
Abstract
How common are cryptic species - those overlooked because of their morphological similarity? Despite its wide-ranging implications for biology and conservation, the answer remains open to debate. Butterflies constitute the best-studied invertebrates, playing a similar role as birds do in providing models for vertebrate biology. An accurate assessment of cryptic diversity in this emblematic group requires meticulous case-by-case assessments, but a preview to highlight cases of particular interest will help to direct future studies. We present a survey of mitochondrial genetic diversity for the butterfly fauna of the Iberian Peninsula with unprecedented resolution (3502 DNA barcodes for all 228 species), creating a reliable system for DNA-based identification and for the detection of overlooked diversity. After compiling available data for European butterflies (5782 sequences, 299 species), we applied the Generalized Mixed Yule-Coalescent model to explore potential cryptic diversity at a continental scale. The results indicate that 27.7% of these species include from two to four evolutionary significant units (ESUs), suggesting that cryptic biodiversity may be higher than expected for one of the best-studied invertebrate groups and regions. The ESUs represent important units for conservation, models for studies of evolutionary and speciation processes and sentinels for future research to unveil hidden diversity.
Similar content being viewed by others

Introduction
Evaluations of cryptic species (those overlooked due to their morphological similarity) are gaining momentum1, fuelled by recent advances in molecular techniques. Several studies suggest that cryptic species may be surprisingly common in certain taxonomic groups2,3,4, but the actual fraction of biodiversity that they represent in different lineages is subject to debate1,5,6. This mainly stems from the fact that ascertaining the incidence of cryptic species across the diversity of life is a challenging, time-consuming task. Yet, the need to gather comprehensive information about overlooked species is higher than ever, not only because of implications for various fields of research7, but also because planetary biodiversity is decreasing at an alarming rate and informed conservation decisions require accurate data. A necessary first step is documenting intraspecific diversity, which in itself is also relevant for the study of evolution, ecology, biogeography and conservation of biodiversity1,8,9.
Short DNA sequences have been used for species identification for more than 20 years10, but DNA barcoding11, the use of sequence variation in a short, standardized DNA marker to assess biodiversity, has only gained broad adoption within the past decade. Although this approach is not sufficient to verify the specific status of taxa, it provides an overview of variability in the mitochondrial genome which is typically accompanied by similar patterns of divergence in the nuclear genome. With comprehensive geographical sampling, this exercise can highlight potential cases of cryptic species that can subsequently be investigated in more detail. The accumulation of more than three million DNA barcode records (648 base pair sequences of the mitochondrial gene cytochrome c oxidase I, COI) (http://www.boldsystems.org/) has created the need for algorithmic approaches for species delimitation, approaches which generate results that are particularly useful for phylogenetic community ecology or for groups lacking a strong taxonomy. The Generalized Mixed Yule-Coalescent (GMYC) model12,13, the Automatic Barcode Gap Discovery (ABGD)14, the Poisson Tree Process (PTP)15 and the Barcode Index Number system (BIN)16 represent four of the most popular approaches. Among these methods, GMYC usually generates the highest number of putative species (i.e. GMYC entities) compared to morphospecies17,18,19. As a consequence, it was adopted to reduce the chance of overlooking potential cryptic species. Based upon phylogenetic and coalescent theory, GMYC defines statistically significant clusters and reports probabilities for the delimitations as a measure of robustness. Thus, resulting entities can be graded according to their GMYC support and minimum inter-entity genetic distance, so that decisions on research and management can be taken objectively. Once phylogenetic significant units have been identified, these can be studied in more detail by targeted studies using multiple data sources, expanding the operational criteria used for species delimitation. It is important to stress that units recovered by GMYC should be regarded as genetically diverged lineages that reflect various evolutionary processes and, in some cases, represent potential cryptic species20.
The Lepidoptera, one of the four most diverse insect orders, are arguably the most intensively studied group of invertebrates and more than one million barcode records have been so far generated for this group (http://www.boldsystems.org/). European butterflies in particular are the focus of monitoring schemes and intensive research on distribution, population dynamics, ecology and biogeography and represent a flagship group for insect conservation efforts21,22,23. Nevertheless, they have been the focus of relatively few DNA barcoding studies24,25,26,27,28, all with rather limited geographical coverage. Even more surprising, no comprehensive study is available for the Mediterranean region, although it was a major glacial refugium, a fact reflected by the high species richness and endemicity of the modern fauna29,30.
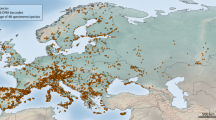
We assembled a DNA barcode dataset for all 228 species of Iberian butterflies, with an average of 15.4 specimens/species from various geographic regions, including mainland Spain, Portugal, Andorra and the Balearic Islands (Supplementary Fig. 1). Comprising about 50% of the European butterfly fauna (sensu22), the Iberian Peninsula is a butterfly diversity hotspot with a fauna representative of the Mediterranean region31. We subsequently merged these sequences with all other public records for European butterflies on the Barcode of Life Datasystems (BOLD)32 (www.boldsystems.org), creating a dataset with 5782 DNA barcodes from 299 species (ca. 60% of the European butterfly fauna) (Fig. 1). We then employed GMYC to identify phylogenetically significant units and highlight deeply diverged lineages which represent potential cases of cryptic species that require further study (Fig. 2). Thus, we present the first continental-scale estimate for the incidence of cryptic diversity in European butterflies. These results provide important direction for future research and have broad implications for the biogeography, conservation, taxonomy and ecology of European butterflies.

Map of Europe illustrating the sampling effort and taxon coverage (percentage of the species present in each country with DNA barcode records).
The distance scale corresponds to longitudinally-oriented distances only. The map was generated using Quantum GIS 1.8.0 based on a map from Natural Earth.

Workflow used to assess recognized biodiversity and potential cryptic biodiversity in European butterflies and percentages obtained in each category.
Results
DNA barcoding performance
For the Iberian dataset we obtained 3502 COI sequences derived from 228 currently recognized species, with an average of 15.4 specimens per species (Supplementary Fig. 1, Supplementary Table 1). Only one species (Azanus jesous) was represented by a single individual. According to our evaluation (see the Methods), DNA sequences for 93.9% of the 228 species were monophyletic, 2.6% were para-/polyphyletic and 3.5% shared barcodes. All species that shared barcodes belonged to the family Lycaenidae: Polyommatus fabressei - P. fulgens, Pseudophilotes baton - P. panoptes, Cupido lorquinii-C. minimus (because of taxon carswelli) and Lysandra caelestissima - L. albicans (due to a potential hybrid between the two species) (Supplementary Figures 2 and 3, Supplementary Table 2). Two of the six Iberian butterfly families barcoded (Pieridae, Papilionidae) showed 100% monophyletic species, while, at the opposite pole, 86.5% of the Lycaenidae were monophyletic.
The European dataset consisted of 5782 sequences with representation for 299 species, with an average of 19.3 specimens per species. Compared to Iberia, this dataset included more geographic and taxon coverage (71 additional species) (Fig. 1, Supplementary Table 1). DNA sequences for 84.6% of the 299 species were monophyletic, 8.0% were para-/polyphyletic and 7.4% shared barcodes. Only the family Papilionidae showed 100% monophyletic species while the most notable decreases in monophyly occurred in the Nymphalidae (83.7% versus 96.8% for Iberia) and the Pieridae (88.6% versus 100% for Iberia) (Supplementary Figures 2 and 4, Supplementary Table 2).
Patterns of genetic diversity in European butterflies
The single-threshold GMYC (ST-GMYC) generated a higher score compared to the multimodel one (MM-GMYC) based on Akaike’s Information Criterion (AIC) and the subsequent analysis of GMYC entities was based on the ST-GMYC model. Depending on the method used and the taxon studied, the estimated values for haploid effective population size (Ne) varied between 4 × 104 and 1.8 × 107, with an average of 1.6 × 106 and the speciation rate (SR) was 0.0455 when considering species based on Fauna Europaea and 0.0473 when considering GMYC entities. Such low values for SR compensate for the rather high Ne and their product (Ne x SR) ranged from 1.8 × 103 to 8 × 105, with an average of 7 × 104, falling within the range found to be optimal for GMYC performance (see33 for details). Because regionally oriented biodiversity surveys typically include very genetically divergent species and a small proportion of closely related species compared to taxon oriented surveys, they should result in datasets with parameters more adequate for GMYC analyses. The ST-GMYC analysis of the full dataset of 1971 haplotypes representing 299 species recovered 55.9% (167 species) as single entities (SE), 16.4% (49 species) were lumped with another species, 21.7% (65 species) were split into two to four entities (ME) and 6% (18 species) were split into multiple entities, but some of these entities were lumped with other species. Overall, 27.7% (83 species) involved ME, indicating potential cryptic biodiversity (Figs 2, 3). These 83 species generated 64 more GMYC entities than the number of recognized species (363 entities versus 299 species, representing an increase of 21.4% compared to the recognized species) (Supplementary Figures 5 and 6 and see Supplementary Table 3 for further details on the GMYC results).

Species distribution by family and the number of species recovered as multiple entities (ME) by the ST-GMYC analysis, or lumped with another species.
Percentages of ME and lumped cases are shown above the bars. The single representative of the Riodinidae (Hamearis lucina), not illustrated in the graph, but counted in the total assessment, was recovered as a single entity.
Cases of ME were detected within five of the six butterfly families, but the sole representative of the Riodinidae, Hamearis lucina, was recovered as a single entity. The highest percentage of ME was found in the Papilionidae (50.0%) followed by the Lycaenidae (33.3% of the species) (Fig. 3).
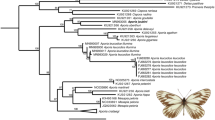
For the 83 species with ME, the minimum p-distance to the nearest conspecific entity (or containing conspecifics) varied from 0.3% to 4.0% and averaged 1.5% (Supplementary Table 4). Fourteen of the ME species displayed a minimum inter-entity p-distance of more than 2.5% and five of these species included entities detected in sympatry (Fig. 4).

Fourteen species split into multiple ST-GMYC entities with a minimum genetic distance of at least 2.5%.
Representative specimens (not drawn to scale) of each species are shown in dorsal view (left side) and ventral view (right side). The ST-GMYC and MM-GMYC supports for entity differentiation are shown. The sympatry/allopatry relationship among conspecific entities is shown based on a 50-km distance threshold. If a species was split in more than two entities, the data presented refer to the entity that was genetically most distant to the others. Specimen photos by R. Vila.
The ST-GMYC and MM-GMYC entity supports were very variable (between 0.24–1.00 for ST-GMYC and between 0.25–1.00 for MM-GMYC) for entities displaying up to 1.5% genetic divergence. When divergence was higher than 1.5% (the average value for all the MEs), both supports rose abruptly, usually exceeding 0.95 (only two cases between 0.85 and 0.9) (Fig. 5, Supplementary Fig. 7).

Bivariate scatter plot illustrating the relationship between minimum genetic divergence (%) among conspecific entities and the ST-GMYC supports for entity differentiation.
ST-GMYC supports rose abruptly over 1.5% divergence, but high support values were also achieved for some p-distances under 1%.
The case of Iphiclides podalirius
The GMYC analysis for I. podalirius included specimens from Iberia and south-western France belonging to the subspecies feisthamelii, which has also been suggested to represent a distinct species by some authors34,35. It also included specimens of nominotypical podalirius from Romania (Supplementary Table 5). All these specimens were recovered as a single highly supported GMYC entity (support 1 for both the ST- and MM-GMYC and maximum within entity p-distance = 0.5%), without support for feisthamelii as an independently evolving lineage (Supplementary Figures 5 and 6). When the dataset was extended with additional samples from southern Greece (one specimen), north-eastern Spain (one specimen) and northern Morocco (three specimens), we found that the three north African specimens were recovered as a distinct COI lineage as illustrated in a neighbor-joining tree (2.1% minimum p-distance to the European specimens) (Fig. 6a). Data from one nuclear marker (ITS2) showed that the Iberian and south-western France specimens clustered with those from Morocco and were distinct from the Romanian and Greek individuals (Fig. 6b). Morphological differences matched the pattern revealed by the nuclear marker (Fig. 6c,d, Supplementary Table 6).

The taxa Iphiclides p. podalirius (in black) and I. p. feisthamelii (in blue) are differentiated morphologically and genetically, but in Europe they share DNA barcodes due to an apparently fixed mitochondrial DNA introgression from podalirius into feisthamelii in the Iberian Peninsula.
(a) Neighbor-joining tree based on DNA barcodes of podalirius and feisthamelii: only the Moroccan specimens form a distinct clade. (b) Neighbor-joining tree based on nuclear ITS2 sequences of podalirius and feisthamelii: with the exception of one specimen from the northern Pyrenees, all specimens from Iberia, south-western France and Morocco form a distinct clade composed of feisthamelii. (c) Results of linear morphometrics for elements of the phallus (phallus width, phallus length and ostium phallus). The morphological separation between podalirius (black circles) and feisthamelii (blue dots) is in agreement with the nuclear marker (ITS2). (d) Phallus of Iphiclides indicating the measurements performed. (e) Sampling localities for the sequenced Iphiclides specimens and taxon assignment based on morphology and the nuclear marker ITS2. The map was generated using Quantum GIS 1.8.0 based on a map from Natural Earth. Specimen photos by R. Vila.
Discussion
Our results showed that a very high proportion (93.9%) of Iberian butterfly species were recovered as monophyletic, indicating that they can be unambiguously identified to a species level using DNA barcodes. When the geographical and taxon coverage was extended across much of Europe (Fig. 1), the overall levels of monophyly decreased to 84.6% (Supplementary Fig. 2, Supplementary Table 2). Many of the paraphyletic species are likely to be diagnosable as well since they displayed well-diverged lineages with respect to their nearest neighbour (e.g. Erebia pronoe, Spialia sertorius) (Supplementary Fig. 4). Some of the few cases where resolution is only possible to a triplet or quartet, such as species in the genus Lysandra, are known for a relatively high frequency of introgression events36. Studies assessing the effects of increased geographical coverage on DNA barcoding are scarce for large faunas. In Eurasian butterflies, for example, only one study has addressed this issue with a dataset on central Asian species37. In contrast to our findings, increasing geographical coverage in central Asian butterflies had little effect on the ability of DNA barcodes to discriminate species. This discrepancy could be caused by the difference in sampling between the above-mentioned study (2.9 specimens/species) and the current study (19.3 specimens/species). Furthermore, the complex local orography in the Mediterranean region, especially in the Iberian Peninsula, in conjunction with complex patterns of population isolation and reconnection throughout the Pleistocene, has created a mosaic of genetic diversity in this area (e.g.38,39). For example, we found that some boreo-alpine taxa such as Aricia nicias, Erebia pandrose and Pyrgus andromedae display more sequence divergence at COI between populations in the Alps and the Pyrenees (separated by ca. 450 km) than between Scandinavia and the Alps (separated by over 1200 km).
Overall, the resolution delivered by the DNA barcode library now available for Iberia and Europe is high enough for numerous applications because most specimens can be identified to a species or at least to a species-pair. Features such as the ability to identify preimaginal stages and to document food webs or host-parasitoid interactions highlight the advantage of rapid DNA-based assessments. Furthermore, the establishment of a comprehensive DNA barcode library for Iberian butterflies covers a key region in the Mediterranean and, together with the data available for regions such as Romania25 and Germany26 provides the basis for the development of an identification system targeting particular geographical areas, maximizing identification success. This capacity for DNA-based identifications is a valuable resource to support implementation of the new European biodiversity strategy focusing on the main drivers of biodiversity loss40.
In addition to creating a tool for DNA-based specimen identification, the Iberian dataset coupled with GMYC analysis permitted the first large-scale assessment of genetic patterns and potential cryptic diversity in European butterflies. Although European butterflies are among the best-studied invertebrate groups and considerable efforts have been devoted towards revealing hidden diversity, recent studies continue to report new cryptic species41,42,43,44. Conversely, some previously accepted species have been shown not to deserve their taxonomic status45. The ST-GMYC analysis revealed that 83 (27.7%) species in our dataset, including representatives from the five main European butterfly families, were split into two to four entities (Fig. 3, Supplementary Table 3, Supplementary Annex 1), that were mapped to facilitate the visualization of spatial genetic patterns (Supplementary Annex 2). In absolute terms, the GMYC analysis recovered 64 more entities than the number of species currently recognized by Fauna Europaea. Thus, the high number of species split in multiple entities by the GMYC model and the complex patterns detected in some cases that were both split and also lumped with another species, suggest that much research is needed even for one of the best studied invertebrate groups such as European butterflies.
The ST-GMYC model also revealed some potential cryptic species with low levels of genetic differentiation (e.g. as low as 0.3% in the case of M. athalia), with high support (Fig. 5, Supplementary Fig. 7, Supplementary Table 4). At the opposite pole, 14 ME species displayed high levels of genetic divergence (more than 2.5%) between conspecific ST-GMYC entities (Fig. 4) and thus represent particularly likely cases of overlooked taxa that require further study. The ST-GMYC model also recovered as distinct entities well-confirmed cryptic species that have been overlooked for centuries, such as the triplet Leptidea sinapis, L. reali and L. juvernica42 as well as Polyommatus icarus and P. celina41. However, the same analysis also lumped a number of recognized species, several of which had and some still have, debated taxonomical status (e.g. Pieris napi - P. bryoniae, Polyommatus ripartii - P. nephohiptamenos, Erebia medusa - E. polaris, Kretania pylaon - K. sephirus etc.) (Fig. 2 and see Supplementary Table 3 and Supplementary Annex 1 for further comments).
It is also worth noting that a series of the ME cases included populations that were sometimes treated as distinct species by various authors. Interestingly, our results for some of these cases did not always fully correspond with current taxonomic views, often highlighting more complex situations than previously recognized (see Supplementary Annex 1 for a brief discussion of each case). Such examples are: Arethusana arethusa (due to taxon boabdil), Coenonympha pamphilus (due to taxon lyllus), Melitaea athalia (due to taxon celadussa), Hipparchia hermione (due to taxon genava) and Cupido minimus (due to taxon carswelli).
Obviously, conclusions cannot be drawn based on a single genetic marker, but these overall patterns highlight the novelty of the results provided by the genetic survey as an alternative view to traditional (mainly) morphology-based taxonomy and call for more detailed studies to clarify the reasons behind a strong or, conversely, a lack of clear differentiation in mitochondrial DNA. The case study on Iphiclides (Fig. 6) further demonstrates that estimates of cryptic biodiversity are dependent on geographic and gene coverage and that more cases will likely emerge with increased sampling.
While species are the standard currency employed to measure biodiversity and to develop conservation measures, the recognition of biological species is, as exemplified above, complicated by operational issues and by introgressive hybridization. As an alternative, the concept of conserving genetic diversity is receiving growing attention in biodiversity studies8. Extinction is not only the loss of species, but is also preceded by the erosion of genetic diversity, which can result in the loss of potentially useful traits (e.g. resistance to parasites). Thus, even if many of the GMYC entities detected by our analysis do not represent cryptic species, they could still be Evolutionary Significant Units (ESUs)46 that can be highly relevant for fields such as phylogeography and nature conservation. For example, as shown by47, the effects of climate change can be much more accurately assessed when based on patterns in mitochondrial DNA instead of morphospecies. The present data will aid the prioritization for conservation of populations that collectively represent most of the genetic diversity in each taxon. As well, it will aid the selection of genetically similar populations for reintroductions, avoiding the risk of mixing potentially incompatible lineages. A recent study on European butterflies has also highlighted the unique properties of the cryptic fraction of butterfly diversity involving original qualitative aspects (influence on the beta-diversity turnover) that can affect ecological and biogeographical studies7. Such results underline the necessity to thoroughly document cryptic diversity at both intra- and interspecific levels. Overall, the presence of both deeply diverged lineages and cases of lumping suggest interesting historical and evolutionary processes that, through further research, may illuminate the functioning of natural populations and species.
European butterflies also include numerous model taxa for biogeography38,48,49, ecology21 and speciation50 and are intensively used as bioindicators and as flagship group for invertebrate conservation efforts22,51,52. As a consequence, any change in their taxonomy and any improvement of our knowledge will have consequences for both research projects and conservation policies. As53 noted, superficial taxonomic decisions may jeopardize an intensively studied system such as European butterflies. Given the alarming rates of global biodiversity loss54,55 and the limited resources available, the exploration of biodiversity through large-scale molecular approaches such as DNA barcoding combined with automated methods of ESU delineation can provide valuable guidelines for future efforts.
Studies assessing levels of potential cryptic diversity in speciose groups on a large geographic scale are rare and the results are not directly comparable due to unequal sampling efforts and different methods employed. Nonetheless, a study on North American birds found that about 2% of the 643 species in the dataset contained lineages displaying at least 2.5% divergence56. By comparison, 4.7% of the species in our dataset matched this criterion (Fig. 4), suggesting a considerably higher proportion of potential cryptic diversity in European butterflies. Another study on freshwater fishes from the Mediterranean area reported up to 64 potential cryptic species detected as GMYC entities in a dataset of 498 species57. This finding matches the increase of 64 GMYC entities identified in our dataset compared to currently recognized species, which represents 12.9% compared to the 21.4% that we obtained. Obviously, there is a critical need for more large-scale studies that, besides improving knowledge on biodiversity, can provide accurate answers to the ongoing debate about the global proportion and distribution of cryptic diversity1,5,6.
DNA barcoding combined with automated methods for species delineation represents a cost-effective, rapid approach that is a powerful first step for exploring cryptic biodiversity across wide geographical areas. This is especially true for poorly studied taxonomic groups and for those with few diagnostic characters, such as numerous invertebrates.
Methods
Sampling and collection data
We gathered over 10,000 specimens that provided coverage for the 228 butterfly species belonging to the six families that occur in the Iberian Peninsula (Spain, Portugal and Andorra, including the Balearics) according to Fauna Europaea. To provide a good representation of intraspecific genetic variability, these samples were collected from hundreds of localities across the Iberian Peninsula (Supplementary Fig. 1, Supplementary Table 1) and a subset of these specimens was selected for barcode analysis (see the Results). Butterfly bodies were stored in 2 ml polypropylene tubes with 100% or 96% ethanol and kept at −20° C, while the wings were detached from the body and kept in glassine envelopes as vouchers. A small percentage of the COI sequences (usually highly localized species) were obtained from dried specimens, most of which were less than ten years old.
The Iberian dataset was expanded to 5782 sequences by including records from several other regions of Europe (Fig. 1, Supplementary Table 1). Romania and southern Germany have been intensively analysed in previous studies25,26, while the other regions of Europe have seen less investigation. Because one source project (“Lepidoptera of Central Asia” - LOWA) contained both European and Asian specimens, we filtered the data by setting the southeastern limit of Europe to 50° long. E and 43.5° lat. N, between the Black Sea and the Caspian Sea, just north of the Caucasus Mountains. The European dataset was compiled by adding 363 complementary sequences from various European countries that were produced either specifically for this study, or were available from previous publications by the authors. We also used previously published DNA barcode data from the following projects publicly available on BOLD: “Butterflies of Romania” (EZROM, 1363 sequences)25, “Lepidoptera of Romania – 2GenBank” (EOEME, 18 sequences)58, “Fauna Bavarica - Lepidoptera Rhopalocera” (FBLRH, 406 sequences)26, European arctic-alpine Lepidoptera (DATASET-AALE1, 55 sequences)28, “Lepidoptera of the Alps 3” (PHLAC, 8 sequences)27 and “Lepidoptera of Central Asia” (LOWA, 67 sequences)37. We preferentially used DNA barcodes from public projects on BOLD because this platform aids both the verification of specimen identifications and an assessment of the quality of the electropherograms. We used sequences that were greater than 400 bp long to allow the inclusion of 407 bp-long amplicons obtained with a primer set often used when the standard Lepidoptera primers failed to recover a full barcode, usually because of DNA degradation in older specimens (25 and references therein). Five sequences from already published BOLD projects were reedited: Aglais urticae (EZROM597-08), Brenthis hecate (EZROM-288-08), Boloria napaea (GWOSA776-10), Boloria thore (PHLAB292-10) and Carcharodus lavatherae (EZROM311-08). One Eumedonia eumedon (FBLMW307-10) and one Polyommatus dorylas (GWORA2454-09) from published BOLD projects were not included in the analyses because inspection of electropherograms revealed their poor quality. All data have been uploaded to the public dataset “Iberia-Europe” (DS-IBEUR, dx.doi.org/10.5883/DS-IBEUR) on BOLD at www.barcodinglife.org.
Sequence analyses, phylogenetic inference and Generalized Mixed Yule-Coalescent (GMYC) models
DNA extraction, amplification and sequencing of the COI fragment followed standard protocols for Lepidoptera25 (and references therein). A 658-bp fragment of COI was targeted for amplification using the primers LepF (5′-ATTCAACCAATCATAAAGATATTGG-3′) and LepR (5′-TAAACTTCTGGATGTCCAAAAAATCA-3′). Samples that did not produce a PCR product with the primers LepF and LepR were amplified with the primers LepF and Enh_LepR (5′-CTCCWC CAGCAGGATCAAAA-3′), which amplify a 609-bp fragment of COI. If this approach failed, we used two primer combinations that amplify shorter overlapping fragments: LepF + MH-MR1 (5′-CCTGTT CCAGCTCCATTTTC-3′) (307-bp amplicon) and MH-MF1 (5′-GCTTTCCCACGAATAAATAATA-3′) + LepR (407-bp amplicon). Sequences were edited in CodonCode Aligner 3.0 or in Geneious Pro 4.7.5 (Biomatters, http://www.geneious.com/) and assembled using the latter.
The use of the Bayesian software BEAST for phylogenetic inference, as well as the removal of identical haplotypes, have been shown to optimize GMYC analysis18,20. Thus, the European dataset was collapsed to 1971 haplotypes under a conservative approach so that, where applicable, sequences that were shorter than 658 bp and/or included ambiguities were assigned to multiple potential haplotypes. BEAST 1.7.459 was used to reconstruct a reference ultrametric phylogenetic tree. Four independent chains were run for 15 million generations. The substitution model was set to GTR + I + G with four gamma rate categories. A strict clock and coalescent tree priors were used. Values were sampled every 10% of the run length and convergence was inspected in Tracer v.1.560. The GMYC method was applied to the resulting tree using the “splits 1.0–11” R package61. The single-threshold (ST-GMYC) (Supplementary Fig. 5) and the multimodel (MM-GMYC)62 (Supplementary Fig. 6) approaches were evaluated and GMYC support values based on the Akaike’s Information Criterion (AIC) were calculated for tree nodes following the approach described by63.
The suitability of our dataset to fit GMYC models was evaluated according to previous findings based on empirical20 and simulated datasets13,33. On one side, the sampling strategy followed guidelines for regional biodiversity surveys designed to capture maximum intraspecific divergences and to maximize taxon coverage. In general, species in our dataset are represented by similar numbers of specimens (the average number of specimens per species in the European dataset was 19.3 and 75.3% of the species were represented by at least ten specimens), typically including phenotypically differentiated populations from a variety of habitats. On the other side, because the accuracy of the GMYC models is dependent on the scaling parameters for both the speciation and coalescent parts, estimates of speciation rate (SR) and haploid effective population size (Ne) were used as a test for data suitability as suggested by simulation studies33. Ne and SR were estimated for both the currently recognized species according to Fauna Europaea and for the resulting GMYC cluster delimitations. For Ne calculations, π for synonymous sites and slow (0.0075 per site per Myr) and fast (0.0115 per site per Myr) substitution rates reported for COI in invertebrates were used for those species represented by at least 10 individuals. For SR calculation, we used the R routine bd.ms in the “geiger” package considering an age of 110 Myr for the Papilionoidea tree root64.
For the case study on Iphiclides podalirius podalirius and I. p. feisthamelii, we used 23 COI sequences from the European dataset and sequenced three other specimens from northern Morocco (using the DNA barcoding protocols described above). A nuclear marker (the internal transcribed spacer 2 - ITS2), was sequenced for all 26 specimens analysed for COI. Two specimens of I. p. podalirius from Greece and Spain possessed both COI and ITS2 sequences in GenBank and these records were added to the dataset (Supplementary Table 5). All 28 Iphiclides specimens analysed (including ITS2 sequences produced for this study) were included in the dataset “Iphiclides” (DS-EUIPHI, dx.doi.org/10.5883/DS-EUIPHI) publicly available on BOLD at www.barcodinglife.org.
A 699–728 bp fragment at the 5′ end of ITS2 was amplified by polymerase chain reaction using the primers ITS3 (5′-GCATCGATGAAGAACGCAGC-3′) and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′)65. Double-stranded DNA was amplified in 25 μl volume reactions: 14.4 μl ultra-pure (HPLC quality) water, 5 μl 5X buffer, 2 μl 25 mM MgCl2, 0.5 μl 10 mM dNTP, 0.5 μl of each primer (10 mM), 0.1 μl Taq DNA Polymerase (Promega) and 2 μl of extracted DNA. The typical thermal cycling profile was: 95 °C for 45 seconds, 51 °C for 60 seconds and 72 °C for 60 seconds, for 40 cycles. ITS2 sequence editing and alignment was done using Geneious Pro 4.7.5. MEGA 566 was used to produce neighbor-joining (NJ) trees for COI and ITS2 and to perform bootstrap analysis (100 replicates). COI and ITS2 sequences for Papilio phorcas and Leptidea sinapis from GenBank were used as outgroup (Supplementary Table 5).
All sequences obtained in this study have been deposited in GenBank (see Supplementary Tables 1 and 5, as well as the DS-IBEUR and DS-EUIPHI public datasets on BOLD).
DNA barcoding performance assessment
To facilitate the visualization of genetic distances, bootstrapped NJ trees (built with MEGA 5, see above) were produced using uncorrected p distances67,68. The NJ trees were based on unique haplotypes: 1264 haplotypes for the Iberian dataset and 1971 haplotypes for the European dataset. The performance of DNA barcodes in species identification at Iberian and European scales was assessed based on the following categories: (1) monophyletic, (2) para- or polyphyletic and (3) species sharing DNA barcodes (Supplementary Figures 2–4).
Morphology examination
Strong effort was made to ensure the correct morphology-based identification of each specimen. Genitalia were examined where external morphology was considered insufficient for reliable identification. For the case study on Iphiclides, we performed linear morphometrics of the male genitalia. Genitalia were prepared for all 15 specimens that were also sequenced for COI and ITS2 (Supplementary Table 6), according to the following protocol: maceration for 20 minutes at 100 °C in 10% potassium hydroxide, dissection and cleaning under a stereomicroscope and storage in 0.5 mL tubes with 70% ethanol. The genitalia were slightly pressed under a cover slip and were photographed in a thin layer of 50% ethanol under a Leica Z16 APO macroscope equipped with a Leica DFC500 digital camera for photomicrography. Measurements were based on digital photographs using the ImageJ software and three elements of the genitalia were measured: ostium phallus length (OP), phallus width (PW) and phallus length (PL). The resultant measurements are provided in Supplementary Table 6.
Defining species and potentially cryptic taxa
Although European butterflies are among the best-studied invertebrates, recognition of species is not always straightforward, as it has been based on different approaches for various taxa. Some species have been defined based on their external morphology, while detailed morphological, molecular and behavioural analyses have been needed in other cases69. In this study, 299 species were recognized based on the checklist of the Fauna Europaea Project70 (www.faunaeur.org), which gathers widely accepted views of the scientific community, with a few additions according to recent publications (see Supplementary Annex 1). We then compared the 299 species with the entities recovered by the ST-GMYC analysis and classified each of them into one of three main categories (see Fig. 2 for a schematic representation of the workflow): 1) Single entity (SE): species that were recovered as a single entity, indicating correspondence with current taxonomy. 2) Lumped: specimens of two or more species that were recovered as a single ST-GMYC entity. 3) Multiple entities (ME): species that were split in two or more ST-GMYC entities, which represent potential cryptic biodiversity.
For species involving ME, entities were considered as sympatric if they were separated by less than 50 km. To facilitate the visualization of spatial genetic patterns, the ME cases are shown on maps in Supplementary Annex 2.
Additional Information
How to cite this article: Dincă, V. et al. DNA barcode reference library for Iberian butterflies enables a continental-scale preview of potential cryptic diversity. Sci. Rep. 5, 12395; doi: 10.1038/srep12395 (2015).
References
Bickford, D. et al. Cryptic species as a window on diversity and conservation. Trends Ecol. Evol. 22, 148–155 (2007).
Knowlton, N. Sibling species in the sea. Annu. Rev. Ecol. Syst. 24, 189–216 (1993).
Hebert, P. D. N. et al. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. USA 101, 14812–14817 (2004).
Smith A. M. et al. Extreme diversity of tropical parasitoid wasps exposed by iterative integration of natural history, DNA barcoding, morphology and collections. Proc. Natl. Acad. Sci. USA 105, 12359–12364 (2008).
Pfenninger, M. & Schwenk, K. Cryptic animal species are homogeneously distributed among taxa and biogeographical regions. BMC Evol. Biol. 7, 121 (2007).
Trontelj, P. & Fišer, C. Cryptic species diversity should not be trivialised. Syst. Biodivers. 7, 1–3 (2008).
Vodă, R., Dapporto, L., Dincă, V. & Vila, R. Cryptic matters: overlooked species generate most butterfly beta-diversity. Ecography 38, 405–409 (2015) (2015).
Fraser, D. J. & Bernatchez, L. Adaptive evolutionary conservation: Towards a unified concept for defining conservation units. Mol. Ecol. 10, 2741–2752 (2001).
Habel, J. C., Rodder, D., Schmitt, T. & Neve, G. Global warming will affect the genetic diversity and uniqueness of Lycaena helle populations. Glob. Change Biol. 17, 194–205 (2011).
Bartlett, S. E. & Davidson, W. S. Identification of Thynnus tuna species by the polymerase chain reaction and direct sequence analysis of their mitochondrial DNA. Can. J. Fish. Aquat. Sci. 48, 309–317 (1991).
Hebert, P. D. N., Cywinska, A., Ball, S. L. & DeWaard, J. R. Biological identifications through DNA barcodes. Proc. R. Soc. B. 270, 313–321 (2003).
Pons, J. et al. Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst. Biol. 55, 595–610 (2006).
Fujisawa, T. & Barraclough, T. G. Delimiting species using single-locus data and the generalized mixed yule coalescent approach: a revised method and evaluation on simulated data sets. Syst. Biol. 62, 707–724 (2013).
Puillandre, N., Lambert, A., Brouillet, S. & Achaz, G. ABGD, automatic barcode gap discovery for primary species delimitation. Mol. Ecol. 21, 1864–1877 (2012).
Zhang, J., Kapli, P., Pavlidis, P. & Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics. 29, 2869–2876 (2013).
Ratnasingham, S. & Hebert, P. D. N. A DNA-Based registry for all animal species: the barcode index number (BIN) system. PLoS ONE 8, e66213 (2013).
Miralles, A. & Vences, M. New metrics for comparison of taxonomies reveal striking discrepancies among species delimitation methods in Madascincus lizards. PLoS ONE 8, e68242 (2013).
Tang, C. Q., Humphreys, A. M., Fontaneto, D. & Barraclough, T. G. Effects of phylogenetic reconstruction on the robustness of species delimitation methods using single-locus data. Methods Ecol. Evol. 5, 1086–1094 (2014).
Kekkonen, M. & Hebert, P. D. N. DNA barcode-based delineation of putative species: efficient start for taxonomic workflows. Mol. Ecol. Resour. 14, 706–715 (2014).
Talavera, G., Dincă, V. & Vila, R. Factors affecting species delimitations with the GMYC model: insights from a butterfly survey. Methods Ecol. Evol. 4, 1101–1110 (2013).
Settele, J., Shreeve, T., Konvička, M. & Van Dyck, H. Ecology of butterflies in Europe (Cambridge University Press, 2009).
Van Swaay, C. et al. European red list of butterflies (Publications office of the European Union, 2010).
Kudrna, O. et al. Distribution atlas of butterflies in Europe (Gesellschaft für Schmetterlingsschutz e.V., 2011).
Wiemers, M. & Fiedler, K. Does the DNA barcoding gap exist?- a case study in blue butterflies (Lepidoptera: Lycaenidae). Front. Zool. 4, 8 (2007).
Dincă, V., Zakharov, E. V., Hebert, P. D. N. & Vila, R. Complete DNA barcode reference library for a country’s butterfly fauna reveals high performance for temperate Europe. Proc. R. Soc. B. 278, 347–355 (2011).
Hausmann, A. et al. Now DNA-barcoded: the butterflies and larger moths of Germany (Lepidoptera: Rhopalocera, Macroheterocera). Spixiana 34, 47–58 (2011).
Huemer, P. & Hebert P. D. N. DNA-Barcoding von Schmetterlingen (Lepidoptera) in Waldstandorten Südtirols (IT01 Ritten und IT02 Montiggl). Forest Observer 6, 75–98 (2012).
Mutanen, M. et al. Allopatry as a gordian knot for taxonomists: patterns of DNA barcode divergence in arctic-alpine Lepidoptera. PLoS ONE 7, e47214 (2012).
Mittermeier, R. A. et al. Hotspots revisited: Earth’s biologically richest and most endangered ecoregions (CEMEX, 2004).
Blondel, J., Aronson, J., Bodiou, J.-Y. & Boeuf, G. The Mediterranean region. Biological diversity in space and time. Second edition (Oxford University Press, 2010).
García-Barros, E., Munguira, M. L., Stefanescu, C. & Vives Moreno, A. in Fauna Ibérica vol. 37 (eds Ramos, M. A. et al. .) 1–1213 (Museo Nacional de Ciencias Naturales Madrid, 2013).
Ratnasingham, S. & Hebert, P. D. N. BOLD: The Barcode of Life Data System (www.barcodinglife.org). Mol. Ecol. Resour. 7, 355–364 (2007).
Esselstyn J. A., Evans B. J., Sedlock J. L., Anwarali Khan F. A. & Heaney L. R. Single-locus species delimitation: a test of the mixed Yule-coalescent model, with an empirical application to Philippine round-leaf bats. Proc. R. Soc. B. 279, 3678–3686 (2012).
Wiemers, M. & Gottsberger, B. Discordant patterns of mitochondrial and nuclear differentiation in the Scarce Swallowtail Iphiclides podalirius feisthamelii (Duponchel, 1832) (Lepidoptera: Papilionidae). Entomol. Z. 120, 111–115 (2010).
Coutsis, J. & Van Oorschot, H. Differences in the male and female genitalia between Iphiclides podalirius and Iphiclides feisthamelii, further supporting species status for the latter (Lepidoptera: Papilionidae). Phegea 39, 12–22 (2011).
Talavera, G., Lukhtanov, V. A., Rieppel, L., Pierce, N. E. & Vila, R. In the shadow of phylogenetic uncertainty: The recent diversification of Lysandra butterflies through chromosomal change. Mol. Phylogenet. Evol. 69, 469–478 (2013).
Lukhtanov, V. A., Sourakov, A., Zakharov, E. V. & Hebert, P. D. N. DNA barcoding Central Asian butterflies: increasing geographical dimension does not significantly reduce the success of species identification. Mol. Ecol. Resour. 9, 1302–1310 (2009).
Schmitt, T. 2007. Molecular biogeography of Europe: Pleistocene cycles and postglacial trends. Front. Zool. 4, 11 (2007).
Gratton, P., Konopinski, M. K. & Sbordoni, V. Pleistocene evolutionary history of the Clouded Apollo (Parnassius mnemosyne): genetic signatures of climate cycles and a ‘time-dependent’ mitochondrial substitution rate. Mol. Ecol. 17, 4248–4262 (2008).
European Commission. The EU biodiversity strategy to 2020 (available at ec.europa.eu/environment/nature/biodiversity/comm2006/2020.htm 2011).
Dincă, V., Dapporto, L. & Vila, R. A combined genetic-morphometric analysis unravels the complex biogeographical history of Polyommatus icarus and Polyommatus celina common blue butterflies. Mol. Ecol. 20, 3921–3935 (2011).
Dincă, V., Lukhtanov, V. A., Talavera, G. & Vila, R. Unexpected layers of cryptic diversity in wood white Leptidea butterflies. Nat. Commun. 2, 324 (2011).
Zinetti, F. et al. When the rule becomes the exception. No evidence of gene flow between two Zerynthia cryptic butterflies suggests the emergence of a new model group. PLoS ONE 8, e65746 (2013).
Sañudo-Restrepo, C., Dincă, V., Talavera, G. & Vila, R. Biogeography and systematics of Aricia butterflies (Lepidoptera, Lycaenidae). Mol. Phylogenet. Evol. 66, 369–379 (2013).
Vila, R., Lukhtanov, V. A., Talavera, G., Gil, T. F. & Pierce, N. E. How common are dot-like distributions? Taxonomical oversplitting in western European Agrodiaetus (Lepidoptera: Lycaenidae) revealed by chromosomal and molecular markers. Biol. J. Linn. Soc. 101, 130–154 (2010).
Moritz, C. Defining ‘Evolutionary Significant Units’ for conservation. Trends Ecol. Evol. 9, 373–375 (1994).
Bálint, M. et al. Cryptic biodiversity loss linked to global climate change. Nature Clim. Change 1, 313–318 (2011).
Habel, J. C., Schmitt, T. & Müller, P. The fourth paradigm pattern of post-glacial range expansion of European terrestrial species: the phylogeography of the marbled white butterfly (Satyrinae, Lepidoptera). J. Biogeogr. 32, 1489–1497 (2005).
Dapporto, L., Vodă, R., Dincă, V. & Vila, R. Comparing population patterns for genetic and morphological markers with uneven sample sizes. An example for the butterfly Maniola jurtina. Methods Ecol. Evol. 5, 834–843 (2014).
Dincă, V. et al. Reproductive isolation and patterns of genetic differentiation in a cryptic butterfly species complex. J. Evol. Biol. 26, 2095–2106 (2013).
Pollard, E. & Yates, T. J. Monitoring butterflies for ecology and conservation (Springer, 1993).
Thomas J. A. et al. Comparative losses of British butterflies, birds and plants and the global extinction crisis. Science 303, 1879–1881 (2004).
Dennis, L. R. H. An inflated conservation load for European butterflies: increases in rarity and endemism accompany increases in species richness. J. Insect Conserv. 1, 43–62 (1997).
Baillie, J. E. M., Hilton-Taylor, C. & Stuart, S. N. 2004 IUCN Red List of threatened species. A global species assessment (IUCN Press, 2004).
Mora, C. & Sale, P. F. Ongoing global biodiversity loss and the need to move beyond protected areas: a review of the technical and practical shortcomings of protected areas on land and sea. Mar. Ecol. Prog. Ser. 434, 251–266 (2011).
Kerr, K. C. R. et al. Comprehensive DNA barcode coverage of North American birds. Mol. Ecol. Notes 7, 535–543 (2007).
Geiger, M. F. et al. Spatial heterogeneity in the Mediterranean Biodiversity Hotspot affects barcoding accuracy of its freshwater fishes. Mol. Ecol. Resour. 14, 1210–1221 (2014).
Dincă, V., Cuvelier, S., Zakharov, E. V., Hebert, P. D. N. & Vila, R. Biogeography, ecology and conservation of Erebia oeme in the Carpathians. Ann. Soc. Entomol. Fr. 46, 486–498 (2010).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012).
Rambaut, A. & Drummond, A. J. Tracer v.1.4 (available at http://beast.bio.ed.ac.uk/Tracer 2007).
Ezard, T., Fujisawa, T. & Barraclough, T. G. SPLITS: SPecies’ LImits by Threshold Statistics (available at http://r-forge.r-project.org/projects/splits/ 2009).
Powell, J. R. Accounting for uncertainty in species delineation during the analysis of environmental DNA sequence data. Methods Ecol. Evol. 3, 1–11 (2012).
Burnham, K. P. & Anderson, D. R. Model selection and multimodel inference: A practical information-theoretic approach. Second edition (Springer, 2002).
Heikkilä, M., Kaila, L., Mutanen, M., Peña, C. & Wahlberg, N. Cretaceous origin and repeated tertiary diversification of the redefined butterflies. Proc. R. Soc. B. 279, 1093–1099 (2012).
White, T. J. et al. in PCR Protocols: A Guide to Methods and Applications (eds Innis, M. A. et al. .) 315–322 (Academic Press, 1990).
Tamura, K. et al. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance and maximum parsimony methods. Mol. Biol. Evol. 28, 2731–2739 (2011).
Collins, R. A., Boykin, L. M., Cruickshank, R. H. & Armstrong, C. F. Barcoding´s next top model: an evaluation of nucleotide substitution models for specimen identification. Methods Ecol. Evol. 3, 457–465 (2012).
Srivathsan, A. & Meier, R. On the inappropriate use of Kimura-2-parameter (K2P) divergences in the DNA-barcoding literature. Cladistics 28, 190–194 (2012).
Descimon, H. & Mallet, J. in Ecology of Butterflies in Europe (eds Settele, J., Shreeve, T. G., Konvicka, M. & Van Dyck, H. ) 219–249 (Cambridge University Press, 2009).
Karsholt, O. & Van Nieukerken, E. J. Lepidoptera, Papilionoidea. Fauna Europaea version 2.6 (available at http://www.faunaeur.org 2013).
Acknowledgements
We are grateful to the many colleagues who collected material used in this study and to L. Dapporto and S. Cuvelier for comments on the manuscript. We also thank the public bodies (environmental agencies of the Comunidades Autónomas, the National Parks Sierra Nevada, Montseny and Picos de Europa and Reserva Natural Illes Columbretes) that granted collecting permits required for this study. Support for this research was provided by the Spanish Ministerio de Economía y Competitividad (project CGL2010-21226/BOS and CGL2013- 48277-P), by Marie Curie International Outgoing Fellowships within the 7th European Community Framework Programme to V. Dincă (project no. 625997) and G. Talavera (project no. 622716), by grant 1.50.1617.2013 at Saint Petersburg State University for G. Talavera and by the government of Canada through Genome Canada and the Ontario Genomics Institute in support of the International Barcode of Life project.
Author information
Authors and Affiliations
Contributions
All authors conceived and designed the research and contributed to sample acquisition. V.D. and S.M. prepared the data set, G.T. and V.D. performed phylogenetic analyses. S.M. and V.D. performed morphometric analyses. V.D. and R.V. wrote the manuscript and all authors discussed the results and commented on the study.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Dincă, V., Montagud, S., Talavera, G. et al. DNA barcode reference library for Iberian butterflies enables a continental-scale preview of potential cryptic diversity. Sci Rep 5, 12395 (2015). https://doi.org/10.1038/srep12395
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep12395
This article is cited by
-
Pronounced mito-nuclear discordance and various Wolbachia infections in the water ringlet Erebia pronoe have resulted in a complex phylogeographic structure
Scientific Reports (2022)
-
Integrative taxonomy reveals cryptic diversity in North American Lasius ants, and an overlooked introduced species
Scientific Reports (2022)
-
Estimating range disjunction time of the Palearctic Admirals (Limenitis L.) with COI and histone H1 genes
Organisms Diversity & Evolution (2022)
-
DNA barcoding in Dorcadionini (Coleoptera, Cerambycidae) uncovers mitochondrial-morphological discordance and the hybridogenic origin of several subspecies
Organisms Diversity & Evolution (2022)
-
High resolution DNA barcode library for European butterflies reveals continental patterns of mitochondrial genetic diversity
Communications Biology (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
