 473 nm, suggesting the excitation light of R-GECO1 at 559 nm activates opto-α1AR. The insets are partially enlarged views in Figure D and E. (F,G) Ca2+ imaging using the Magnet based approach with pulsed blue light illumination. Time-lapse fluorescence imaging showed that repeatable response of R-GECO1 was evoked upon activation light at 473 nm. Blue bars indicate 473 nm laser illumination.
Next we compared the Magnet-based approach with the corresponding Ca2+ modulating tool of optoXRs called opto-α1AR. Live cell imaging was conducted after R-GECO1 and opto-α1AR were coexpressed in HEK 293 cells. Similarly, 473 nm laser light was illuminated to the specially designated regions as the stimulation source of opto-α1AR. We recorded the time-lapse cytosolic fluorescence intensity of R-GECO1. As a result, the response of R-GECO1 was observed with or without 473 nm light illumination in opto-α1AR (Fig. 2D,E). This experiment yields that opto-α1AR is activated by the excitation light of R-GECO1 because rhodopsin covers a broad absorption band, thereby resulting in undesired Ca2+ oscillations.
To evaluate the repeatability of this approach, 473 nm laser light was illuminated to the same cell scheduling 5–10 minutes interval for a total of 100 minutes and every time Ca2+ signal was captured by R-GECO1 (Fig. 2F,G). The blue light-dependent oscillations show that the regulation of the membrane recruitment of Gαq and the subsequent release of Ca2+ can be repeatedly manipulated with blue light without losing the efficiency. We further certify that the manipulation of intracellular Ca2+ level is spatially photo-regulated (Supplementary Fig. S4). Upon blue light illumination, the fluorescence intensity of R-GECO1 in the specified cell was increased, indicating a higher intracellular Ca2+ level upon blue light stimulation. While the fluorescence brightness of R-GECO1 generally remained unchanged in the other cells without exposure to blue light, showing that the present system is suitable to space-resolved activation of Ca2+ release. Additionally, the amplitude of calcium spikes after blue light illumination showed comparable to that elicited by ligand-induced activation of endogenous histamine receptor (Supplementary Fig. S5).
By the direct comparison between the Magnet-based approach and opto-α1AR, we conclude that our strategy affords advantage in selectable usage of photoswitchable dimerization systems having narrow absorption spectrums, such as the Magnet system in this case, which indeed leads to the incorporation with R-GECO1 rending it more implementable for Ca2+ imaging.
Red light-induced Ca2+ release
Another advantage of the present strategy over the optoXRs is that Gαq can also be manipulated by different wavelength of light via replacing the Magnet system to other types of dimerization system.
Here we present another optogenetic approach to manipulate Gαq based on the red light-inducible PhyB/PIF6 system14,15. The PhyB/PIF6 system is sensitive to red light for binding, and shows low binding affinity in darkness or under far-red illumination. FusionRed-tagged PhyB is anchored to the plasma membrane by tethering to CAAX while PIF6 linked with Gαq is targeted to the cytoplasm. After red light stimulation, the plasma membrane-anchored PhyB binds to the Gαq-linked PIF6, and thus leads to the translocation of PIF6-Gαq from the cytoplasm to the plasma membrane, where the Gαq activates PLCβ and subsequently evokes the Ca2+ releasing to the cytosol. When under far-red condition, PIF6-Gαq dissociates from the plasma membrane to the cytoplasm, blocking the Ca2+ release (Fig. 3A).

Control of Ca2+ release with red light.
(A) Red light-inducible dimerization of the PhyB/PIF6 system allows the plasma membrane recruitment of Gαq and triggers cytosolic Ca2+ release. (B) Constructs for PhyB-FusionRed-CAAX and PIF6-Gαq. (C) Homogeneous cellular diffusion of GCaMP3 in HeLa cell (upper panel) and the plasma membrane localization of PhyB-FusionRed-CAAX (lower panel). (D) Ca2+ imaging of the PhyB/PIF6-based approach with pulsed red light illumination. Time-lapse fluorescence imaging showed that repeatable response of GCaMP3 was evoked upon activation light at 635 nm (red bars), 735 nm far-red (deep red bars) illumination was used as the inactivation light. (E) Negative control in the absence of PhyB-FusionRed-CAAX did not show red light-dependent response. (F) Negative control in the absence of PIF6-Gαq did not show red light-dependent response.
For confocal imaging of the plasma membrane translocation of the PhyB/PIF6-based photoswitch, PhyB-FusionRed-CAAX and PIF6-mYFP-Gαq were transfected to HeLa cells. Upon stimulation with red light (635 nm), the calculated fluorescence intensity of mYFP in the cytosol was decreased, while it was increased in the plasma membrane (Supplementary Fig. S6 and Supplementary movie S1), suggesting the red light-induced recruitment of PIF6-mYFP-Gαq to the plasma membrane. Previous researches show that the dissociation of PhyB/PIF6 interaction not only occurs under far-red illumination but also takes place in darkness with very likely different kinetics14,15, and it is clear that far-red light-induced PhyB/PIF6 dissociation is much faster than the dark reversion rate (Supplementary Fig. S6C,D).
We employed GCaMP316 as the Ca2+ indicator and excited it at 473 nm to avoid the interference to the PhyB/PIF6 interaction. PhyB-FusionRed-CAAX, PIF6-Gαq and GCaMP3 were transfected to HeLa cells. It is evident that GCaMP3 was homogeneously dispersed in the cytoplasm and the PhyB domain was anchored to the plasma membrane (Fig. 3C). During the Ca2+ imaging with GCaMP3 at 473 nm, far-red (735 nm) and red (635 nm) light was alternatively used to control the dissociation and association of PhyB/PIF6. In accordance with the result of the Magnet-based approach, Ca2+ signal could be repeatedly evoked in the cells upon red light illumination at 635 nm (Fig. 3D). Negative controls in the absence of PhyB-FusionRed-CAAX or PIF6-Gαq did not show any red light-dependent Ca2+ response (Fig. 3E,F). These results demonstrate that our strategy allows selective utilization of different photoswitchable dimerization systems to manipulate the membrane recruitment of Gαq and light-induced Ca2+ release.
Red light-induced cAMP increase through membrane recruitment of Gαs
Varieties of Gα appropriate different downstream effectors in the plasma membrane. As already mentioned, the manipulation of the Gαq leads to the release of cytosolic Ca2+. We sought to extend our strategy by the replacement of PIF6-linked Gαq to PIF6-linked Gαs (Fig. 4A). Gαs activates the downstream adenylyl cyclase (AC) after its localization at the plasma membrane and catalyzes the conversion of adenosine triphosphate (ATP) to 3′,5′-cyclic AMP (cAMP) and pyrophosphate3,17,18.

Regulation of cAMP increase with red light.
(A) Red light-inducible dimerization of the PhyB/PIF6 system allows Gαs to translocate to the plasma membrane and trigger cAMP release owning to the activation of adenylyl cyclase (AC). (B) Constructs for PhyB-FusionRed-CAAX and PIF6-Gαs. (C) Time-lapse luminescence intensity of samples under red illumination at 660 nm (red) or far-red illumination at 735 nm (far-red), with (PCB (+)) or without (PCB (−)) addition of PCB. The increment of luminescence intensity was only observed in the samples under red illumination in the presence of PCB. The upper table means the normalized intensity ratio changes between red PCB (+) and far-red PCB (+). (D) Relationship between PCB concentration and the luminescence intensity. (E) Power dependency of the luminescence intensity under illumination at 660 nm. (F) Result of the bioluminescence assay after optimization of several parameters including PCB concentration (50 μM), illumination time (14 hours) and light power (red: 0.15 mW/cm2; far-red: 1.5 mW/cm2). The inset is a partially enlarged view of mock, which means a negative control in the absence of PhyB-FusionRed-CAAX. The error bars indicate standard deviation from three individual samples (n = 3).
We employed cAMP response element (CRE) driven secretory luciferase construct PCRE-luc19 as a reporter to test whether light-actuated recruitment of Gαs to the plasma membrane will increase the cAMP level in living cells. cAMP regulates the transcription of the downstream luciferase gene via a conserved gene promoter element CRE (Fig. 4A). Theoretically, a higher intracellular cAMP level leads to a greater expression of luciferase, which can be sensitively measured by the luminescence intensity.
PhyB-FusionRed-CAAX, PIF6-Gαs and PCRE-luc were transfected to HEK293 cells, and the cells were kept in a dark incubator for protein expression before exposure to light. We measured the cAMP level by tracing the luminescence intensity of the CRE-driven gene expression products. The luminescence intensity of samples under the far-red light (735 nm) condition remains constant (Fig. 4C), which reveals far-red illumination kept the intracellular cAMP in a roughly invariant level. Conversely, the luminescence intensity of the samples under the red light (660 nm) condition significantly increased after the beginning of the illumination and reached a maximum value in about 15 hours. This result indicates that red light-triggered recruitment of Gαs generates a higher intracellular cAMP level that is sufficient to facilitate the gene expression.
The incorporation of cofactor phytochromobilin (PΦB) or phycocyanobilin (PCB) is required for the light sensitivity of PhyB20. As expected, the luminescence intensity of samples in absence of cofactor PCB did not increase even under red light illumination (Fig. 4C). Further analysis about the correlation between PCB concentration and the PhyB/PIF6-induced cAMP level suggested that 50 μM is preferable under the conditions we used (Fig. 4D). Moreover, we measured the red power dependency against the luminescence intensity. The result indicates that the cAMP level is precisely fine-tuned by varying the red light power between 0.01 mW/cm2 and 1 mW/cm2 (Fig. 4E).
Finally optimization of several parameters including PCB concentration, illumination time and light power allowed a 14-fold bioluminescence intensity difference between the samples under red (660 nm) and far-red (735 nm) light conditions (Fig. 4F). Besides, the samples kept in darkness showed a similar result to those illuminated by far-red light (Fig. 4F), which is consistent with the fact that both dark and far-red conditions make PhyB binding with its partner PIF6 in a low affinity. As a negative control, cells without expressing PhyB-FusionRed-CAAX showed very faint luminescence and appeared no appreciable difference between red and far-red conditions (Fig. 4F). Additionally, we showed that the red light-induced cAMP increase with the PhyB/PIF6-based Gαs system is more significant than cAMP increase stimulated by endogenous GPCR ligands and the effect is approximately equivalent to the result when 5 μM foskolin was tested (Supplementary Fig. S7). Based on all the calculated results, it is proved that optical manipulation of Gα using our strategy is not merely applicable to Gαq but also suited for Gαs.
Discussion
In this study, we developed a strategy for light-actuated recruitment of Gαq and Gαs to the plasma membrane using either the Magnet system or the PhyB/PIF6 system. We demonstrated the achievement of light-dependent regulation of second messengers including Ca2+ and cAMP in mammalian cells.
Our strategy provides multiple selections of dimerization system to manipulate Gα in order to meet the demands of different applications. We demonstrate that the wavelength used in light-triggered recruitment of the Gαq can be easily shifted from blue (473 nm) to red (635 nm) by replacing the dimerization systems. Both two approaches achieve temporal and repeatable regulation of cytosolic Ca2+ release upon photoirradiation. These two different dimerization systems have their own features. The Magnet system has high interaction affinity, fast switch-off kinetic, and an exogenous addition of cofactor is not required. The PhyB/PIF6 system should be superior when applied to in vivo studies because of the better tissue penetration of red light. Regardless of which approach is used, it should be emphasized that the most remarkable characteristic for manipulation of Gαq with the Magnet system and the PhyB/PIF6 system is the competitive advantage over the opto-α1AR. Opto-α1AR getting broad absorption band suffers from the perturbation by the excitation light for Ca2+ indicators. The use of the Magnet system or the PhyB/PIF6 system that has narrow absorption band overcomes this fundamental difficulty. These photoswitching systems are supposed to free up vacant spectrum that provide more selection of fluorescent proteins for labeling. This distinct spectral property also can be harnessed to combine with other optogenetic tools such as channelrhodpsin-2 (refs 21,22).
The successful manipulation of both Gαq and Gαs demonstrates that our strategy is feasible to optically manipulate different classes of Gα. Light-dependent control of Ca2+ and cAMP has been achieved through the corresponding activation of Gαq and Gαs. Optical regulation of these second messenger molecules provides extensive applications to mediate biological process. Furthermore, optical recruitment of other classes of Gα as well as Gαq and Gαs to the plasma membrane may also be applicable by indiscriminately apply this strategy, and the property of ‘auto-activation’ upon the plasma membrane recruitment can be shared to other classes of Gα, because they are generally believed to carry out signal functions at the plasma membrane. Therefore, diversified downstream pathways presumably can be regulated so as to mediate various patterns of signaling events in mammalian cells.
In summary, we provide a strategy to construct highly versatile approaches to optically manipulate the Gα. Various types of Gα can be recruited to the plasma membrane using different dimerization systems so as to reach the activation. As the functional application of this strategy, it enables optical switchable regulation of different second messengers in mammalian cells.
Methods
DNA constructions
The humanized genes encoded PhyB construct harboring tandem PAS (1–908) and 100-residue N-terminal phytochrome binding domain of PIF6 were synthesized by Eurofins Genemics (Tokyo, Japan). cDNAs encoding human Gαq and Gαs were gifted from Dr. Putyrski. We used the C9S and C10S mutants in Gαq. Because the wild type Gαq is palmitoylated at these two cysteine residues for membrane localization23,24, and these two mutations prevent membrane association of the protein25. The Q209L mutant lacking the constitutive GTPase activity in Gαq26 was also used to improve the present Gαq-based optogenetic approach. The present mutagenesis was performed with an overlap extension technique and Multi Site-Directed Mutagenesis Kit (MBL, Nagoya, Aichi, Japan) according to the manufacturer’s instructions.
All other plasmid constructions appeared in this study were created by standard molecular biology techniques and separately subcloned into the mammalian expression vector pcDNA3.1. All cloning enzymes were obtained from Takara Biomedical (Tokyo, Japan) and used according to the manufacturer’s instructions. The constructions were confirmed by sequencing the cloned fragments.
Protein purification and spectral analysis
nMagHigh1 with an N-terminal six-residue histidine tag was expressed in Escherichia coli DH5α cells with the pCold I vector and cultured in 200 mL LB medium containing 40 mg/ml of ampicillin. The bacterial cells were grown at 37 °C until they reached a density of approximately OD600 = 0.5. Protein expression was induced by addition of isopropyl β-d-1-thiogalactopyranoside (IPTG) at a final concentration of 0.1 mM following a temperature downshift from 37 °C to 15 °C. The bacterial cells were cultured for 48 hours. The histidine-tagged proteins were purified using TALON Spin Columns (Clontech, Palo Alto, CA) and eluted with an imidazole solution (500 mM imidazole, 50 mM sodium phosphate, 300 mM NaCl, pH 7.0). Absorption spectrometry was performed at room temperature using an Evolution Array spectrophotometer (Thermo Scientific, Waltham, MA, USA).
Cell culture
COS-7 and HEK293 cells were cultured at 37 °C under 5% CO2 in Dulbecco’s Modified Eagle Medium (DMEM; Invitrogen). HeLa cells were cultured under 5% CO2 in Μinimum Essential Medium Eagle (MEM; SIGMA). Both mediums were supplemented with 10% fetal bovine serum (GIBCO, Carlsbad, CA, USA), 100 unit/ml of penicillin and 100 μg/ml of streptomycin (GIBCO). These cells were used for translocation assay, Ca2+ imaging and bioluminescence assay.
TIRF imaging
To conduct the plasma membrane-translocation assay of the activator construct using TIRF imaging, COS-7 cells were plated at 1.0 × 104 cells per dish on glass-bottomed dishes and cultured for 24 hours at 37 °C in 5% CO2. The cells were transfected with cDNAs encoding DsRedEx2-pMagFast1-Gαq and nMagHigh1-mKikGR-CAAX at a 1:1 ratio using X-tremeGENE 9 DNA transfection reagent (Roche Diagnostics GmbH, Mannheim, Germany) according to manufacturer’s protocol. The total amount of DNA was 1 μg per dish. Twenty-four hours after transfection, the medium was replaced by the DMEM culture medium supplemented with 10% FBS. The cells were maintained for 24 hours at 28 °C. Before imaging, the culture medium was replaced with Hanks’ Balanced Salt Solution (HBSS, Grand Island Biological Co., Grand Island, NY, USA) containing 10 mM HEPES. Imaging was performed at room temperature with a 100× oil objective on the stage of an ECLIPSE Ti TIRF microscope (Nikon, Tokyo, Japan). Fluorescence images of DsRedEx2 were taken using an optically pumped semiconductor laser at 561 nm (Coherent, CA, USA). Blue light illumination was conducted using an optically pumped semiconductor laser (488 nm) at 1 mW for 50 ms.
Live cell imaging
The 35 mm glass-bottomed dishes (AGC TECNO GLASS Co., Shizuoka, Japan) were coated with poly-l-lysine (Sigma-Aldrich Co., Missouri, USA) at room temperature for 60 minutes, washed twice with 2 ml Milli-Q for use. In Fig. 2 and Supplementary Fig. S3, HEK293 cells were plated at approximately 2.0 × 104 cells/dish and cultured for 24 hours at 37 °C in 5% CO2. Cells were transfected with cDNAs encoding nMagHigh1-mKikGR-CAAX, pMagFast1-Gαq and R-GECO1 or substitutions using X-tremeGENE 9 DNA transfection reagent. Twenty-four hours after transfection, the culture media were replaced with HBSS containing 10 mM HEPES. Live cell imaging was conducted at 37 °C with a heated stage adaptor (Tokai Hit Co., Fujinomiya, Shizuoka, Japan). Fluorescence images of mKikGR and R-GECO1 were taken using a solid-state laser at 515 nm (Coherent) and a laser diode at 559 nm (NTT electronics, Yokohama, Japan) respectively. Blue light illumination was completed with a Laser diode at 473 nm.
In Fig. 3, HeLa cells were plated in 35 mm glass bottom dish (MATSUNAMI, Tokyo, Japan) and seeded for 24 hours, achieving to approximately 70% confluence of the plate. cDNAs encoding PhyB-FusionRed-CAAX, PIF6-Gαq and GCaMP3 were transfected to cells at a ratio 3:1:1 using the Lipofectamine 3000 Transfection Kit (Invitrogen, CA, USA) for 24 hours at 37 °C. The total amount of DNA was 0.5 μg. The cells were further incubated for 12 hours at 28 °C. PCB (Frontier Scientific, Logan, UT, USA) was added to the cells under dark condition to give a final concentration of 20 μM and incubated another 60 minutes. Then the culture media were washed and replaced with HBSS. Live cell imaging was conducted at 28 °C with the heated stage adaptor. Fluorescence images of FusionRed and GCaMP3 were taken using a solid-state laser at 559 nm (NTT electronics, Yokohama, Japan) and a laser diode at 473 nm (Olympus, Tokyo, Japan) respectively. Red light illumination was completed with a laser diode at 635 nm (Olympus). LED array (735 nm ± 20 nm, 1.5 mW/cm2) was used as the inactivation light. In Supplementary Fig. S6, to monitor the plasma membrane recruitment of PIF6-Gαq, mYFP (Q70K) was inserted between PIF6 and Gαq. Translocation assay was performed under a same condition after PhyB-FusionRed-CAAX and PIF6-mYFP-Gαq were transfected to HeLa cells at a 9:1 ratio.
FV1200 confocal laser scanning microscope (Olympus) and 60× oil immersion objective were used in live cell fluorescence imaging and translocation assay.
Bioluminescence assay
HEK 293 cells were plated at approximately 1.0 × 105 cells/well in a 24-well plate (AGC TECNO GLASS Co., Shizuoka, Japan), and cultured for 24 hours at 37 °C in 5% CO2. cDNAs encoding PhyB-FusionRed-CAAX, PIF6-Gαs and the reporter pGL4.29 [luc2P/CRE/Hygro] were transfected to cells at a ratio 3.5:0.5:1 using the Lipofectamine 3000. The total amount of DNA was 0.5 μg/well. Then samples were covered with foil to ensure dark conditions and kept in 37 °C incubator for 12 hours, an additional 12 hours incubation at 28 °C was executed for enough protein accumulation and better membrane localization. PCB was dissolved in DMEM and added to the samples half an hour before illumination. We used LED light sources (CCS Inc., Kyoto, Japan) for red (660 ± 20 nm) and far-red (735 ± 20 nm) light illumination respectively. The culture medium was removed after a period of illumination. The cells were treated with 200 μl/well passive lysis buffer (Promega, Madison, WI, USA) for 15 minutes at room temperature. 500 μl HBSS culture medium containing 200 μM d-luciferin pottasium salt (Wako Pure Chemistry Industries, Ltd., Osaka, Japan) was prepared as a substrate. Bioluminescence measurements were immediately performed after mixing the d-luciferin solution with the lysed cells using a Glomax 20/20 Luminometer at room temperature (Promega).
Additional Information
How to cite this article: Yu, G. et al. Optical manipulation of the alpha subunits of heterotrimeric G proteins using photoswitchable dimerization systems. Sci. Rep. 6, 35777; doi: 10.1038/srep35777 (2016).
References
Wettschureck, N. & Offermanns, S. Mammalian G proteins and their cell type specific functions. Physiol. Rev. 85, 1159–1204 (2005).
Banaszynski, L. A., Liu, C. W. & Wandless, T. J. Characterization of the FKBP Rapamycin·FRB Ternary Complex. J. Am. Chem. Soc. 127, 4715–4721 (2005).
Putyrski, M. & Schultz, C. Switching heterotrimeric G protein subunits with a chemical dimerizer. Chem. Biol. 18, 1126–1133 (2011).
Putyrski, M. & Schultz, C. Protein translocation as a tool: The current rapamycin story. FEBS Lett. 586, 2097–2105 (2012).
Airan, R. D., Thompson, K. R., Fenno, L. E., Bernstein, H. & Deisseroth, K. Temporally precise in vivo control of intracellular signalling. Nature 458, 1025–1029 (2009).
Kawano, F., Suzuki, H., Furuya, A. & Sato, M. Engineered pairs of distinct photo-switches for optogenetic control of cellular proteins. Nat. Commun. 6, 6256 (2015).
Schwerdtfeger, C. & Linden, H. VIVID is a flavoprotein and sever as a fungal blue light pgotoreceptor for pgotoadaptation. EMBO J. 22, 4846–4855 (2003).
Zoltowski, B. D. et al. Conformational switching in the fungal light sensor Vivid. Science 316, 1054–1057 (2007).
Vaidya, A. T. et al. Structure of a light-activated LOV protein dimer that regulates transcription. Sci. Signaling 4, ra50 (2011).
Steven, C. Protein isoprenylation and methylation at carboxyl terminal-cysteine residues. Annu. Rev. Biochem. 61, 355–386 (1992).
Waldo, G. L. et al. Kinetic scaffolding mediated by a phospholipase C-beta and Gq signaling complex. Science 330, 974–980 (2010).
Kurosaki, T. & Baba, Y. Ca2+ signaling and STIM1. Prog. Biophys. Mol. Biol. 103, 51–58 (2010).
Zhao, Y. et al. An expanded palette of genetically encoded Ca2+ indicators. Science 33, 1888–1891 (2011).
Levskaya, A., Weiner, O. D., Lim, W. A. & Voigt, C. A. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 461, 997–1001 (2009).
Konrad, M. et al. A red/far-red light-responsive Bi-stable toggle switch to control gene expression in mammalian cells. Nucleic Acids Research 41, e77 (2013).
Tian, L. et al. Imaging neural activity in worms, flies and mice with improved GCaMP calcium indicators. Nat. Methods 6, 875–881 (2009).
Kandel, E. R. The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol. Brain 5, 14–25 (2012).
Sassone-Corsi, P. The cyclic AMP pathway. Cold Spring Harbor Perspect. Biol. 4, a011148 (2012).
Fan, F. & Wood, K. F. Bioluminescent assays for high-throughput screening. Assay Drug Dev. Technol. 5, 127–136 (2007).
Anjia, R., Antje, R., Silvia, E. B., Wolfgang, G. & Kurt, S. Chromophore incorporation, Pr to Pfr kinetics, and Pfr thermal reversion of recombinant N-terminal fragments of Phytochrome A and B chromoproteins. Biochemistry 37, 9983–9990 (1998).
Nagel, G. et al. Channelrhodopsin-2, a directly light-gated cation-selective membrane channel. Proc. Natl. Acad. Sci. USA 100, 13940–13945 (2003).
Leifer, A. M. et al. Optogenetic manipulation of neural activity in freely moving Caenorhabditis elegans. Nat. Methods 8, 147–152 (2011).
Lind, M. E. et al. Lipid modifications of G proteins: alpha sununits are palmitoylated. Proc. Natl. Acad. Sci. USA 90, 3675–3679 (1993).
Parenti, M., Vigano, M. A., Newman, C. M., Milligan, G. & Magee, A. I. A novel N-terminal motif for palmitoylation of G-protein alpha subunits. Biochem. J. 291, 349–353 (1993).
Wedegaertner, P. B., Chu, D. H., Wilson, P. T., Levis, M. J. & Bourne, H. R. Palmitoylation is required for signaling functions and membrane attachment of Gqα and Gsα. J. Biol. Chem. 268, 25001–25008 (1993).
Kalinec, G., Nazarali, A. J., Hermouet, S., Xu, N. & Gutkind, J. S. Mutated α sununit of the Gq protein induces malignant transformation in NIH 3T3 cells. Mol. Cell. Biol. 12, 4687–4693 (1992).
Acknowledgements
This work was supported by Platform for Dynamic Approaches to Living System from the Ministry of Education, Culture, Sports, Science and Technology, Japan (MEXT) and Japan Agency for Medical Research and Development (AMED), and by grants from Japan Society for the Promotion of Science (JSPS). This work was also supported by grants from Yamada Science Foundation and Japan Foundation for Applied Enzymology to M.S.
Author information
Authors and Affiliations
Contributions
G.Y., F.K., H.S. and M.S. designed the experiments. G.Y. and H.O. conducted the main experiments. A.F. performed the TIRF imaging. G.Y., H.O., Y.A., F.K. and Y.U. analyzed the data. G.Y. and M.S. wrote the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Yu, G., Onodera, H., Aono, Y. et al. Optical manipulation of the alpha subunits of heterotrimeric G proteins using photoswitchable dimerization systems. Sci Rep 6, 35777 (2016). https://doi.org/10.1038/srep35777
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep35777
This article is cited by
-
Astrocytes: new evidence, new models, new roles
Biophysical Reviews (2023)
-
Reversible photocontrol of oxidase activity by inserting a photosensitive domain into the oxidase
Bioresources and Bioprocessing (2019)
-
Near-infrared light–controlled systems for gene transcription regulation, protein targeting and spectral multiplexing
Nature Protocols (2018)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
