
Abstract
Carbon-ion radiotherapy (CIRT) holds promise to treat inoperable locally-advanced non-small cell lung carcinoma (NSCLC), a disease poorly controlled by standard chemoradiotherapy using X-rays. Since CIRT is an extremely limited medical resource, selection of NSCLC patients likely to benefit from it is important; however, biological predictors of response to CIRT are ill-defined. The present study investigated the association between the mutational status of EGFR and KRAS, driver genes frequently mutated in NSCLC and the relative biological effectiveness (RBE) of carbon-ion beams over X-rays. The assessment of 15 NSCLC lines of different EGFR/KRAS mutational status and that of isogenic NSCLC lines expressing wild-type or mutant EGFR revealed that EGFR-mutant NSCLC cells, but not KRAS-mutant cells, show low RBE. This was attributable to (i) the high X-ray sensitivity of EGFR-mutant cells, since EGFR mutation is associated with a defect in non-homologous end joining, a major pathway for DNA double-strand break (DSB) repair and (ii) the strong cell-killing effect of carbon-ion beams due to poor repair of carbon-ion beam-induced DSBs regardless of EGFR mutation status. These data highlight the potential of EGFR mutation status as a predictor of response to CIRT, i.e., CIRT may show a high therapeutic index in EGFR mutation-negative NSCLC.
Similar content being viewed by others


Introduction
Locally-advanced non-small cell lung carcinoma (NSCLC) has poor prognosis. The 5 year overall survival rate of standard chemoradiotherapy using X-rays is 15–20%1,2,3. The local recurrence rate of approximately 30% in this population highlights the necessity for increased treatment intensity at primary disease sites; however, the characteristics of X-ray dose distribution limits further dose escalation in tumors with keeping tolerance dose in the surrounding organs such as the lung4,5. Therefore, a treatment modality with a higher efficacy than X-rays in primary tumors is required in locally-advanced NSCLC.
Carbon-ion radiotherapy (CIRT) has been provoking interest as a highly intensive local therapy. Carbon-ion beams have advantages over X-rays: a superior dose distribution associated with the sharp penumbra and the Bragg peak and a strong cell-killing effect6,7. In early NSCLC, CIRT demonstrates a 5 year local control rate of 90–95%8,9. Based on these promising results, a clinical trial on CIRT in inoperable locally-advanced NSCLC was launched at Gunma University in 2013 (protocol number: GUNMA1201). Nevertheless, CIRT is currently an extremely limited medical resource, with fewer than ten facilities in the world; this situation may not be substantially improved in the next few decades because of high costs. Therefore, the selection of locally-advanced NSCLC cases in which CIRT is beneficial is of great importance.
Recent genome-wide mutation analyses revealed that NSCLCs possess genetic alterations, called “driver gene mutations”, that play significant roles in carcinogenesis by abnormally activating oncogenes10,11,12. In most cases, these driver gene mutations are mutually exclusive13,14; in other words, NSCLCs can be classified based on driver gene mutation status. Drugs that target activated oncogene products have begun to replace conventional cytotoxic chemotherapy, even for first-line use10. However, little is known about the association between driver gene mutation status and relative biological effectiveness (RBE) of carbon-ion beams in NSCLC. If it affects RBE, the driver gene mutation status may be a useful predictor of response to CIRT. To investigate this issue, we analyzed the sensitivity of 15 NSCLC lines with known mutations in EGFR and KRAS, genes frequently mutated in NSCLC13,14, to X-rays and carbon-ion beams.
Results and Discussion
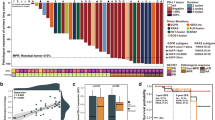
We first examined the sensitivity of 15 NSCLC lines with different EGFR and KRAS mutational statuses to X-rays or carbon-ion beams by clonogenic survival assay (Fig. 1a, Supplementary Fig. 1). The mutation status of these lines is listed in Supplementary Table 115,16,17,18,19,20. The X-ray dose producing 10% survival (D10) varied widely among the cell lines (3.8–10.9). The D10 of EGFR-mutant lines was significantly lower than that of EGFR wild-type lines (Fig. 1c, upper left panel). These data indicate that NSCLC cells show varying degrees of sensitivity to X-rays and that EGFR-mutant cells are more sensitive to X-rays than EGFR wild-type cells. The D10 achieved by carbon-ion beams was lower than that achieved by X-rays in all of the cell lines and was scored within a relatively narrow range (1.5-4.0) (Fig. 1a). No significant difference was observed between the carbon-ion beam D10 in EGFR-mutant lines and that in EGFR wild-type lines (Fig. 1c, upper middle panel). These data indicate that carbon-ion beams have a higher cell-killing effect than X-rays regardless of EGFR mutation status. Carbon-ion beam RBEs calculated from the D10 values obtained for X-rays and carbon-ion beams ranged from 1.5 to 3.8 (Fig. 1b). The RBEs in EGFR-mutant lines were significantly lower than those in wild-type lines (Fig. 1c, upper right panel). In contrast to EGFR, there was no significant difference in X-ray D10, carbon-ion beam D10 and the RBE between the 4 KRAS-mutant and the 11 KRAS wild-type lines (Fig. 1a,b,c, lower panels).

Sensitivity of EGFR-mutant, KRAS-mutant, or EGFR/KRAS wild-type NSCLC lines to X-rays or carbon-ion beams assessed by clonogenic survival assay.
C-ion, carbon-ion; mut, mutant; wt, wild-type. The original survival curves are shown in Supplementary Figure 1. (a) D10 for X-rays and carbon-ion beams. (b) RBE of carbon-ion beams at D10. (c) Statistical analysis of the difference in D10 for X-rays or carbon-ion beams, or RBE of carbon-ion beams in NSCLC lines based on EGFR or KRAS mutation status. P values on the significant differences in the mean values (black lines) between mutant and wild-type lines were shown.
To confirm the greater X-ray sensitivity and RBE of EGFR-mutant cells compared to EGFR wild-type cells, we compared the sensitivity to X-rays and carbon-ion beams using isogenic A549 cells stably expressing wild-type or mutant EGFR proteins. A549-WT, A549-ΔE746-A750 and A549-L858R cells, which express, respectively, wild-type EGFR, a ΔE746-A750 deletion mutant and a L858R point mutant, were used. Among the various EGFR mutations identified in human NSCLC, ΔE746-A750 and L858R are the most frequent (39.4%) and second most frequent (37.5%), respectively21. A549-ΔE746-A750 and A549-L858R cells showed higher sensitivity to X-rays than A549-WT cells (Fig. 2), but all three cell lines showed similar sensitivity to carbon-ion beams. Thus, the RBE observed in A549-ΔE746-A750 and A549-L858R cells was lower than that observed in A549-WT cells (Supplementary Table 2). Taken together, these data suggest that EGFR-mutant NCSLC cells show a low RBE due to their high sensitivity to X-rays and to the strong EGFR mutation-independent cell-killing effect of carbon-ion beams.

Sensitivity of isogenic A549 cells stably expressing wild-type or mutant (ΔE746-A750 or L858R) EGFR proteins to X-rays or carbon-ion beams assessed by clonogenic survival assay.
Data are represented as mean ± standard deviations. C-ion, carbon-ion; wt, wild-type.
DNA double-strand breaks (DSBs) are most critical lesions contributing to the cell-killing effect of ionizing irradiation. Therefore, we investigated the association between the EGFR mutation status and the capacity for repair of X-ray- or carbon-ion beam-induced DSBs. DSB repair was assessed by scoring the number of Ser139-phosphorylated histone H2AX (γH2AX) foci, which are markers for DSBs, 24 h post-irradiation using immunofluorescence staining22. H1299, H1703 and A549 cells, which showed the three highest D10 values for X-rays and HCC827, H1650 and Ma-24 cells, which showed the three lowest, were used. In EGFR wild-type lines, the numbers of γH2AX foci after X-ray irradiation were slightly higher than those in non-treated controls, indicating that a major proportion of X-ray-induced DSBs were repaired 24 h post-irradiation (Fig. 3a). In EGFR-mutant lines, the numbers of γH2AX foci after X-ray irradiation were significantly higher than those in X-ray-irradiated EGFR wild-type lines (Fig. 3a, Supplementary Table 3). These data suggest that the capacity of EGFR-mutant cells for repair of X-ray-induced DSBs is lower than that of EGFR wild-type cells.

Repair of X-ray- or carbon-ion beam-induced DSBs in EGFR-mutant or wild-type NSCLC lines assessed by immunofluorescence staining of γH2AX.
Cells were exposed to X-rays (6 Gy) or carbon-ion beams (2 Gy) in the presence or absence of the DNA-PKcs inhibitor NU7441 (10 μM) and stained with an antibody to γH2AX 24 h post-irradiation. The number of γH2AX foci per nucleus was scored in 30-50 cells for each experimental condition using a fluorescence microscope at ×100 magnification. The results of a representative experiment are shown as box plots. (a) X-rays. (b) Carbon-ion beams (C-ion).
Previous studies showed that the EGFR ΔE746-A750 deletion mutant and L858R point mutant are defective in translocation to the nucleus and in binding to the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs) in response to ionizing irradiation23,24,25. DNA-PKcs plays central roles in non-homologous end joining (NHEJ), a major DSB repair pathway26. After ionizing irradiation, DNA-PKcs is recruited to DSB sites and autophosphorylated. Then DNA-PKcs contributes DNA end ligation through the recruitment of x-ray cross-complementing gene 4 (XRCC4) and DNA ligase IV (LIG4)27. Together, we investigated NHEJ in the EGFR-mutant lines using NU7441, which inhibits DNA-PKcs activity28. In the presence of NU7441, the number of X-ray-induced γH2AX foci was comparable regardless of the EGFR mutation status, i.e., the additive effect of NU7441 on the increase in X-ray-induced γH2AX foci number was smaller in EGFR-mutant lines than in wild-type lines (Fig. 3a, Supplementary Table 3). If the mutant EGFRs function in homologous recombination, an alternative to NHEJ in DSB repair, the additive effect of NU7441 should not depend on the EGFR mutation status. Therefore, these data indicate that EGFR-mutant cells are defective in NHEJ.
Finally, we examined the repair of carbon-ion beam-induced DSB in EGFR-mutant and wild-type cells. No significant difference in the number of γH2AX foci was observed after irradiation between EGFR-mutant and wild-type lines (Fig. 3b, Supplementary Table 3). Meanwhile, in EGFR wild-type cells, the box plots of irradiation-alone and that of irradiation plus NU7441 were closer in carbon-ion beams than in X-rays. Furthermore, when the cells were irradiated with X-rays or carbon-ion beams for the same dose of 2 Gy, the number of γH2AX foci was significantly smaller in X-rays than in carbon-ion beams, in all the EGFR-mutant and the EGFR wild-type cells examined (Supplementary Fig. 2). Taken together, these data indicate that the repair efficacy for carbon-ion beam-induced DSBs is lower than that of X-ray-induced DSBs regardless of the EGFR mutation status. The low repair efficacy of carbon-ion beam-induced DSBs may be attributable in part to the structural complexity of the DSB ends29,30.
The results of the present study highlight the potential value of the EGFR mutation status as a predictor of CIRT RBE. To the best of our knowledge, this study is the first to report the association of driver gene mutation status with the RBE of carbon-ion beams. The results suggest that NSCLCs driven by mutations in oncogenes other than EGFR, including KRAS, show high RBE and thus should be selected as candidates for CIRT. Validation in animal models should be conducted. Nevertheless, previous research demonstrates an excellent agreement between the radiosensitivity of cancer cells assessed by clonogenic survival assay and the clinical response to radiotherapy31,32,33. Therefore, our results provide a valuable biological basis for selecting NSCLC patients for CIRT.
The present study focused on EGFR and KRAS, however, alterations in other genes were identified as drivers of NSCLC13,14. For example, H1299 cells carry a Q61K mutation in the oncogene NRAS18. Moreover, recent genome-wide analyses and functional validation demonstrated that genes that have not been recognized as classical oncogenic drivers, including genes involved in chromatin remodeling and DNA damage responses, are frequently mutated in human cancers, underscoring the pathogenic significance of these mutations34,35. Studies assessing the association between the mutation status of a wide panel of cancer-related genes and the sensitivity of cancer cells to X-rays and carbon-ion beams will further elucidate genetic profiles that affect radiosensitivity and RBE and will provide biological basis for the establishment of useful predictors for personalized radiotherapy. To this end, the mutational analysis of 409 known cancer-related genes in the 15 NSCLC lines used in the present study is ongoing.
In summary, EGFR mutation-negative NSCLCs show a high RBE compared to EGFR-mutant NSCLCs and may, therefore, benefit from CIRT.
Methods
Cell lines
The human NSCLC lines A427, A549, H1299, H1650, H1703, H1975, H460, H520, H522 and HCC827 were purchased from ATCC (Manassas, VA, USA). LK2 and II-18 were purchased from JCRB Cell Bank. H157, Ma-24 and PC9 were provided by Dr. Harris (National Institute of Health), Dr. Shimizu (Tokushima University) and Dr. Kato (Tokyo Medical Collage), respectively. A549-WT, −ΔE746-A750 and −L858R cells were established as described previously23,36. All cell lines were cultured in RPMI-1640 (Sigma-Aldrich, St. Louis, MO, USA) supplemented with 10% fetal bovine serum (Life Technologies, Carlsbad, CA, USA).
Irradiation
X-ray irradiation was performed using a Faxitron RX-650 (100 kVp, 1.14 Gy/min; Faxitron Bioptics, Tucson, AZ, USA). Carbon-ion beam irradiation was performed at the Gunma University Heavy Ion Medical Center using the same beam specifications used in clinical settings (290 MeV/nucleon and an average linear energy transfer at the center of a 6 cm spread-out Bragg peak of approximately 50 keV/μm). Carbon-ion beams were delivered in a vertical direction so that cells on culture plates could receive the dose evenly.
Clonogenic survival assay
Cells were seeded into 6-well plates and exposed to X-rays or carbon-ion beams. After incubation for 10 days, the cells were fixed with methanol and stained with crystal violet. Colonies of at least 50 cells were counted. The surviving fraction was normalized to the corresponding controls. The D10 was calculated using the linear-quadratic model as described previously37.
Immunofluorescence staining
Cells were seeded on glass coverslips in 35 mm dishes and incubated overnight. The culture medium was changed to that containing 10 μM of the DNA-PKcs inhibitor NU7441 (R&D Systems, Minneapolis, MN, USA). After incubation for 1 h, the cells were exposed to X-rays or carbon-ion beams. After incubation for 24 h, the coverslips were stained with antibodies against γH2AX (Merck Millipore, Billerica, MA, USA) as described previously38. The number of γH2AX foci per nucleus was scored in sequential 2D images captured from multiple focal planes using a fluorescence microscope (Eclipse Ni, Nikon, Tokyo, Japan) at ×100 magnification.
Statistical analysis
Statistical analysis was performed using SigmaPlot 12.0 (Hulinks, Tokyo, Japan)28. Normality was tested by Shapiro-Wilk test. For the data sets that followed a normal distribution, variance was further tested by Levene Median test; if the variance was equal, significance was then tested by unpaired Student’s t-test. For the data sets that did not follow a normal distribution, significance was tested by Man-Whitney U test. P < 0.05 was considered significant.
Additional Information
How to cite this article: Amornwichet, N. et al. The EGFR mutation status affects the relative biological effectiveness of carbon-ion beams in non-small cell lung carcinoma cells. Sci. Rep. 5, 11305; doi: 10.1038/srep11305 (2015).
References
Furuse, K. et al. Phase III study of concurrent versus sequential thoracic radiotherapy in combination with mitomycin, vindesin and cisplatin in unresectable stage III non small-cell lung cancer. J Clin Oncol 17, 2692–2699 (1999).
Yamamoto, N. et al. Phase III study comparing second- and third-generation regimens with concurrent thoracic radiotherapy in patients with unresectable stage III non-small-cell lung cancer: West Japan Thoracic Oncology Group WJTOG0105. J Clin Oncol 28, 3739–3745 (2010).
Curran, W. J. et al. Sequential vs. concurrent chemoradiation for stage III non-small cell lung cancer: randomized phase III trial RTOG 9410. J Natl Cancer Inst 103, 1452–1460 (2011).
Sause, W. et al. Final results of phase III trial in regionally advanced unresectable non-small cell lung cancer: Radiation Therapy Oncology Group, Eastern Cooperative Oncology Group and Southwest Oncology Group. Chest 117, 358–364 (2000).
Cox J. D. Are the results of RTOG 0617 mysterious? Int J Radiat Oncol Biol Phys 82, 1042–1044 (2012).
Schulz-Ertner, D. & Tsujii, H. Particle radiation therapy using proton and heavier ion beams. J Clin Oncol 25, 953–964 (2007).
Loeffler, J. S. & Durante. M. Charged particle therapy--optimization, challenges and future directions. Nat Rev Clin Oncol 10, 411–424 (2013).
Miyamoto, T. et al. Carbon ion therapy for stage I non-small cell lung cancer. Radiother Oncol 66, 127–140 (2003).
Miyamoto, T. et al. Curative treatment of stage I non-small-cell lung cancer with carbon ion beams using hypofractionated regimen. Int J Radiat Oncol Biol Phys 67, 750–758 (2007).
Oxnard, G.R., Binder, A. & Jänne, P. A. New targetable oncogenes in non-small-cell lung cancer. J Clin Oncol 31, 1097–1104 (2013).
Pao, W. & Hutchinson, K. E. Chipping away at the lung cancer genome. Nat Med 18, 349–351 (2012).
Imielinski, M. et al. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell 150, 1107–1120 (2012).
Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 489, 519–525 (2012).
Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 (2014).
Blanco, R. et al. A gene-alteration profile of human lung cancer cell lines. Hum Mutat 30, 1199–1206 (2009).
Sakai, K. et al. Pertuzumab, a novel HER dimerization inhibitor, inhibits the growth of human lung cancer cells mediated by the HER3 signaling pathway. Cancer Sci 98, 1498–1503 (2007).
Kawahara, A. et al. Molecular diagnosis of activating EGFR mutations in non-small cell lung cancer using mutation-specific antibodies for immunohistochemical analysis. Clin Cancer Res 16, 3163–3170 (2010).
Oike, T. et al. A synthetic lethality-based strategy to treat cancers harboring a genetic deficiency in the chromatin remodeling factor BRG1. Cancer Res 73, 5508–5518 (2013).
Helfrich, B. A. et al. Antitumor activity of the epidermal growth factor receptor (EGFR) tyrosine kinase inhibitor gefitinib (ZD1839, Iressa) in non-small cell lung cancer cell lines correlates with gene copy number and EGFR mutations but not EGFR protein levels. Clin Cancer Res 12, 7117–7125 (2006).
Spoerke, J. M. et al. Phosphoinositide 3-kinase (PI3K) pathway alterations are associated with histologic subtypes and are predictive of sensitivity to PI3K inhibitors in lung cancer preclinical models. Clin Cancer Res 18, 6771–6783 (2012).
Seigelin, M. D. & Borczuk, A. C. Epidermal growth factor receptor mutations in lung adenocarcinoma. Lab Invest 94, 129–137 (2014).
Löbrich, M. et al. gammaH2AX foci analysis for monitoring DNA double-strand break repair: strengths, limitations and optimization. Cell Cycle 9, 662–669 (2010).
Das, A. K. et al. Somatic mutations in the tyrosine kinase domain of epidermal growth factor receptor (EGFR) abrogate EGFR-mediated radioprotection in non-small cell lung carcinoma. Cancer Res 67, 5267–5274 (2007).
Dittmann, K. et al. Radiation-induced epidermal growth factor receptor nuclear import is linked to activation of DNA-dependent protein kinase. J Biol Chem 280, 31182–31189 (2005).
Huang, S. M. & Harari, P. M. Modulation of radiation response after epidermal growth factor receptor block- ade in squamous cell carcinomas: inhibition of damage repair, cell cycle kinetics and tumor angiogenesis. Clin Cancer Res 6, 2166–2174 (2000).
Burma, S., Chen, B. P. & Chen, D. J. Role of non-homologous end joining (NHEJ) in maintaining genomic integrity. DNA Repair 5, 1042–1048 (2006).
Wang, C. & Lees-Miller S. P. Detection and repair of ionizing radiation-induced DNA double strand breaks: new developments in nonhomologous end joining. Int J Radiat Oncol Biol Phys 86, 440–449 (2013).
Shibata, A. et al. DNA double-strand break repair pathway choice is directed by distinct MRE11 nuclease activities. Mol Cell 53, 7–18 (2014).
Shibata, A. et al. Factors determining DNA double-strand break repair pathway choice in G2 phase. EMBO J 30, 1079–1092 (2011).
Terato, H. & Ide, H. Clustered DNA damage induced by heavy ion particles. Biol Sci Space 18, 206–215 (2004).
Eschrich, S. A. et al. Validation of a radiosensitivity molecular signature in breast cancer. Clin Cancer Res 18, 5134–5143 (2012).
Eschrich, S. et al. Systems biology modeling of the radiation sensitivity network: a biomarker discovery platform. Int J Radiat Oncol Biol Phys 75, 497–505 (2009).
Eschrich, S. A. et al. A gene expression model of intrinsic tumor radiosensitivity: prediction of response and prognosis after chemoradiation. Int J Radiat Oncol Biol Phys 75, 489–496 (2009).
Oike, T. et al. Chromatin-regulating proteins as targets for cancer therapy. J Radiat Res 55, 613–628 (2014).
Yap, K. L. et al. Whole-exome sequencing of muscle-invasive bladder cancer identifies recurrent mutations of UNC5C and prognostic importance of DNA repair gene mutations on survival. Clin Cancer Res 20, 6605–6617 (2014).
Das, A. K. et al. Non-small-cell lung cancers with kinase domain mutations in the epidermal growth factor receptor are sensitive to ionizing radiation. Cancer Res 66, 9601–9608 (2006).
Oike, T. et al. Garcinol, a histone acetyltransferase inhibitor, radiosensitizes cancer cells by inhibiting non-homologous end joining. Int J Radiat Oncol Biol Phys 84, 815–821 (2012).
Nakajima, N. I. et al. Visualisation of γH2AX foci caused by heavy ion particle traversal; distinction between core track versus non-track damage. PLoS One 8, e70107 (2013).
Acknowledgements
Funding: This work was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology of Japan for programs for Leading Graduate Schools, Cultivating Global Leaders in Heavy Ion Therapeutics and Engineering and for Strategic Young Researcher Overseas Visits Program for Accelerating Brain Circulation. This work was also supported by Grants-in-Aid from the Japan Society for the Promotion of Science for Young Scientists (B) KAKENHI [10643471] and NIH CA 129364 to CN.
Author information
Authors and Affiliations
Contributions
T. Oike, T. Ohno, T.K. and T.N. designed the study. N.A., T. Oike, A.S., C.S.N., H.O., H.M., Y.K., Y.H, M.I. and Y.Y. performed experiment. N.A., T. Oike and A.S. wrote the main manuscript and prepared figures and tables. All authors reviewed the manuscript.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Amornwichet, N., Oike, T., Shibata, A. et al. The EGFR mutation status affects the relative biological effectiveness of carbon-ion beams in non-small cell lung carcinoma cells. Sci Rep 5, 11305 (2015). https://doi.org/10.1038/srep11305
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep11305
This article is cited by
-
Prognostic impact of EGFR/ALK alterations in leptomeningeal metastasis from lung adenocarcinoma treated with whole-brain radiotherapy
Clinical & Experimental Metastasis (2023)
-
Radiosensitizing Effect of Gadolinium Oxide Nanocrystals in NSCLC Cells Under Carbon Ion Irradiation
Nanoscale Research Letters (2019)
-
Visualization of complex DNA double-strand breaks in a tumor treated with carbon ion radiotherapy
Scientific Reports (2016)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
