
Abstract
Biodiversity hotspots are centers of biological diversity and particularly threatened by anthropogenic activities. Their true magnitude of species diversity and endemism, however, is still largely unknown as species diversity is traditionally assessed using morphological descriptions only, thereby ignoring cryptic species. This directly limits evidence-based monitoring and management strategies. Here we used molecular species delimitation methods to quantify cryptic diversity of the montane amphipods in the Irano-Anatolian and Caucasus biodiversity hotspots. Amphipods are ecosystem engineers in rivers and lakes. Species diversity was assessed by analysing two genetic markers (mitochondrial COI and nuclear 28S rDNA), compared with morphological assignments. Our results unambiguously demonstrate that species diversity and endemism is dramatically underestimated, with 42 genetically identified freshwater species in only five reported morphospecies. Over 90% of the newly recovered species cluster inside Gammarus komareki and G. lacustris; 69% of the recovered species comprise narrow range endemics. Amphipod biodiversity is drastically underestimated for the studied regions. Thus, the risk of biodiversity loss is significantly greater than currently inferred as most endangered species remain unrecognized and/or are only found locally. Integrative application of genetic assessments in monitoring programs will help to understand the true magnitude of biodiversity and accurately evaluate its threat status.
Similar content being viewed by others


Introduction
Biodiversity is a key resource of our planet providing important ecosystem functions thereby ensuring sustainable life on Earth1,2,3. However, the dramatic loss of biodiversity is proceeding at a striking pace and has become a major concern for human well-being4,5,6. Environmental legislation and management actions have been implemented at both international and national levels to counteract biodiversity loss. The establishment of national parks, the convention on biological diversity, the United Nations decade on biodiversity from 2011–2020 or the declaration of so-called biodiversity hotspots are just a few instances of such recognition. The concept of biodiversity hotspots involves defined regions of high conservation priority referring to both, a high threat to their natural intact vegetation (NIV) and an exceptional richness of endemic (by definition: vascular plant) species7,8,9. On a global scale, 35 biogeographic regions qualify as biodiversity hotspots10, covering 17.3% of the Earth’s land surface (excluding Antarctica) and being home for more than 70% of the known animal fauna10,11. Biodiversity hotspots are critically prone to biodiversity loss as they harbour many endemic species, but likewise unique ecosystems and gene variants7,8. Despite of their negligible surface area (<1% on a global scale), freshwater ecosystems are of particular concern as they provide habitat to an over-proportionally great number of species12. The efficiency of traditional morphology-based methods in monitoring biodiversity changes is widely accepted, however, it is also well known that these methods lack the ability to identify cryptic species and cannot inform on the loss of genetic variation. Hence, they underestimate the “true” biological diversity present in an ecosystem, which adds an additional level of uncertainty for monitoring and management actions13. Therefore, it is of utmost necessity to employ fast and efficient tools to monitor the ongoing biological diversity loss in particular in freshwater ecosystems and genetic tools have been proven to be extremely effective14,15,16,17,18,19,20,21,22. One ecologically highly important animal group for which recent studies have shown that species assignments are difficult is amphipod crustaceans23,24,25,26,27. They play an important functional role in freshwater food webs28,29 by i) serving as prey for fishes30,31, ii) acting as intermediate and definitive hosts for parasites29,32,33 and iii) regulating organic litter breakdown34 and must be considered as ecological keystone engineers.
Here, we used freshwater amphipods of the genus Gammarus sampled from freshwater ecosystems of two mountain ranges in the Caucasus (Northern slopes of the Alborz Mountains, Iran) and Irano-Anatolian biodiversity hotspot (Zagros Mountains and southern slopes of the Alborz, both Iran) as a model to test whether the true species diversity is accurately reflected by the present morphology-based estimates. To this aim, amphipod species were genetically surveyed and linked to morphologically known species for the broader region of Iran, with three and 16 morphologically defined species present in the Alborz and Zagros Mountains, respectively35. We expect our molecular-based approach to reveal a more realistic pattern of higher biological diversity in those two critically endangered biodiversity hotspots. Furthermore, based on recent data from other regions23,24,25,26,27,36 we expect that several of the reported widespread species will consist of regional endemics.
Results
Species diversity based on morphology
The following freshwater species have been identified morphologically: Gammarus lacustris s. str. Sars37, (4 specimens from 2 localities), Gammarus cf. lacustris Sars37, (=G. lacustris complex) (12 specimens, 6 localities), Gammarus lordeganensis Khalaji-Pirbalouty and Sari38, (7 specimens, 3 localities), Gammarus balcanicus Schäferna39, (1 specimen, 1 locality), Gammarus hegmatanensis Hekmatara et al.40, (2 specimens, 1 locality) and Gammarus cf. komareki Schäferna39, (=G. komareki complex) (154 specimens, 49 localities) (Table 1; for type localities see Supplementary Table 1). The latter was by far the most abundant morphospecies in the studied freshwater systems. The designation “cf.” was applied to species when taxonomic assignment was uncertain as many of the surveyed individuals showed ambiguous diagnostic characters, i.e. largely corresponding to the respective morphospecies description but not allowing explicit species assignment. Furthermore, two brackish water species were collected serving as outgroups for the molecular analyses: Obesogammarus acuminatus Stock et al.41, (2 specimens, 1 locality) and Gammarus aequicauda (Martynov)42 (2 specimens, 1 locality).
Species diversity based on molecular data
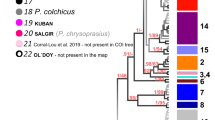
The final COI alignment included 184 specimens trimmed to 465 bps. All COI-sequences were checked against the NCBI database for possible contaminations, which were not present. The four conceptually different molecular species delimitation methods revealed 44–58 groups. The Automatic Barcode Gap Discovery (ABGD) approach delineated 44 groups (Fig. 1) with a proposed barcoding gap at 7.9% Kimura-2-Parameter (K2P) genetic distance. Of the 44 groups, 35 were morphologically identified as belonging to the Gammarus komareki complex and six to the G. lacustris complex. The reversed Statistical Parsimony (SP) approach revealed 53 groups. Among those groups, 41 were within the G. komareki complex and nine belonged to G. lacustris. The Bayesian General Mixed Yule-Coalescent (bGMYC) model split the dataset into 51 groups, with 40 and eight within the G. komareki and G. lacustris species complex, respectively. Finally, the delimitation approach based on the Bayesian implementation of the Poisson Tree Process (bPTP) model yielded the largest number of groups (58). Of those, 46 were within the G. komareki complex and nine in G. lacustris. Obesogammarus acuminatus, G. balcanicus, G. aequicauda and G. hegmatanensis were identified as single delineated groups each in all four approaches. Gammarus lordeganensis and G. hegmatanensis were always found to be part of the G. lacustris and G. komareki complex, respectively.

Neighbor-Joining tree visualizing the results of the four different molecular species delimitation methods based on COI.
ABGD: Automatic Barcode Gap Discovery; SP: reversed Statistical Parsimony; bPTP: Bayesian Poisson tree process model; bGMYC: Bayesian General Mixed Yule-Coalescent model. Localities for each species are color-coded in green (Alborz Mountains, in Caucasus biodiversity hotspot), orange (Alborz, Irano-Anatolian), red (Zagros, Irano-Anatolian) and black (Central Iranian Plateau or brackish water localities at the Caspian Sea; 31 and 41). Bootstrap support values are provided at the branches. Gk1-35: Cryptic species within the G. komareki complex; Gl1-4: Cryptic species within the G. lacustris complex. The depicted specimen is a representative of the Gammarus komareki complex.
As the four delimitation approaches yielded different numbers of groups, we here refer to the 44 groups revealed by AGBD as the most conservative approach. This is reasonable since i) by referring to the lowest species estimates, we follow a most parsimonious decision avoiding an over-splitting of lineages and an associated overestimation of amphipod species richness and endemism, ii) none of the lineages proposed by ABGD is lumped by any of the other three methods and iii) 35 of the ABGD lineages (80%) are congruently detected in all four approaches. Four more lineages are supported by ABGD and at least one other delimitation approach (9%). Only five lineages (11%) are exclusively found by ABGD but distinctly split in the other three approaches.
Six of the 44 putative species could be morphologically linked to a species name: Gammarus hegmatanensis, G. balcanicus, G. lacustris s. str., G. lordeganensis, G. aequicauda and Obesogammarus acuminatus, comprising four freshwater and two brackish water species, respectively. The remaining 38 groups represented cryptic species within the two species complexes of G. komareki (n = 34) and G. lacustris s. l. (n = 4). Groups within the G. komareki complex were consistently named Gk1 to Gk35, with Gk1 and Gk2 already introduced by Hou et al.43. Only Gk1 was found in our study. Groups of the G. lacustris complex were named Gl1-Gl4.
In order to test for congruency with the COI delimitation results, nuclear 28S rDNA data was successfully obtained for 87 specimens covering all putative species except Gl4. The 28S alignment was trimmed at both ends to a final length of 798 bp for all but four sequences, which were shorter at the 3′ end (YB1 = 795 bp, BIL5 = 789 bp, QCQ1 = 765 bp and JLR5 = 759 bp). ABGD groupings are supported by nuclear data in most of the cases (36 out of 43; 84%), except: i) the specimens L1, LH2, SRD1, SRD2 and KLD6 showed a highly similar 28S sequence but belonged to four different ABGD (COI) groups, ii) the specimen NZH5 had a highly distinct 28S sequence (>10 mutations difference) when compared with the nuclear sequences of the six other specimens of the ABGD group Gk21 for which nuclear data was obtained, iii) the two specimens of Gk4 (JR1 and JR2) possess nuclear sequences with seven mutations difference and iv) the three specimens of the sampling site Biledargh (BIL5, BIL9 and BIL10) clustered into two COI-groups (Gk11, Gk12), but all exhibit 28S sequences with 3–4 mutations difference, not allowing a clear distinction of the two COI-groups with nuclear data (Supplementary Fig. 1). However, the latter two cases do not contradict the initial delimitation results.
Discussion
It is a generally accepted fact that biodiversity is in decline due to anthropogenic land use and climate change. However, the true magnitude of biodiversity loss is a topic of central debate. Our results underscore the benefit of molecular tools in capturing the “true” biological diversity present in an ecosystem. The montane freshwater amphipod fauna of the Alborz and Zagros Mountains, situated within the biodiversity hotspots of the Caucasus and Irano-Anatolian region, comprises over an order of magnitude more species than previously known, meaning that most species were overlooked by traditional morphology-based assessments. In line with our expectations, the 42 gammarid freshwater species detected in this study can be linked to just five morphospecies. More than 90% of the found species are cryptic and cluster within the two species complexes of Gammarus komareki (34 cryptic species) and Gammarus lacustris (four cryptic species). Only four freshwater species (10%) could be unambiguously linked with morphology, i.e. a single morphospecies comprising a single genetically identified species: Gammarus hegmatanensis, Gammarus balcanicus, Gammarus lacustris s. str. and Gammarus lordeganensis. Although our sampling covers 154 freshwater sites, there exists an obvious geographical sampling bias towards freshwater systems in the Alborz and Northern Zagros Mountains. In general, sampling effort affects the number of species and the level of intraspecific genetic diversity to be discovered44,45,46 and it is likely that further unrecognized members of both widespread species complexes, G. komareki and G. lacustris, also occur in the Southern Zagros Mountains. Gammarus balcanicus might be another candidate species complex comprising cryptic lineages within the studied region, as it has been revealed for montane freshwater systems in Europe25. However, our sampling was not appropriate to assess hidden species diversity for this species in Iran.
The observed high level of cryptic species diversity becomes even more intriguing when compared to the number of amphipod freshwater species previously reported for the study area, specifically for the Alborz Mountains at the junction of the Caucasian and Irano-Anatolian biodiversity hotspots. So far, three species have been reported from that region, however our results imply that species diversity is drastically higher, i.e. by more than one order of magnitude. After integration of the amphipod species data presented in this study with the known faunal record, the Alborz Mountains harbor 34 species and the Zagros Mountains 25 species (16 previously reported species), respectively. The observed pattern of a high number of unrecognized species is consistent with other studies on montane freshwater amphipod communities in Europe (e.g.25,36). On the one hand, many of the newly recognized species of the G. komareki and G. lacustris complexes fit to the assumed intraspecific morphological variability of their respective type species. On the other hand, some species demonstrate substantial morphological differences, e.g. in the degree of setation of the telson/second antenna, shape and size of the gland cone and shape of the third epimeral plate. As already noted, morphology alone often does not seem to be sufficient in describing Gammarus species (e.g.24,47,48,49). For each new species, taxonomists have to question whether the (sometimes ambiguous) combination of conserved and variable morphological traits are diagnostic or reflect the natural intraspecific variability of a species.
The delimitation of species based solely on molecular data likewise can sometimes also be problematic50. In general, single-gene approaches are error-prone if incomplete lineage sorting or introgression occur. Furthermore, single threshold delimitation estimates (e.g. genetic distance values, transition points or connection limits) can also be misleading when different species exhibit variable diversification rates. Therefore, to interpret results more carefully, a suitable analytical framework based on more than one gene and including a combination of delimitation approaches should be applied. In our study, the majority of species is supported by both mtDNA and nuclear data (84%) and by all four delimitation approaches (80%). There are many possible reasons that can lead to the discrepancies between different delimitation methods. However, in our case these discrepancies are mostly likely due to the known tendency of coalescent-based approaches (i.e. PTP and GMYC) to overestimate the number of lineages if only a few specimens per species are investigated and/or the proportion of singletons in the dataset is high51,52. Both these scenarios lower the portion of coalescing lineages in the analysis and produce younger transition point estimates, which will finally result in the delimitation of more recently diverged lineages. Incongruencies between both molecular markers can be likely attributed to the retention of ancestral polymorphisms (groups Gk1, 11, 12, 15, 16 and 35), which is fourfold more likely in nuclear as compared to mitochondrial genes and to an mtDNA introgression event (specimen NZH5, group Gk21). For the future, an integrative taxonomic approach is needed to delimit and describe species with independent lines of evidence.
While the number of unrecognized species already documents the potential of molecular methods to rapidly quantify the status quo and threat status of a region, knowledge about a high level of unrecognized biodiversity is only the first step. The inferred patterns on species distributions add another layer of information. Contrary to the assumed wide distribution of some amphipod species present within the study region (e.g. G. komareki and G. lacustris), 69% of the genetically identified species only occurred at a single site. Even localities separated by only a few tens of kilometers were most often inhabited by distinct species. In cases where a species was present at multiple sites, localities were generally in close geographical proximity and/or a clear phylogeographical pattern with exclusive spatial lineages exists. Only specimens of a single group, i.e. Gk21, occurred in both the Alborz and Zagros mountain ranges. Passive transportation by humans, e.g. between the abundant trout farms, or by birds such as the mountain stream inhabiting dipper (Cinclus cinclus Linnaeus, 1758) may be responsible for the presence of this species in streams approximately 800 km apart. An increased sampling may reveal this species to be even more common and/or other more widespread species.
The occurrence of such a high level of narrow endemism as reported here can be explained by a combination of two evolutionary processes. First, diversification rate can be increased through the emergence of new ecological opportunities in general but specifically in gammaridean amphipods43. In particular, the transition from a saline to a freshwater environment is regarded as an evolutionary trigger for increased diversification rates observed in freshwater gammarids43. Second, when compared to marine habitats, freshwater ecosystems are much more isolated. This micro- and macro vicariance is likely the main driver further accelerating diversification of lineages. A similar scenario may be likely for marine Gammarus-ancestors from the Paratethys Sea colonizing freshwater habitats in Iran. The biogeographic setting of the studied mountain regions promote the formation of narrow endemism as rivers of the Alborz Mountains predominantly flow either north- or southwards and thus either directly end up in the Caspian Sea (northern slopes) or dry out in the Central Iranian Plateau and Dasht-e Kavir desert (southern slopes). The latter is also true for rivers in the eastern Zagros Mountains53. Hence, after the uplift of both mountain ranges during the Miocene and Pliocene, multiple hydrologically separated freshwater habitats were available for initial colonization, population differentiation and finally species diversification43. At this point, it has to be noted that our sampling scheme along both mountain ranges (with only a few specimens analyzed per sampling site) systematically favors the discovery of narrow endemism, in particular the degree of species found at a single site. A denser sampling (i.e. within a single river or catchment) may reveal broader distribution ranges of many species. The results, however, also suggest that further sampling, especially in yet sparsely sampled areas or during different seasons at temporally desiccated sites, will most likely reveal more endemic species.
Knowledge about the distinct pattern of small-scale endemism in freshwater amphipod species is of paramount importance for conservation and management strategies: since these amphipods primarily demonstrate a shredding feeding type, they will heavily rely on coarse organic material input, e.g. provided by the NIV54. As such, species are most likely co-affected by the continuing rapid decline of NIV observed for both biodiversity hotspots55. Considering the high probability that the rate of narrow endemism found for Iranian freshwater amphipods is the rule rather than the exception, this situation becomes even more severe. Endeavors in protecting and sustaining the NIV and accompanied faunal biological diversity within biodiversity hotspots should become another prime target.
Freshwater ecosystems and their biodiversity are endangered due to many factors, such as habitat degradation, overexploitation, flow modification and water pollution56. We showed that montane freshwater ecosystems in two highly endangered biodiversity hotspots harbor a drastically underestimated amphipod diversity, both at the species and genetic level. Many of the newly discovered species comprise narrow range endemics rather than widespread lineages. Although species extinction is globally perceived to a high degree, the dimension of biological diversity loss may be even greater than currently inferred given that most endangered species may still be unrecognized and/or only locally found. Finally, our study provides further support for the urgently needed and integrative implementation of genetic monitoring strategies57, which can make a plethora of information on the status quo and threat status of biodiversity, from the level of genes to ecosystems, available for researchers, stakeholders and policymakers alike.
Methods
Sampling and morphological identification
We sampled 154 freshwater sites throughout the Alborz Mountains, Zagros Mountains and the Central Iranian Plateau, covering montane, arid and semi-arid areas. Most of the sampling sites are situated in the Irano-Anatolian and Caucasus biodiversity hotspots (Table 1 and Fig. 2). Amphipod specimens were found and collected from 67 of the 154 localities (43.5%) using dipping nets. Furthermore, two brackish water sites (Gomishan lagoon; GLA, and, Chaf lagoon; TCH) were visited to collect amphipod specimens as outgroups for the molecular analyses. Sampling was performed on macrophytes in the water, beneath stones and within sand. Freshly collected samples were immediately preserved in 96% ethanol and morphologically identified based on the most validated and recent freshwater amphipod keys35,41,58,59,60 as well as original species descriptions.

Overview of sampling localities.
Shown are all localities where living amphipods have been found. Red area: Irano-Anatolian biodiversity hotspot; green area: Caucasus biodiversity hotspot. Localities are color coded in green (Alborz Mountains, in Caucasus biodiversity hotspot), orange (Alborz, Irano-Anatolian), red (Zagros, Irano-Anatolian) and black (Central Iranian Plateau or brackish water localities at the Caspian Sea; 31 and 41). The map was created using DIVA-GIS 7.5 (www.diva-gis.org).
DNA extraction, PCR, sequencing
Pereopods from one side of the body were used for DNA extraction by a salt precipitation method (modified after61, following27) and for at least two individuals per locality. The standard DNA barcoding locus—a 658 bp fragment of the Cytochrome c oxidase subunit I gene (COI)—was amplified using the modified primer pair LCO1490-JJ and HCO2198-JJ62. Polymerase chain reaction (PCR) was carried out in a total volume of 25 μL containing 2.5 μL 10X PCR buffer, 2.5 μL dNTPs (2 mM), 0.125 μL of each primer (100 pmol/μL), 0.125 μL of Hotmaster Taq (5 U/μL, 5 PRIME GmbH, Hamburg, Germany), 1 μL of template DNA (20–60 ng/μL) and molecular grade water. For samples which did not amplify under standard PCR settings, illustra PuReTaq Ready-To-Go PCR beads (GE Healthcare, Freiburg, Germany) were used with the following protocol: 0.125 μL of each primer, 1 μL of DNA, filled up to 25 μL with water. The PCR setting for COI amplification was: initial denaturation at 94 °C for 60 s; 36 cycles of denaturation at 94 °C for 30 s, annealing at 51 °C for 45 s, extension at 65 °C for 60 s; final extension at 65 °C for 5 min. We used a similar PCR setting for illustra PuReTaq Ready-To-Go PCR beads, with only the extension temperature adjusted to 72 °C. As an additional nuclear marker, the ribosomal DNA (rDNA) locus 28S was amplified. The 28S rDNA PCR reaction mix was the same as for COI, containing the primer pair 28Sa and 28Sb63. PCR settings were: initial denaturation at 94 °C for 60 s; 36 cycles of denaturation at 94 °C for 30 s, annealing at 48 °C for 45 s, extension at 65 °C for 60 s; final extension at 65 °C for 5 min. PCR setting in using illustra PuReTaq Ready-To-Go PCR beads was similar, but the extension temperature was adjusted to 72 °C. For DNA sequencing, 10 μL of PCR product was enzymatically purified with 0.5 μL ExoI (20 U/μL) and 1 μL FastAP (1 U/μL) (both Thermo Fisher Scientific, Schwerte, Germany). The reaction mix was incubated at 37 °C for 25 min and 85 °C for 15 min. The purified products were sequenced at GATC-Biotech (Cologne, Germany), Macrogen Inc. (Korea) or on an ABI 3130xl sequencer (Dept. of Receptor Biochemistry, Faculty of Chemistry, Ruhr University Bochum). Sequence chromatograms were edited and assembled in Geneious 6.0.664. The COI-alignment was constructed using the MUSCLE algorithm plugin in Geneious with eight iterations65. The 28S rDNA alignment was generated using the Geneious MAFFT-plugin66, automatically selecting for the most appropriate algorithm.
Molecular species delimitation
We used four molecular species delimitation approaches to delineate freshwater amphipod species with the standard barcode marker COI67. Our delimitation strategy consists of approaches based on three different delineation concepts: i) distance-based (ABGD), ii) network-based (SP) and iii) topology-based (bPTP, bGMYC). This combination was applied thus to compare results of conceptually different methods and to lower difficulties when relying on single parameter estimates only, i.e. genetic distance thresholds, topology-based transition points or connection limits in haplotype networks to distinguish intra- and interspecific diversity.
The Automatic Barcode Gap Discovery (ABGD68) method is based upon pairwise genetic distance calculations. Sequences are semi-automatically grouped in order that distances between sequences of two groups are always larger than a certain genetic distance threshold value, i.e. the barcode gap68. We tested the COI dataset with a combination of ABGD settings within the parameter range of Pmin = 0.001, Pmax = 0.08–0.10 and gap width = 0.5–0.8. A Kimura-2-parameter (K2P) corrected genetic distance matrix was used as it is the standard model proposed for DNA barcoding analyses69. K2P-distances were calculated using MEGA v6.070.
The reversed Statistical Parsimony (SP) method71 delineates species based on the network topology. The calculation of a statistical parsimony network was performed in TCS v1.2172. Sequences were collapsed into haplotypes and connected based on a given connection probability of parsimony. If mutational steps between two haplotypes exceed the connection probability, i.e. a certain number of mutational steps, the haplotypes are clustered into two separate networks (or putative species). A 95% connection probability threshold was applied to delineate putative species.
The Bayesian Poisson Tree Process (bPTP)73 model approach delineates species based on a topology. The bPTP webserver (http://www.species.h-its.org) was used with a COI topology produced by BEAST v1.8.274 as an input tree. The bPTP method was run under default settings. BEAST settings were computed in BEAUTi v1.8.274: 30 million chain length, sampling each 3000th tree, standard coalescent model, GTR + G + I substitution model with four gamma categories and a strict clock. The GTR + G + I model was proposed by model selection in MEGA v6.070 and jModeltest75. Appropriateness of parameters (effective sample size >200) was tested with Tracer v1.676. Results were visualized with TreeAnnotator v1.8.277 with a 10% burn-in rate, posterior probability of 0.9 and under the maximum clade credibility option for the consensus tree.
The Bayesian General Mixed Yule-Coalescent (bGMYC) model approach is conceptually similar to bPTP, but needs an ultrametric input tree78. The run parameters were mcmc = 100,000, burn-in: 90,000 and thinning = 100.
A Neighbor-joining (NJ) tree was calculated using MEGA based on K2P-distances and branch support assessed using 2000 bootstrap replicates. The 28S rDNA network was calculated in SplitsTree79 using uncorrected p-distances.
Ethical approval
All applicable international, national, and/or institutional guidelines for the care and use of animals were followed. No experiments were done on living animals in this study. The Research and Ethics Committee of the College of Science, University of Tehran approved the experimental protocol.
Additional Information
Data Availability: COI sequences: GenBank accession numbers: KT778323–KT778506. 28S rDNA sequences: GenBank accession numbers: KT827482–KT827554, KU513423–KU513436. Trees have been deposited in TreeBASE under submission number 18324. Voucher specimens and their comparative material are deposited at Zoological Museum, University of Tehran (ZUTC).
How to cite this article: Katouzian, A.-R. et al. Drastic underestimation of amphipod biodiversity in the endangered Irano-Anatolian and Caucasus biodiversity hotspots. Sci. Rep. 6, 22507; doi: 10.1038/srep22507 (2016).
References
Chapin, F. S. III et al. Consequences of changing biodiversity. Nature 405, 234–242 (2000).
Humphries, C. J., Williams, P. H. & Vane-Wright, R. I. Measuring biodiversity value for conservation. Ann. Rev. Ecol. Syst., 93–111 (1995).
Tilman, D. Causes, consequences and ethics of biodiversity. Nature 405, 208–211 (2000).
Butchart, S. H. et al. Global biodiversity: indicators of recent declines. Science 328, 1164–1168 (2010).
Rockström, J. et al. A safe operating space for humanity. Nature 461, 472–475 (2009).
Steffen, W. et al. Planetary boundaries: Guiding human development on a changing planet. Science 347, 1259855 (2015).
Myers, N. Threatened biotas: “Hot spots” in tropical forests. Environmentalist 8, 187–208, 10.1007/BF02240252 (1988).
Myers, N., Mittermeier, R. A., Mittermeier, C. G., da Fonseca, G. A. B. & Kent, J. Biodiversity hotspots for conservation priorities. Nature 403, 853–858 (2000).
Reid, W. V. Biodiversity hotspots. Trends Ecol. Evol. 13, 275–280 (1998).
Sloan, S., Jenkins, C. N., Joppa, L. N., Gaveau, D. L. A. & Laurance, W. F. Remaining natural vegetation in the global biodiversity hotspots. Biol. Cons. 177, 12–24 (2014).
Mittermeier, R., Turner, W., Larsen, F., Brooks, T. & Gascon, C. Global Biodiversity Conservation: The Critical Role of Hotspots (eds. Zachos, F. et al.) (Springer Berlin Heidelberg, 2011).
Strayer, D. L. & Dudgeon, D. Freshwater biodiversity conservation: recent progress and future challenges. J. N. Am. Benthol. Soc. 29, 344–358 (2010).
Adams, M., Raadik, T. A., Burridge, C. P. & Georges, A. Global Biodiversity Assessment and Hyper-Cryptic Species Complexes: More Than One Species of Elephant in the Room? Syst. Biol. 63, 518–533, 10.1093/sysbio/syu017 (2014).
Bálint, M. et al. Cryptic biodiversity loss linked to global climate change. Nature Clim. Change 1, 313–318 (2011).
Dincă, V. et al. DNA barcode reference library for Iberian butterflies enables a continental-scale preview of potential cryptic diversity. Sci. Rep. 5, 12395, 10.1038/srep12395 http://www.nature.com/articles/srep12395#supplementary-information (2015).
Féral, J.-P. How useful are the genetic markers in attempts to understand and manage marine biodiversity? J. Exp. Mar. Biol. Ecol. 268, 121–145 (2002).
Geist, J. Integrative freshwater ecology and biodiversity conservation. Ecol. Indic. 11, 1507–1516 (2011).
Karp, A., Ingram, D. S. & Isaac, P. G. Molecular tools for screening biodiversity: plants and animals (Springer Science & Business Media, 2012).
Mutanen, M., Kaila, L. & Tabell, J. Wide-ranging barcoding aids discovery of one-third increase of species richness in presumably well-investigated moths. Sci. Rep. 3, 2901, 10.1038/srep02901 http://www.nature.com/articles/srep02901#supplementary-information (2013).
Pauls, S. U., Nowak, C., Bálint, M. & Pfenninger, M. The impact of global climate change on genetic diversity within populations and species. Mol. Ecol. 22, 925–946 (2013).
Vieites, D. R. et al. Vast underestimation of Madagascar’s biodiversity evidenced by an integrative amphibian inventory. Proc. Natl. Acad. Sci. USA 106, 8267–8272 (2009).
Yannic, G. et al. Genetic diversity in caribou linked to past and future climate change. Nature Clim. Change 4, 132–137 (2014).
Esmaeili-Rineh, S., Sari, A., Delić, T., Moškrič, A. & Fišer, C. Molecular phylogeny of the subterranean genus Niphargus (Crustacea: Amphipoda) in the Middle East: a comparison with European Niphargids. Zool. J. Linnean Soc, 10.1111/zoj.12296 (2015).
Hou, Z. & Sket, B. A review of Gammaridae (Crustacea: Amphipoda): the family extent, its evolutionary history and taxonomic redefinition of genera. Zool. J. Linnean Soc, 10.1111/zoj.12318 (2015).
Mamos, T., Wattier, R., Majda, A., Sket, B. & Grabowski, M. Morphological vs. molecular delineation of taxa across montane regions in Europe: the case study of Gammarus balcanicusSchäferna, (Crustacea: Amphipoda). J. Zool. Syst. Evol. Res. 52, 237–248, 10.1111/jzs.12062 (2014).
Müller, J. C., Schramm, S. & Seitz, A. Genetic and morphological differentiation of Dikerogammarus invaders and their invasion history in Central Europe. Freshwater Biol. 47, 2039–2048 (2002).
Weiss, M., Macher, J., Seefeldt, M. & Leese, F. Molecular evidence for further overlooked species within the Gammarus fossarum complex (Crustacea: Amphipoda). Hydrobiologia 721, 165–184, 10.1007/s10750-013-1658-7 (2014).
MacNeil, C., Dick, J. T. A. & Elwood, R. W. The trophic ecology of freshwater Gammarus spp.(Crustacea: Amphipoda): problems and perspectives concerning the functional feeding group concept. Biol.Rev. 72, 349–364 (1997).
Väinölä, R. et al. Global diversity of amphipods (Amphipoda; Crustacea) in freshwater. Hydrobiologia 595, 241–255 (2008).
Andersson, K. et al. Presence of sculpins (Cottus gobio) reduces drift and activity of Gammarus pulex (Amphipoda). Hydrobiologia 133, 209–215 (1986).
Newman, R. M. & Waters, T. F. Size-selective predation on Gammarus pseudolimnaeus by trout and sculpins. Ecology, 1535–1545 (1984).
Conlan, K. E. Amphipod crustaceans and environmental disturbance: a review. J. Nat. Hist. 28, 519–554, 10.1080/00222939400770241 (1994).
Grabner, D. et al. Invaders, natives and their enemies: distribution patterns of amphipods and their microsporidian parasites in the Ruhr Metropolis, Germany. Parasit. Vectors 8, 1–15, 10.1186/s13071-015-1036-6 (2015).
Nelson, D. Gammarus-Microbial Interactions: A Review. Int. J. Zool. 2011, 1–6 (2011).
Zamanpoore, M., Grabowski, M. & Schiemer, M. P. F. Taxonomic review of freshwater Gammarus (Crustacea: Amphipoda) from Iran. Zootaxa 3140, 1–14 (2011).
Copilaş‐Ciocianu, D. & Petrusek, A. The south‐western Carpathians as an ancient centre of diversity of freshwater gammarid amphipods: insights from the Gammarus fossarum species complex. Mol. Ecol. 24, 3980–3992, 10.1111/mec.13286 (2015).
Sars, G. Beretning om en i Sommeren 1863 foretagen Reise i Christiania Stift. Johan Dahl, Christiania (1864).
Khalaji-Pirbalouty, V. & Sari, A. Biogeography of amphipods (Crustacea: Amphipoda: Gammaridae) from the central Zagros Mountains, Iran, with descriptions of two new species. J. Nat. Hist. 38, 2425–2445 (2004).
Schäferna, K. Amphipoda balcanica, spolu s poznámkami o jiných sladkovodních Amphipodech. Vĕstnik Královske České společnosti Náuk 2, 1–111 (1922).
Hekmatara, M., Sari, A. & Heidary Baladehi, M. H. Two new Gammarus species (Crustacea: Amphipoda: Gammaridae) from Zagros Mountains, Iran. Zootaxa 2894, 39–57 (2011).
Stock, J. H., Mirzajani, A. R., Vonk, R., Naderi, S. & Kiabi, B. H. Limnic and brackish water Amphipoda (Crustacea) from Iran. Beaufortia 48, 173–234 (1998).
Martynov, A. Zur Kenntnis der Amphipoden der Krim. Zoologische Jahrbücher, Abteilung für Systematik, Ökologie Und Geographie der Tiere 60, 5–6 (1931).
Hou, Z., Sket, B., Fišer, C. & Li, S. Eocene habitat shift from saline to freshwater promoted Tethyan amphipod diversification. Proc. Natl. Acad. Sci. USA 108, 14533–14538 (2011).
Purvis, A. & Hector, A. Getting the measure of biodiversity. Nature 405, 212–219 (2000).
Bergsten, J. et al. The Effect of Geographical Scale of Sampling on DNA Barcoding. Syst. Biol. 10.1093/sysbio/sys037 (2012).
Hill, J. L., Curran, P. J. & Foody, G. M. The Effect of Sampling on the Species-Area Curve. Global Ecol. Biogeogr. 4, 97–106, 10.2307/2997435 (1994).
Hou, Z., Fu, J. & Li, S. A molecular phylogeny of the genus Gammarus (Crustacea: Amphipoda) based on mitochondrial and nuclear gene sequences. Mol. Phylogenet. Evol. 45, 596–611 (2007).
Mamos, T., Wattier, R., Burzyński, A. & Grabowski, M. The legacy of a vanished sea: a high level of diversification within a European freshwater amphipod species complex driven by 15 My of Paratethys regression. Mol. Ecol., 10.1111/mec.13499 (2015).
Pinkster, S. The value of morphological characters in taxonomy of Gammarus. Beaufortia 33, 15–28 (1983).
Moritz, C. & Cicero, C. DNA Barcoding: Promise and Pitfalls. PLoS Biol. 2, e354, 10.1371/journal.pbio.0020354 (2004).
Lim, G. S., Balke, M. & Meier, R. Determining species boundaries in a world full of rarity: singletons, species delimitation methods. Syst. Biol. 61, 165–169 (2012).
Talavera, G., Dincă, V. & Vila, R. Factors affecting species delimitations with the GMYC model: insights from a butterfly survey. Methods Ecol. Evol. 4, 1101–1110 (2013).
Fisher, W. B., Avery, P., Hambly, G. R. & Melville, C. The cambridge history of Iran. Vol. 7, (Cambridge University Press, 1991).
Graca, M. A. The role of invertebrates on leaf litter decomposition in streams-a review. Int. Rev. Hydrobiol. 86, 383–393 (2001).
Dudgeon, D. et al. Freshwater biodiversity: importance, threats, status and conservation challenges. Biol. Rev. 81, 163–182, 10.1017/S1464793105006950 (2006).
Dudgeon, D. Prospects for sustaining freshwater biodiversity in the 21st century: linking ecosystem structure and function. Curr. Opin. Environ. Sustain. 2, 422–430 (2010).
Brodersen, J. & Seehausen, O. Why evolutionary biologists should get seriously involved in ecological monitoring and applied biodiversity assessment programs. Evol. Appl. 7, 968–983, 10.1111/eva.12215 (2014).
Karaman, G. S. & Pinkster, S. Freshwater Gammarus species from Europe, north Africa and adjacent regions of Asia (Crustacea-Amphipoda)- Part 1—Gammarus Pulex group and related species. (Commissie voor de artis bibliotheek, 1977).
Karaman, G. S. & Pinkster, S. Freshwater Gammarus species from Europe, north Africa and adjacent regions of Asia (Crustacea-Amphipoda)- Part 2—Gammarus Roeseli group and related species. (Commissie voor de artis bibliotheek, 1977).
Karaman, G. S. & Pinkster, S. Freshwater Gammarus species from Europe, North Africa and adjacent regions of Asia (Crustacea-Amphipoda). Part III. Gammarus balcanicus-group and related species. Bijdragen tot de Dierkunde 57, 207–260 (1987).
Sunnucks, P. & Hales, D. F. Numerous transposed sequences of mitochondrial cytochrome oxidase I-II in aphids of the genus Sitobion (Hemiptera: Aphididae). Mol. Biol. Evol. 13, 510–524 (1996).
Astrin, J. J. & Stüben, P. E. Phylogeny in cryptic weevils: molecules, morphology and new genera of western Palaearctic Cryptorhynchinae (Coleoptera: Curculionidae). Invertebr. Syst. 22, 503–522 (2008).
Cristescu, M. E. & Hebert, P. D. The” Crustacean Seas” an evolutionary perspective on the Ponto Caspian peracarids. Can. J. Fish. Aquat. Sci. 62, 505–517 (2005).
Kearse, M. et al. Geneious Basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649, 10.1093/bioinformatics/bts199 (2012).
Edgar, R. C. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797, 10.1093/nar/gkh340 (2004).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Raupach, M. J. & Radulovici, A. E. Looking back on a decade of barcoding crustaceans. ZooKeys 539, 53 (2015).
Puillandre, N., Lambert, A., Brouillet, S. & Achaz, G. ABGD, Automatic Barcode Gap Discovery for primary species delimitation. Mol. Ecol. 21, 1864–1877, 10.1111/j.1365-294X.2011.05239.x (2012).
Hebert, P., Cywinska, A., Ball, S. & DeWaard, J. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. Ser. B-Biol. Sci. 270, 313–321 (2003).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis Version 6.0. Mol. Biol. Evol. 30, 2725–2729, 10.1093/molbev/mst197 (2013).
Hart, M. W. & Sunday, J. Things fall apart: biological species form unconnected parsimony networks. Biol. Lett. 3, 509–512, 10.1098/rsbl.2007.0307 (2007).
Clement, M., Posada, D. & Crandall, K. A. TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9, 1657–1659 (2000).
Zhang, J., Kapli, P., Pavlidis, P. & Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 29, 2869–2876, 10.1093/bioinformatics/btt499 (2013).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian Phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973, 10.1093/molbev/mss075 (2012).
Posada, D. jModelTest: Phylogenetic Model Averaging. Mol. Biol. Evol. 25, 1253–1256, 10.1093/molbev/msn083 (2008).
Rambaut, A., Suchard, M., Xie, D. & Drummond, A. Tracer v1.6. (2014) Available at: http://beast.bio.ed.ac.uk/Tracer. (Accessed 27th July 2014).
Rambaut, A. & Drummond, A. TreeAnnotator. (2007) Available at: http://beast.bio.ed.ac.uk/TreeAnnotator. (Accessed 27th July 2014).
Reid, N. & Carstens, B. Phylogenetic estimation error can decrease the accuracy of species delimitation: a Bayesian implementation of the general mixed Yule-coalescent model. BMC Evol. Biol. 12, 196 (2012).
Huson, D. H. & Bryant, D. Application of Phylogenetic Networks in Evolutionary Studies. Mol. Biol. Evol. 23, 254–267, 10.1093/molbev/msj030 (2006).
Acknowledgements
We thank Ralph Tollrian, Ruhr University Bochum, for providing laboratory infrastructure. The Iran Department of Environment and Golestan National Park (GNP) authorities are acknowledged for sampling permission. We express our gratitude to Abbas Kazemi, Kaveh Darabi-Darestani, Hasan Mirmonsef and Robabeh Latif for providing some samples. Alimorad Sarafrazi (Institute of Plant Protection, Tehran) is acknowledged for his contribution in technical and scientific issues with whom we had fruitful discussions. We are also grateful to Hassan Salehi, Zoological Museum, University of Tehran and Oliver Coleman, Museum für Naturkunde Berlin for providing us with museum material. A visiting scholarship to ARK was provided by International office, University of Tehran. Their help is greatly acknowledged. This study was funded in part by a grant of the Kurt Eberhard Bode Foundation to FL and partly by the Research Council, University of Tehran and the Scientific and Technological Department of Iran Presidential Office to ASari.
Author information
Authors and Affiliations
Contributions
Designed the study: A.R.K., A.S., A.M.W., A.S. and F.L. Sampling: A.R.K. and A.S. Performed the laboratory work: A.R.K., A.M.W. and M.W. Contributed reagents: A.S. and F.L. Data analysis: A.R.K., A.M.W., J.N.M. and F.L. Discussion and interpretation of the results: A.R.K., A.M.W., A.S., J.N.M., F.L. and M.W. Wrote the manuscript: A.R.K., A.M.W., J.N.M., F.L. and A.S. All authors have read and approved the final version of the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Katouzian, AR., Sari, A., Macher, J. et al. Drastic underestimation of amphipod biodiversity in the endangered Irano-Anatolian and Caucasus biodiversity hotspots. Sci Rep 6, 22507 (2016). https://doi.org/10.1038/srep22507
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep22507
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
