
Abstract
Triggering receptor expressed on myeloid cells 1 (TREM-1) increases the expression of TGF-β family genes, which are known as profibrogenic cytokines in the pathogenesis of pulmonary fibrosis. In this study, we determined whether TGF-β1 regulated the expression of TREM-1 in a mouse model of pulmonary fibrosis. The expression of TGF-β1 and TREM-1 was increased on day 7, 14 and 21 after single intratracheal injection of bleomycin (BLM). And there was positive correlation between the expression of TGF-β1 and TREM-1. TGF-β1 increased expression of TREM-1 mRNA and protein in a time- and dose-dependent manner in mouse macrophages. The expression of the activator protein 1 (AP-1) was increased in lung tissues from mouse after BLM injection and in mouse macrophages after TGF-β1 treatment, respectively. TGF-β1 significantly increased the relative activity of luciferase in the cells transfected with plasmid contenting wild type-promoter of TREM-1. But TGF-β1 had no effect on the activity of luciferase in the cells transfected with a mutant-TREM1 plasmid carrying mutations in the AP-1 promoter binding site. In conclusion, we found the expression of TREM-1 was increased in lung tissues from mice with pulmonary fibrosis. TGF-β1 increased the expression of TREM-1 in mouse macrophages partly via the transcription factor AP-1.
Similar content being viewed by others


Introduction
The term “genomic storm” describes a new paradigm in human immune and inflammatory responses at the time of serious injury1. Triggering receptor expressed on myeloid cells 1 (TREM-1) expression is increased significantly during “genomic storms”2. As a cell-membrane surface receptor belonging to the IgG super family, TREM-1 is selectively expressed in mononuclear macrophages and neutrophils3,4. Synergistic activation of TREM-1, toll-like receptor and nod-like receptor results in the activation of pro-inflammatory factors such as TNF-α and IL-1β, as well as the inhibition of the anti-inflammatory factor IL-105, which prolongs macrophage survival6 and ultimately leads to excessive inflammatory responses. TREM-1 is reported to amplify inflammatory responses and aggravate acute lung injury and acute respiratory distress syndrome4,7. However, there is little research on the role of TREM-1 in fibrotic disease. A recent study found that the expression of TREM-1 was significantly increased in renal fibrosis8, while our previous observations also shown that TREM-1 was expressed in lung tissues4. It has been shown that TREM-1 activation increases the expression of transforming growth factor (TGF)-β family genes by 96.7-fold9. These previous findings prompted us to speculate that there is a putative link between TREM-1 and pulmonary fibrosis.
Pulmonary fibrosis is a chronic, progressive and devastating interstitial lung disease10,11,12. Clinically, pulmonary fibrosis manifests as a continuous decline in lung function, which eventually leads to respiratory failure13. The morbidity and mortality of pulmonary fibrosis, which is similar to lung cancer, are increasing14. The median survival period of pulmonary fibrosis is 2–5 years after diagnosis15 and patients often have poor prognosis and impairment of health-related quality of life. Currently, there is no effective drug treatment for late stage pulmonary fibrosis and the exponential increase in the number of clinical trials for the treatment of pulmonary fibrosis has shown few promising results16. Although the origin and pathogenesis of pulmonary fibrosis is complex, TGF-β1 has been implicated as a key player in the pathogenesis of pulmonary fibrosis, as it may induce proliferation and differentiation of fibroblasts, epithelial-mesenchymal transition (EMT) and transformation of lung fibroblasts to myofibroblasts, which eventually lead to serious pulmonary fibrosis in vivo and in vitro17,18,19.
TGF-β1 is a central node in the pulmonary fibrosis pathogenesis pathway, while TREM-1 activation dramatically increases the expression of TGF-β family genes. Conversely, it remains unknown whether the increase of TGF-β1 could affect the expression of TREM-1. This study was designed to determine the effect of TGF-β1 on the expression of TREM-1 in lungs and to explore the transcriptional mechanism at both the organismal and cellular levels, which will provide novel insights into the regulatory mechanisms in the pathogenesis of pulmonary fibrosis and therefore the potential therapeutic targets for the treatment of pulmonary fibrosis.
Materials and Methods
Establishment of a Mouse Pulmonary Fibrosis Model
All animal care and experimental protocols were in compliance with the Animal Management Rules of the Ministry of Health of the People’s Republic of China. The animal experimental protocols were approved by the Ethics Committee of Xiangya Hospital of Central South University. All mouse experiments were performed under anesthesia and we made every effort to minimize pain during the experiments.
Male Swiss mice weighing 20 ± 2 g were purchased from the Laboratory Animal Department of Central South University, fed in a clean animal room maintained at 23 to 25 °C, 50% to 60% relative humidity and 12 h circadian rhythm. Mice were randomly divided into two groups (n = 24/group): bleomycin (BLM)-treated and control. The BLM-treated group was intratracheally administered with BLM (5 mg/kg, Nippon Kayaku, Japan) to establish a model of pulmonary fibrosis, while the control group was administered with equal amount of normal saline19. The 8 mice of the each group were randomly selected and sacrificed respectively on day 7, 14 and 21 after administration of BLM or saline for further experiments.
Histological Examination of Lung Tissues
The lower right lobe of the lungs was fixed in 4% paraformaldehyde solution and embedded in paraffin. Pathological changes in lung tissues and collagen deposition were observed by staining lung sections with hematoxylin-eosin (HE) staining or Masson triple staining.
RNA Extraction and Real-Time PCR
RNA was extracted from lung tissues or mouse macrophages using the TRIzol method (Invitrogen, USA). The cDNA was synthesized using RevertAid First Strand cDNA short Kit (Thermo Scientific, USA) according to the manufacturer’s protocol. Transcriptional levels of procollagen types I and III, TGF-β1, TREM-1 and AP-1 were determined using UltraSYBR Mixture (cwbiotech, China) and a Real-time PCR Detection System (Bio-Rad, USA). The mRNA expression of our genes of interest was calculated by 2−ΔΔCt method normalized to housekeeper GAPDH.
Western Blotting
The lung tissues were homogenized, separated on 10% SDS-PAGE gel (Bio-Rad, USA), then transferred onto a PVDF membrane using the semi-dry method20,21,22. After blocking, membranes were incubated with mouse anti-TREM-1 antibodies (R&D Systems, USA) overnight. Horseradish peroxidase-conjugated secondary antibodies (Santa Cruz, USA) were applied and enhanced chemiluminescence to detect protein content. Images were collected using a FluorChemQ system (Alpha Innotech Corporation, USA).
Cell Cultures
Mouse macrophage cell line RAW264.7 was purchased from Xiangya Central Experiment Laboratory of Central South University. Cells were cultured in DMEM (Sigma Chemical company, USA) supplemented with 10% Fetal Bovine Serum (HyClone, USA) and incubated at 37 °C in a 5% CO2 incubator.
Primary alveolar macrophages isolation
Bronchoalveolar lavage fluid (BALF) were flushed three times with 1 mL ice-cold PBS in Swiss mice after anesthetization and arteria femoralis bloodletting23. Primary alveolar macrophages were isolated from BALF by centrifugation method and adherence-changing culture method24.
Flow Cytometry
Mouse macrophages (a cell line and primary alveolar macrophages) were plated into 6-well cell culture plates and stimulated with 10 ng/mL TGF-β1 for various periods of time (0, 1, 2, 5, 10, 20 and 40 h). Different concentrations (0, 1, 2, 5, 10 and 20 ng/mL) of TGF-β1 were tested for their stimulatory effect on mouse macrophages at the 20 h time point.
Flow Cytometry was used for testing protein level of TREM-1 on the surface of cells25,26,27. After incubating cells for 30 min using mouse anti-TREM-1 antibody (Source: Monoclonal Rat IgG) (R&D Systems, USA) and chicken anti-rat IgG-FITC antibody (Santa Cruz, USA), the percentage of cells positive for TREM-1 was measured using a Moflo XDP type flow cytometry instrument (Beckman Coulter, USA), revealing the amount of surface-expressed TREM-1 protein. The data were analyzed with Summit 5.0 or FlowJo 7.6.5.
Site-Directed Mutagenesis and Plasmid Construction
The TREM-1 promoter sequence in mouse found by analyzing the sequence of chromosome 17 and the EST database. The TREM-1 promoter sequence was amplified by PCR from mouse genomic DNA using Pfu polymerase (Sangon, China) and the following oligonucleotide PCR amplification primers: sense primer, 5′-caaGGTACCTGTATGTGGGCAAATGTAGTGTGTGTGGCGGG-3′ and anti-sense primer, 5′-cacacgtctcAAGCTTCCTTCAAGCTCAGCTCCAACGACTGCCTCTG-3′. PCR products were digested with KpnI and HindIII and subcloned into the firefly luciferase expression plasmid pGL3-basic (Promega, USA) to produce the wild type (WT)-TREM1 plasmid.
The TFSEARCH database was used to find transcriptional factor binding sites located within the TREM-1 5′-upstream promoter region. The promoter sequence was found to contain two AP-1 binding sites, which were mutated to adenines via site directed mutagenesis. The WT-TREM1 plasmid was used as the template with Pfu polymerase (Sangon, China) and site-directed mutagenesis oligonucleotide primers (Table 1) for PCR amplification of the entire plasmid. PCR products were purified using E.Z.N.A.TM Gel Extraction Kit (Omega, USA), then ligated with T4 DNA ligase (Thermo Scientific, USA).
Construction of the plasmid containing wild type (WT) or mutation of the activator protein 1 (AP-1) promoter sequence using site-directed mutagenesis were based on the Hosoda study (Table 1)28. WT-TREM-1 and mutant-TREM-1 plasmid sequences were confirmed using BigDye® Terminator v3.1 sequencing kit and 3730xl DNA Analyzer (Applied Biosystems, USA).
Transfection and Dual-Luciferase Reporter Assay
Mouse macrophages (1 × 105) per well were plated into a 24-well cell culture plate, transfected with either the firefly luciferase expression plasmid pGL3-basic (500 ng), WT-TREM-1 (500 ng), or mutant-TREM-1 (500 ng). The renilla luciferase expression plasmid pRL-TK (1 ng) (Promega, USA) was transfected into all cells as an internal control. All transfections were performed using Lipofetamine® 2000 (Invitrogen, USA) and cells were incubated for 5 h following transfection. Thereafter, mouse macrophages were stimulated with TGF-β1 (10 ng/mL) for 20 h, washed twice with PBS and lysed in 100 uL Passive Lysis Buffer (Promega, USA). Following this, firefly and renilla luciferase activities were detected using a Dual-luciferase® Reporter Assay System (Promega, USA) and Varioskan Flash (Thermo SCIENTIFIC, USA). Relative luciferase activities were standardized to renilla luciferase activity and expressed as a ratio of firefly luciferase activity to renilla luciferase activity.
Statistical Analysis
The data were analyzed using SPSS 17.0 software and expressed as mean ± standard deviation (SD). The difference between two groups was tested using the t-test, while difference between multiple groups was tested using one-way analysis of variance. The SNK test served as the post hoc test for multiple comparisons. Correlations were analyzed using Pearson Correlation analysis. A p-value < 0.05 was considered to be statistically significant.
Results
Establishment of Pulmonary Fibrosis Model in Mice
To verify the establishment of our mouse pulmonary fibrosis model, morphological changes in the mouse lung tissues was observed. HE staining results showed that lung tissues in mice from the control group had intact structure, clear alveolar outlines, thin alveolar septum and no sign of inflammation (Fig. S1a–c). In the BLM-treated group, there was extensive infiltration of inflammatory cells on day 7 (Fig. S1d). The destruction of alveolar structure, formation of fibrotic lesions and persistent inflammation were present on day 14 (Fig. S1e). Further serious damage to the alveolar structure and extensive fibrosis were observed on day 21 (Fig. S1f). The mRNA levels of procollagen types I and III in lung tissues were dramatically ascended on day 14 and 21 when compared to control group (p < 0.05) (Fig. S1m,n). The deposition of collagen in lung tissues was detected by Masson staining and we found the quantity of collagen increased dramatically and extensive pulmonary fibrosis was formed on days 7, 14 and 21 in mice from the BLM-treated group in comparison with the control group (Fig. S1g–l).
TGF-β1 Expression Positively Correlated with TREM-1 Overexpression in Pulmonary Fibrosis
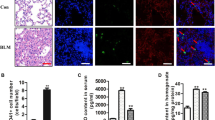
To observe the expression changes of TGF-β1 and TREM-1 in pulmonary fibrosis, TGF-β1 and TREM-1 mRNA levels were measured by real-time PCR. The results showed that both TGF-β1 and TREM-1 mRNA levels were significantly increased on day 7, 14 and 21 in the BLM-treated group (p < 0.05) (Fig. 1a,b). The TREM-1 protein detected by western-blot in mouse lung tissues was also remarkably increased (p < 0.05) (Fig. 1d,e). To further analyze the relationship between TGF-β1 and TREM-1, we applied Pearson Correlation analysis and showed that the change of TGF-β1 mRNA expression was positively correlated with change of TREM-1 mRNA expression in murine lungs (r = 0.787, p < 0.05) (Fig. 1c).

TGF-β1 expression was positive correlated with TREM-1 upregulation in mouse lungs.
The mRNA levels of TGF-β1 (a) and TREM-1 (b) in mouse lungs were detected by real-time PCR. The correlation between TGF-β1 and TREM-1 (c) was analyzed by Pearson correlation analysis and the correlation coefficient was 0.787, p < 0.01. The controls for each time-point were normalized as 1 in (e). Expression of TREM-1 protein (d,e) in lung tissues was detected by Western blot. Data were expressed as the mean ± SD with 8 mice per group, **p < 0.01.
TGF-β1 increased TREM-1 Expression in a Time- and Dose-Dependent Manner in Mouse Macrophages
To test whether TGF-β1 can affect the expression of TREM-1 in mouse macrophages, we measured TREM-1 mRNA level by real-time PCR after stimulation with 10 ng/mL TGF-β1 for various periods of time (0, 1, 2, 5, 10, 20 and 40 h). TREM-1 mRNA level was increased notably after treatment with TGF-β1 (10 ng/mL) for 1, 2, 5, 10, 20 and 40 h and the most notable increase was observed at 20 h (p < 0.05) (Fig. 2a). The TREM-1 protein in mouse macrophages cell line was measured through Flow cytometry. These results showed that the expression of TREM-1 protein did not change significantly after 1, 2, 5 and 10 h incubation with TGF-β1 (p > 0.05). However, there was a significant increase in TREM-1 protein expression after 20 and 40 h, with the highest expression level of TREM-1 at 20 h after TGF-β1 stimulation in mouse macrophages cell line (p < 0.05) (Figs 2b and S2a).

TGF-β1 increased TREM-1 expression in a time- and dose-dependent manner in mouse macrophages line.
TREM-1 mRNA expression (a) of mouse macrophages line following stimulation with 10 ng/mL TGF-β1 for various periods of time (0, 1, 2, 5, 10, 20 and 40 h) was examined by real-time PCR. TREM-1 protein level (b) on the surface of mouse macrophages line after stimulation with 10 ng/mL TGF-β1 for various periods of time was determined by Flow cytometry. TREM-1 mRNA expression (c) of macrophages line after incubation with TGF-β1 at various concentrations (0, 1, 2, 5, 10 and 20 ng/mL) for 20 h was detected by real-time PCR. The protein content of TREM-1 (d) on the surface of mouse macrophages line following incubation with TGF-β1 at various concentrations for 20 h was examined by Flow cytometry. Data were expressed as mean ± SD and were representative of 5 separate experiments performed in quadruplicate, **p < 0.01.
To assess the effect of different concentrations of TGF-β1 on TREM-1 expression, mouse macrophages cell line was incubated with TGF-β1 at various concentrations (0, 1, 2, 5, 10 and 20 ng/mL) for 20 h and TREM-1 mRNA level was measured by real-time PCR at the end of each incubation time. Expression of the TREM-1 mRNA was increased prominently when 2, 5, 10 and 20 ng/mL TGF-β1 were used and expression of TREM-1 mRNA reached its peak level at 10 ng/mL TGF-β1 (p < 0.05) (Fig. 2c). TREM-1 protein in mouse macrophages cell line was detected by Flow cytometry analysis. It revealed that expression of the TREM-1 protein was increased following stimulation with all tested concentrations of TGF-β1 in a dose-dependent manner, with a peak in expression when 10 ng/mL TGF-β1 was used to stimulate mouse macrophages cell line (p < 0.05) (Figs 2d and S2b).
In order to further validate the effect of TGF-β1 on TREM-1 expression, the mouse primary alveolar macrophages were isolated and treated with 10 ng/mL TGF-β1 for various periods of time (0, 1, 2, 5, 10, 20 and 40 h) as well as TGF-β1 at various concentrations (0, 1, 2, 5, 10 and 20 ng/mL) for 20 h. The results showed that TREM-1 protein level was not altered following 1, 2, 5 and 10 h incubation with 10 ng/ml TGF-β1 (p > 0.05), but there was a distinct increase at 20 h and 40 h, with the highest expression level at 20 h after TGF-β1 stimulation in primary alveolar macrophages (p < 0.05) (Fig. 3a,b). Also, TREM-1 protein level elevated following treatment with all tested concentrations of TGF-β1 in a dose-dependent manner, with a peak expression at 10 ng/mL TGF-β1 in primary alveolar macrophages (p < 0.05) (Fig. 3c,d). These results indicated that TGF-β1 increases TREM-1 protein expression in a time- and dose-dependent manner in mouse primary alveolar macrophages, which were consistent with the effect of TGF-β1 on TREM-1 expression in mouse macrophages cell line.

TGF-β1 increased TREM-1 expression in a time- and dose-dependent manner in mouse primary alveolar macrophages.
(a,b) TREM-1 protein level on the surface of mouse primary alveolar macrophages after stimulation with 10 ng/mL TGF-β1 for various periods of time (0, 1, 2, 5, 10, 20 and 40 h) was tested by Flow cytometry. (c,d) The protein content of TREM-1 of macrophages following incubation with TGF-β1 at various concentrations (0, 1, 2, 5, 10 and 20 ng/mL) for 20 h was examined by Flow cytometry. Data were expressed as mean ± SD and were representative of 2 separate experiments performed in triplicate, **p < 0.01.
TGF-β1-Dependent Increase in TREM-1 Expression may be linked to AP-1 Expression in Mouse Macrophages
In order to assess the transcriptional level of AP-1 in the lungs of mice with pulmonary fibrosis, AP-1 mRNA in lung tissues was detected by real-time PCR. We found the expression of AP-1 mRNA levels was increased significantly on day 7, 14 and 21 in the BLM-treated group (p < 0.05) (Fig. 4a). In order to further analyze the relationship between AP-1 and TGF-β1 or TREM-1 in pulmonary fibrosis, Pearson Correlation analysis was performed and the results showed that the AP-1 mRNA expression was positively related to TGF-β1 mRNA expression (r = 0.713, p < 0.05) (Fig. 4b) and TREM-1 mRNA expression (r = 0.906, p < 0.05) (Fig. 4c), respectively. In order to study any putative effect of TGF-β1 on AP-1 transcription levels, the expression of AP-1 mRNA was measured by real-time PCR after mouse macrophages were stimulated with 10 ng/ml TGF-β1 for various periods of time (0, 1, 2, 5, 10, 20 and 40 h), or various concentrations (0, 1, 2, 5, 10 and 20 ng/mL) for 20 h. Our results showed that expression of AP-1 mRNA was increased significantly 1 h after stimulation with 10 ng/ml TGF-β1 and increased expression of AP-1 mRNA was sustained up to 40 h post-stimulation (p < 0.05). Meanwhile, expression of AP-1 mRNA was increased significantly when 2, 5, 10 and 20 ng/mL of TGF-β1 were used to stimulate macrophages for 20 h and the maximal induction of AP-1 mRNA expression was observed when 10 ng/mL TGF-β1 was used (Fig. 4d,e).

The TGF-β1 mediated increase in TREM-1 expression might be associated with AP-1 in mouse macrophages.
Expression of AP-1 mRNA in lung tissues (a) was determined by real-time PCR. Correlations between AP-1 and TGF-β1 (b), between AP-1 and TREM-1 (c) by Pearson correlation analysis, the correlation coefficients were 0.773 and 0.773 (p < 0.01), respectively. The expression of AP-1 mRNA in 10 ng/ml TGF-β1 stimulated mouse macrophages at different time points (0, 1, 2, 5, 10, 20 and 40 h) and different concentrations for 20 h (0, 1, 2, 5, 10 and 20 ng/mL) (d,e) was detected by real-time PCR. Data were expressed as mean ± SD and were representative of 5 separate experiments performed in 8 mice or quadruplicate per group, **p < 0.01.
Transcription Factor AP-1 was Involved in TGF-β1 Induced TREM-1 Expression in Mouse Macrophages
To further confirm the transcriptional control of AP-1 in the upregulation of TREM-1 by TGF-β1 in mouse macrophages, WT- and mutant-TREM-1 plasmids (containing mutations at the AP-1 promoter binding site) were constructed and the binding activity of AP-1 with TREM-1 promoter under TGF-β1 stimulation was measured by the dual-luciferase reporter assay system. The results showed that TGF-β1 did not stimulate luciferase expression in the pGL3-basic plasmid (empty plasmid) (p > 0.05), but significantly increased the relative luciferase activity in WT-TREM-1 plasmid (p < 0.05). No increase in relative luciferase activity was observed when the mutant-TREM–1 plasmid was used (p > 0.05) (Fig. 5).

TGF-β1 stimulation increased the transcriptional activity of AP-1 at the TREM-1 promoter.
Mouse macrophages were transfected with pGL3-basic vector (500 ng), or WT-TREM1 reporter plasmid (500 ng), or mutant-TREM1 reporter plasmid (500 ng) and the renilla luciferase expressing pRL-TK plasmid (1 ng, an internal control) and were incubated for 5 h. Cells were then incubated in the absence or presence of TGF-β1 (10 ng/mL) for 20 h. Firefly and renilla luciferase activities were detected using Varioskan Flash. The relative luciferase activities were standardized to renilla luciferase activity. Data were expressed as mean ± SD and were representative of 5 separate experiments performed in quadruplicate, **p < 0.01.
Discussion
The concept of the “genome storm” is a novel paradigm of human immune reactions after serious injury1. Research has found that over 80% of the transcriptome of white blood cell (WBC) changed within 4 to 12 h after severe injury and that the changes were maintained for several weeks and even months after injuries2. The expression of genes involved in innate immunity as well as systemic immune and anti-inflammatory reactions was increased, while the expression of genes regulating adaptive immunity was decreased. These observations are in line with the concept of “nonresolving inflammation” and challenge the traditional “two-hit” model1. This new paradigm clearly suggests that therapies that limit the initial “genomic storm” within WBCs may be valuable in improving the prognosis of patients with severe injury2,29. Multiple proteins play their parts in amplifying the initial inflammatory response, including the upregulated TREM-1 during the “genomic storm” research, which may be attractive therapeutic targets. As an inflammatory amplificational factor30, TREM-1 comes into play in a variety of diseases, such as sepsis31, pneumonia32, lung cancer33, periodontitis34 and relapsing polychondritis35. However, there has been little study on TREM-1 in the pathogenesis of pulmonary fibrosis.
Pulmonary fibrosis is an irreversible and lethal interstitial lung disease with no effective pharmacological treatment36,37. BLM, an antibiotic, is commonly used for studying human pulmonary fibrosis in mouse and rat models. Following BLM treatment the lung undergoes significant biochemical, histological and physiological changes, which are similar to those of humans, leading to pulmonary fibrosis12,36. We found that TGF-β1 expression was positively correlated to the expression of TREM-1 significantly and expression of the TREM-1 was upregulated in the lungs of mice with BLM-induced pulmonary fibrosis. These observations leaded us to further investigate the role of macrophages in this possible cross-talk feedback interaction between TGF-β1 and TREM-1 in BLM-induced pulmonary fibrosis. We observed the changes in TREM-1 expression following stimulation with TGF-β1 and found that the low levels of TREM-1 expression in the unstimulated state, while the expression of TREM-1 was increased significantly in a time- and dose-dependent manner upon stimulation with TGF-β1. The previous studies have shown that TGF-β1 is a central regulator in the processes of pulmonary fibrosis38, while TREM-1 can markedly increase the expression of TGF-β family genes. Therefore, the expression of TGF-β1 and TREM-1 is mutually synergistic and a positive feedback can be formed that speeds up the development and progression of pulmonary fibrosis. It is worth noting that TGF-β1 is historically considered as a typical anti-inflammatory factor39,40, but our experiments showed that TGF-β1 exhibits pro-inflammatory effects by dramatically increasing the expression of the inflammatory amplifier TREM-1. This effect may be attributed to the different effects of TGF-β1 in distinct cell types and disease models.
AP-1 is a transcription factor, a heteromultimeric protein that is composed of proteins from the Jun (c-Jun, Jun-B, Jun-D) and Fos (c-Fos, Fos-B, Fra-1, Fra-2) families of proteins. AP-1 works by controlling a broad range of responses to different stimuli and regulating the expression of genes involved in oxidative stress, inflammatory reactions, cell growth and cell remodeling41,42. One previous study has shown that AP-1 can increase TREM-1 transcription induced by LPS in mouse macrophages28, indicating that AP-1 may be a transcription regulator in LPS-induced expression of TREM-1. AP-1 transcriptional activity is regulated by TGF-β1 in vivo and in vitro43. These findings suggest that AP-1 may be related to the upregulation of TREM-1 upon stimulation by TGF-β1. Therefore, we determined the expression of AP-1 in BLM-induced pulmonary fibrosis and we found that the expression of AP-1 mRNA was increased significantly in BLM-treated mice as well as macrophages suggesting an important role of AP-1 in BLM-induced pulmonary fibrosis. We also found that the binding activity of AP-1 with TREM-1 promoter is increased significantly upon TGF-β1 stimulation, but this pattern was not observed when the AP-1 binding sites in the TREM-1 promoter were mutated to alanines. To our knowledge, this is the first observation that reveals AP-1-mediated transcriptional control in TGF-β1-induced upregulation of the TREM-1 in cultured macrophages.
It should be noted that no functional data are performed in this mechanistic study. It is not clear whether modification of the expression of TREM-1 and TGF-β1 affects pulmonary function or progression of the pulmonary fibrosis. Further studies are warranted to illustrate the functional consequences of this interplay between TREM-1 and TGF-β1. Interestingly, a new TREM-1 inhibitor LR12 has just been discovered29, it is worthwhile to investigate whether LR12 can ameliorate BLM-induced pulmonary fibrosis and therefore improve functional outcome in this animal model.
In conclusion, we found that 1) the expression of TREM-1 was increased significantly in a mouse model of BLM-induced pulmonary fibrosis; 2) TGF-β1 increased expression of the TREM-1 by interfering AP-1 transcriptional pathway in mouse macrophages. TGF-β1 may form a positive feedback with TREM-1 and accelerate the progression of pulmonary fibrosis. These findings may provide new mechanistic insights into the pathogenesis of pulmonary fibrosis.
Additional Information
How to cite this article: Peng, L. et al. TGF-β1 Upregulates the Expression of Triggering Receptor Expressed on Myeloid Cells 1 in Murine Lungs. Sci. Rep. 6, 18946; doi: 10.1038/srep18946 (2016).
References
Leavy, O. Inflammation: Trauma kicks up a storm. Nat Rev Immunol 12, 3 (2012).
Xiao, W. et al. A genomic storm in critically injured humans. J Exp Med 208, 2581–90 (2011).
Bostanci, N. et al. Porphyromonas gingivalis Regulates TREM-1 in Human Polymorphonuclear Neutrophils via Its Gingipains. PLoS One 8, e75784 (2013).
Sun, G. Y. et al. Vasoactive intestinal peptide re-balances TREM-1/TREM-2 ratio in acute lung injury. Regul Pept 167, 56–64 (2011).
Bostanci, N., Ozturk, V. O., Emingil, G. & Belibasakis, G. N. Elevated oral and systemic levels of soluble triggering receptor expressed on myeloid cells-1 (sTREM-1) in periodontitis. J Dent Res 92, 161–5 (2013).
Yuan, Z. et al. Triggering receptor expressed on myeloid cells 1 (TREM-1)-mediated Bcl-2 induction prolongs macrophage survival. J Biol Chem 289, 15118–29 (2014).
Liu, N., Gu, Q. & Zheng, Y. S. Expression of triggering receptor-1 in myeloid cells of mice with acute lung injury. World J Emerg Med 1, 144–8 (2010).
Campanholle, G. et al. TLR-2/TLR-4 TREM-1 signaling pathway is dispensable in inflammatory myeloid cells during sterile kidney injury. PLoS One 8, e68640 (2013).
Dower, K., Ellis, D. K., Saraf, K., Jelinsky, S. A. & Lin, L. L. Innate immune responses to TREM-1 activation: overlap, divergence and positive and negative cross-talk with bacterial lipopolysaccharide. J Immunol 180, 3520–34 (2008).
Lin, C. et al. Targeting protease activated receptor-1 with P1pal-12 limits bleomycin-induced pulmonary fibrosis. Thorax 69, 152–60 (2014).
Olsen, K. C. et al. Inhibition of transglutaminase 2, a novel target for pulmonary fibrosis, by two small electrophilic molecules. Am J Respir Cell Mol Biol 50, 737–47 (2014).
Song, N. et al. Vagotomy attenuates bleomycin-induced pulmonary fibrosis in mice. Sci Rep 5, 13419 (2015).
Schmidt, S. L. et al. Predicting pulmonary fibrosis disease course from past trends in pulmonary function. Chest 145, 579–85 (2014).
Ley, B. & Collard, H. R. Epidemiology of idiopathic pulmonary fibrosis. Clin Epidemiol 5, 483–492 (2013).
Raghu, G., Anstrom, K. J., King, T. E., Jr., Lasky, J. A. & Martinez, F. J. Prednisone, azathioprine and N-acetylcysteine for pulmonary fibrosis. N Engl J Med 366, 1968–77 (2012).
Shi, K. et al. Pathogenesis pathways of idiopathic pulmonary fibrosis in bleomycin-induced lung injury model in mice. Respir Physiol Neurobiol 190C, 113–117 (2013).
Song, X. et al. All-transretinoic acid ameliorates bleomycin-induced lung fibrosis by downregulating the TGF-beta1/Smad3 signaling pathway in rats. Lab Invest 93, 1219–31 (2013).
Lepparanta, O. et al. Regulation of TGF-beta storage and activation in the human idiopathic pulmonary fibrosis lung. Cell Tissue Res 348, 491–503 (2012).
Zhou, Y. et al. Soluble epoxide hydrolase inhibitor 1-trifluoromethoxyphenyl-3- (1-propionylpiperidin-4-yl) urea attenuates bleomycin-induced pulmonary fibrosis in mice. Cell Tissue Res, doi: 10.1007/s00441-015-2262-0 (2015).
Brea, D. et al. Study of Protein Expression in Peri-Infarct Tissue after Cerebral Ischemia. Sci Rep 5, 12030 (2015).
Diaz, L. et al. Bryostatin activates HIV-1 latent expression in human astrocytes through a PKC and NF-kB-dependent mechanism. Sci Rep 5, 12442 (2015).
Kreisig, T., Prasse, A. A., Zscharnack, K., Volke, D. & Zuchner, T. His-tag protein monitoring by a fast mix-and-measure immunoassay. Sci Rep 4, 5613 (2014).
Nakayama, Y. et al. Oral administration of Lactobacillus gasseri SBT2055 is effective for preventing influenza in mice. Sci Rep 4, 4638 (2014).
Hancock, A., Armstrong, L., Gama, R. & Millar, A. Production of interleukin 13 by alveolar macrophages from normal and fibrotic lung. Am J Respir Cell Mol Biol 18, 60–5 (1998).
Prufer, S. et al. Distinct signaling cascades of TREM-1, TLR and NLR in neutrophils and monocytic cells. J Innate Immun 6, 339–52 (2014).
Qian, L., Weng, X. W., Chen, W., Sun, C. H. & Wu, J. TREM-1 as a potential therapeutic target in neonatal sepsis. Int J Clin Exp Med 7, 1650–8 (2014).
del Fresno, C. et al. Monocytes from cystic fibrosis patients are locked in an LPS tolerance state: down-regulation of TREM-1 as putative underlying mechanism. PLoS One 3, e2667 (2008).
Hosoda, H., Tamura, H., Kida, S. & Nagaoka, I. Transcriptional regulation of mouse TREM-1 gene in RAW264.7 macrophage-like cells. Life Sci 89, 115–22 (2011).
Derive, M., Boufenzer, A. & Gibot, S. Attenuation of responses to endotoxin by the triggering receptor expressed on myeloid cells-1 inhibitor LR12 in nonhuman primate. Anesthesiology 120, 935–42 (2014).
Bouchon, A., Facchetti, F., Weigand, M. A. & Colonna, M. TREM-1 amplifies inflammation and is a crucial mediator of septic shock. Nature 410, 1103–7 (2001).
van Bremen, T. et al. Triggering receptor expressed on myeloid cells-1 (Trem-1) on blood neutrophils is associated with cytokine inducibility in human E. coli sepsis. Diagn Pathol 8, 24 (2013).
Rivera-Chavez, F. A. et al. A TREM-1 Polymorphism A/T within the Exon 2 Is Associated with Pneumonia in Burn-Injured Patients. ISRN Inflamm 2013, 431739 (2013).
Ho, C. C. et al. TREM-1 expression in tumor-associated macrophages and clinical outcome in lung cancer. Am J Respir Crit Care Med 177, 763–70 (2008).
Bisson, C. et al. Increased gingival crevicular fluid levels of soluble triggering receptor expressed on myeloid cells (sTREM) -1 in severe periodontitis. J Clin Periodontol 39, 1141–8 (2012).
Sato, T. et al. Serum level of soluble triggering receptor expressed on myeloid cells-1 as a biomarker of disease activity in relapsing polychondritis. Mod Rheumatol 24, 129–36 (2014).
Ji, X. et al. The Anti-fibrotic Effects and Mechanisms of MicroRNA-486-5p in Pulmonary Fibrosis. Sci Rep 5, 14131 (2015).
Huan, C. et al. Methylation-mediated BMPER expression in fibroblast activation in vitro and lung fibrosis in mice in vivo. Sci Rep 5, 14910 (2015).
Bringardner, B. D., Baran, C. P., Eubank, T. D. & Marsh, C. B. The role of inflammation in the pathogenesis of idiopathic pulmonary fibrosis. Antioxid Redox Signal 10, 287–301 (2008).
Sugimoto, K. et al. Activated microglia in a rat stroke model express NG2 proteoglycan in peri-infarct tissue through the involvement of TGF-beta1. Glia 62, 185–98 (2014).
Abutbul, S. et al. TGF-beta signaling through SMAD2/3 induces the quiescent microglial phenotype within the CNS environment. Glia 60, 1160–71 (2012).
Davies, C. C., Chakraborty, A., Diefenbacher, M. E., Skehel, M. & Behrens, A. Arginine methylation of the c-Jun coactivator RACO-1 is required for c-Jun/AP-1 activation. EMBO J 32, 1556–67 (2013).
Babu, R. L. et al. Effect of estrogen and tamoxifen on the expression pattern of AP-1 factors in MCF-7 cells: role of c-Jun, c-Fos and Fra-1 in cell cycle regulation. Mol Cell Biochem 380, 143–51 (2013).
Rajasekaran, S., Vaz, M. & Reddy, S. P. Fra-1/AP-1 transcription factor negatively regulates pulmonary fibrosis in vivo. PLoS One 7, e41611 (2012).
Acknowledgements
This work was supported by the Special Funds for Major State Basic Research Projects (No. 2012CB518104), National Natural Science Foundation of China (No. 81170059), NINDS (No. K02NS081000) and the Specialized Research Fund for the Doctoral Program of Higher Education of China (No. 20130162110052).
Author information
Authors and Affiliations
Contributions
Conceived and designed the experiments: C.X.G., J.X.J., L.P. and Y.Z. Performed the experiments: L.P., Y.Z., L.D., R.Q.C. and T.L. Analyzed the data: Y.Z., G.Y.S. and W.Z.R. Contributed reagents/materials/analysis tools: Y.Z., J.X.J. and C.X.G. Wrote the paper: L.P., Y.Z., C.X.G. and J.X.J. Critically reviewed the manuscript: X.F., J.X.J. and C.X.G.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Peng, L., Zhou, Y., Dong, L. et al. TGF-β1 Upregulates the Expression of Triggering Receptor Expressed on Myeloid Cells 1 in Murine Lungs. Sci Rep 6, 18946 (2016). https://doi.org/10.1038/srep18946
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep18946
This article is cited by
-
Rheumatoid arthritis synovial fibroblasts promote TREM-1 expression in monocytes via COX-2/PGE2 pathway
Arthritis Research & Therapy (2019)
-
Soluble Epoxide Hydrolase Inhibitor Suppresses the Expression of Triggering Receptor Expressed on Myeloid Cells-1 by Inhibiting NF-kB Activation in Murine Macrophage
Inflammation (2017)
-
Blocking triggering receptor expressed on myeloid cells-1 attenuates lipopolysaccharide-induced acute lung injury via inhibiting NLRP3 inflammasome activation
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
