
Abstract
Some long noncoding RNAs (lncRNAs) play important roles in the regulation of gene expression by acting as competing endogenous RNAs (ceRNAs). However, the roles of lncRNA associated-ceRNAs in oncogenesis are not fully understood. Here, based on lncRNA microarray data of gastric cancer, bioinformatic algorithm miRcode and microRNA (miRNA) targets database TarBase, we first constructed an lncRNA-miRNA-mRNA network. Then, we confirmed it by data of six types of other cancer including head and neck squamous cell carcinoma, prostate cancer, papillary thyroid carcinoma, pituitary gonadotrope tumors, ovarian cancer and chronic lymphocytic leukemia. The results showed a clear cancer-associated ceRNA network. Eight lncRNAs (AC009499.1, GACAT1, GACAT3, H19, LINC00152, AP000288.2, FER1L4 and RP4-620F22.3) and nine miRNAs (miR-18a-5p, miR-18b-5p, miR-19a-3p, miR-20b-5p, miR-106a-5p, miR-106b-5p, miR-31-5p, miR-139-5p and miR-195-5p) were involved. For instance, through its miRNA response elements (MREs) to compete for miR-106a-5p, lncRNA-FER1L4 regulates the expression of PTEN, RB1, RUNX1, VEGFA, CDKN1A, E2F1, HIPK3, IL-10 and PAK7. Furthermore, cellular experimental results indicated that FER1L4-small interfering RNA (siRNA) simultaneously suppressed FER1L4 and RB1 mRNA level. These results suggest that lncRNAs harbor MREs and play important roles in post-transcriptional regulation in cancer.
Similar content being viewed by others

Introduction
MicroRNAs (miRNAs) play important roles in gene expression regulation1. Each miRNAs may repress up to hundreds of transcripts, while each transcript may be targeted by multiple miRNAs2. Their regulatory networks participate in a variety of biological processes, including development, oncogenesis and tumor metastasis3,4,5.
Salmena and colleagues previously proposed a competing endogenous RNA (ceRNA) hypothesis6, which was supported by numerous evidences7,8,9,10,11. The hypothesis described a complex post-transcriptional regulatory network mediated by miRNAs: by sharing one or more miRNA response elements (MREs), protein-coding and noncoding RNAs compete for binding to miRNAs and then regulate each other's expression (Fig. 1). Later, more studies provided convincing evidences for this hypothesis. Pandolfi group focused on phosphatase and tensin homologue (PTEN) and its ceRNAs12,13. They demonstrated the existence of ceRNA interaction among mRNAs in vitro and in vivo. Sumazin et al. analysed gene expression data in glioblastoma and found more than 7,000 transcripts acting as ceRNAs14. Further study showed that ceRNAs are not limited to mRNAs. Linc-MD1, a long noncoding RNA (lncRNA), regulates myoblast differentiation by competing for binding to miR-133 and miR-13515.

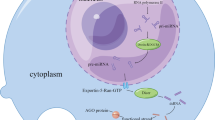
Competing endogenous RNAs (ceRNAs) function as microRNA (miRNA) sponges sequester miRNAs to regulate expression level of other transcripts sharing common miRNA response elements (MREs).
(A) Downregulation of long noncoding RNAs (lncRNA) leads more miRNA molecules free to bind to mRNA that contain the same MREs, thus its protein expression level decreases. (B) Overexpression of lncRNA leads fewer miRNA molecules to bind to mRNA, thus its protein expression level increases.
To annotate the associations between noncoding RNAs and diseases, several disease-associated bioinformatic methods and databases including LncRNADisease16, HMDD17 and dbDEMC18 have been developed. Otherwise, computational tools for miRNA-target interaction prediction have been developed19.
The above studies showed a new layer of post-transcriptional regulation. However, to understand the roles of ceRNA networks in pathological conditions, more work is required. It is found that active translation of mRNA impedes interaction of miRNAs and their targets20. Contrast to mRNAs, noncoding RNAs are more effective ceRNAs without any interference from translation6. Considering these findings and the fact that gastric cancer is one of the most frequent causes of mortality in the world21, in the current study, we first focused on gastric cancer associated-lncRNAs and constructed a ceRNA network in silico. Then, to confirm this network, we performed a regression analysis using several sets of gene expression data of other types of cancer including head and neck squamous cell carcinoma, prostate cancer, papillary thyroid carcinoma, pituitary gonadotrope tumors, chronic lymphocytic leukaemia and ovarian cancer. Finally, we used several experimental data to prove the ceRNA network. This new approach of predicting cancer associated-ceRNA network might help us easily searching for candidate cancer-associated ceRNAs.
Results
Aberrantly expressed lncRNAs in gastric cancer
From the lncRNAs expression profiles (data accessible at NCBI GEO database, accession GSE47850, Guo, 2013), we selected 53 lncRNAs differentially expressed between gastric cancer tissues and paracancerous tissues (fold change ≥ 3.022, P-value < 0.05). To enhance the data reliability, we only retained lncRNAs included in Encyclopedia of DNA Elements (ENCODE)23. Finally, 17 lncRNAs (9, up-regulated; 8 down-regulated) were selected (Table 1).
Gastric cancer associated-lncRNAs' MREs predicted by miRcode
Our previous study has found several gastric cancer-associated miRNAs that include miR-17-5p, miR-18a-5p, miR-18b-5p, miR-19a-3p, miR-20a-5p, miR-20b-5p, miR-21-5p, miR-31-5p, miR-106a-5p, miR-106b-5p, miR-133b, miR-139-5p, miR-195-5p, miR-340-3p, miR-378a-3p, miR-421, miR-497-5p and miR-65821. Here, we focused on whether these miRNAs are targeted on above lncRNAs. Since miRNAs are interacting with lncRNAs through their MREs within ceRNA network, the first thing we should do was to search for the potential MREs in lncRNAs. MREs predicted by miRcode showed that 13 miRNAs may interact with nine of seventeen lncRNAs (Table 2).
mRNAs targeted by miRNAs
To establish lncRNA-miRNA-mRNA network (ceRNA network), the next step we should do was to search for miRNAs' mRNA targets. Based on those miRNAs that might interact with lncRNAs (Table 2), we searched miRNAs' mRNA targets with experimental support using TarBase24. The results showed that nine miRNAs including miR-18a-5p, miR-18b-5p, miR-19a-3p, miR-20b-5p, miR-106a-5p, miR-106b-5p, miR-31-5p, miR-139-5p and miR-195-5p may be included (Table 3). Most of their targets are cancer-associated genes such as PTEN, TNF-α, STAT3, VEGFA, E2F1, RB1, p21, MMP16, BCL2, CCND1, CDK6, CyclinD1, etc. Their functions are involved in cell proliferation, cell cycle, apoptosis, invasion and metastasis.
ceRNA network
Based on the above data from gastric cancer (Table 2 and Table 3), we constructed an lncRNA-miRNA-mRNA ceRNA network. To get more robust results, we assigned a P-value to each relationship by re-sampling analysis (see Methods). As shown in Figure 2, in this ceRNA network, eight lncRNAs and nine miRNAs were involved.

ceRNA network in gastric cancer.
Coloured nodes represent miRNAs; black nodes represent lncRNAs; gray nodes represent mRNAs; coloured edges indicate miRNA-target interactions. mRNAs' names are not shown.
Positive correlation between ceRNAs' expression levels
Our constructed ceRNA network shows that lncRNAs could interact with mRNA in gastric cancer (Fig. 2). To confirm this finding, we performed a regression analysis using several sets of gene expression data of other types of cancer including head and neck squamous cell carcinoma, prostate cancer, papillary thyroid carcinoma, pituitary gonadotrope tumors, chronic lymphocytic leukemia and ovarian cancer. The results reveal a very good or perfect positive correlation between ceRNAs' expression levels (Fig. 3). For example, FER1L4 interacted with RUNX1 mediated by miR-106a-5p (Fig. 3A and B); LINC00152 interacted with THBS1 mediated by miR-18a-5p (Fig. 3C and D); while H19 interacted with MYCN mediated by miR-19a-3p (Fig. 3E and F).

Linear regression of ceRNAs' expression level.
Dashed lines represent 95% confidence interval. (A) FER1L4 vs RUNX1 (head and neck squamous cell carcinoma, n = 36). (B) FER1L4 vs RUNX1 (prostate cancer, n = 15). (C) LINC00152 vs THBS1 (papillary thyroid carcinoma, n = 18). (D) LINC00152 vs THBS1 (pituitary gonadotrope tumors, n = 23). (E) H19 vs MYCN (chronic lymphocytic leukemia, n = 52). (F) H19 vs MYCN (ovarian cancer, n = 15).
FER1L4-RB1 is one pair of ceRNAs associated by miR-106a-5p
RB1 is one of miR-106a-5p's targets and has been validated by dual luciferase reporter assay25. In this study, the interaction between FER1L4 and miR-106a-5p was first predicted by miRcode (Table 2). Then dual luciferase reporter assay showed that the luciferase activity of the mutant FER1L4 plasmid was about 56% higher than that of the wild-type plasmid (Fig. 4). This indicated that the mutations introduced in the seed matches impair the ability of miR-106a-5p to bind to FER1L4. Take together, both FER1L4 and RB1 are targets of miR-106a-5p. This is the basic condition of ceRNAs6.

Relative Firefly/Renilla luminescence (mean ± SD, n = 3) mediated by luciferase plasmid harboring the wild-type or mutant FER1L4 sequence upon transfeciton with miR-106a-5p expression plasmid.
It indicates the direct interaction between FER1L4 and miR-106a-5p. ***P < 0.001.
Next, we observed whether FER1L4 and RB1 expression levels are associated. Quantitative reverse transcription-polymerase chain reaction (qRT-PCR) analysis revealed that transfection of small interfering RNA (siRNA) against FER1L4 not only reduced FER1L4 levels in GES-1, AGS, MGC-803 and SGC-7901, but also reduced RB1 levels in all cells tested (Fig. 5). Since FER1L4 is the target of miR-106a-5p (Table 2 and Fig. 4), FER1L4 knockdown led to more miR-106a-5p free to bind to other targets; and RB1 mRNA is one of them.

qRT-PCR analysis of expression levels (mean ± SD, n = 3) of FER1L4 and RB1 in GES-1, AGS, MGC-803 and SGC-7901 treated with siRNA against FER1L4.
NC, negative control. *P < 0.05; **P < 0.01; ***P < 0.001.
Discussion
Studies revealed that lncRNAs play important regulatory roles in gene expression regulation and contribute to oncogenesis and tumor metastasis26,27. LncRNAs can function as endogenous miRNA sponges as a part of ceRNA network15,28,29. In current study, based on lncRNA microarray and miRNA microarray data, we constructed the ceRNA network jointed by lncRNAs, miRNAs and mRNAs. Previous works have proposed several different strategies, such as RNA22 and miRanda12,30, to predict miRNA-target interactions. However, these algorithms were mainly used in the prediction of miRNA-mRNA interaction. Since that lncRNAs have recently emerged as major players in governing fundamental biological processes and disease occurrence; and that ~18% of the protein-coding genes that produce lncRNAs are associated with cancers, whereas only 9% of all human protein-coding genes are associated with cancers31, here, we focused on searching for lncRNA-miRNA interactions. We used miRcode, an lncRNA-miRNA interaction prediction algorithm, to predict MREs in lncRNAs. In addition, to enhance the data reliability, validated mRNA targets were acquired from TarBase only with effective experimental supporting data. These defined rules are beneficial to screening for candidate ceRNAs.
Our analysis suggested lncRNAs harbor potential MREs and participate in a complex ceRNA network (Fig. 2). The network brings to light an unknown miRNA regulatory network in gastric cancer. It also suggests that lncRNAs may play crucial roles in gastric cancer occurrences and development (Table 1). As Sumazin et al. proved, ceRNA network is a scale-free network in which miRNAs mediated interactions exhibit symmetric behavior14. We found that many oncogenes and tumor suppressors participate in the ceRNA network in gastric cancer (Table 3). For instance, mediated by miR-106a-5p, PTEN, RB1, RUNX1, VEGFA, CDKN1A, E2F1, HIPK3, IL-10, or PAK7 may interact with lncRNA-FER1L4, GACAT1 and H19. Previous study has showed that miR-106a-5p, the core element in this network, is one of the typical onco-miRNAs32. FER1L4 and RB1 are targeted by miR-106a-5p. siRNA silencing of FER1L4 led to RB1 mRNA level decrease (Fig. 5).
In addition, we analysed ceRNAs' expression in several sets of data from cancers including solid cancers and leukemia. We found that there were excellent positive correlations between their expression levels (Fig. 3). This coexpression character corresponds with ceRNA hypothesis.
Recently, two groups have identified a circular RNA (circRNA) that contains ~70 MREs with miR-733,34. The circRNA sequesters miR-7 away from its targets and suppresses its function. Thus circular RNAs are a new class of ceRNAs35,36. On the other hand, to predict ceRNAs' effect on transcriptional network, Ala and colleagues developed a computational model of ceRNA37. Further investigations of ceRNA have implications in many fields. For instant, to block the functions of miRNAs, Tang et al. constructed an artificial ceRNA named short tandem target mimic (STTM)38. STTM is similar to miRNA sponge, but contains different kinds of MREs that bind various miRNAs. Based on the ceRNA hypothesis, Liu et al. further presented a long intergenic noncoding RNA (lincRNA) database named Linc2GO39. Li et al. also developed a database named starBase v2.0 for predicting miRNA-ceRNA interactions40. Moreover, ceRNA might be a new therapeutic approach to diseases41.
In conclusion, we proposed a new approach to construct ceRNA network. It might help us to easily searching for ceRNAs involved in cancer-associated lncRNAs. Our findings suggest that lncRNAs may harbor MREs and participate in a complex ceRNA network.
Methods
The methods were carried out in “accordance” with the approved guidelines.
Tissue samples
Biopsy samples of gastric cancer tissues and paracancerous tissues were obtained from the Affiliated Hospital of Ningbo University School of Medicine. The paracancerous tissues were 5 cm from the edge of tumor; and there were no obvious tumor cells, as evaluated by an experienced pathologist. There was no radiotherapy or chemotherapy prior to the endoscopy examination. The Human Research Ethics Committee from Ningbo University approved all aspects. Written informed consent was taken from all subjects.
RNA extraction
Total RNA was extracted by using TRIzol Reagent (Life Technologies, Carlsbad, CA, USA) according to the manufacturer's protocol. RNA quantity was measured by a SmartSpec Plus spectrophotometer (Bio-Rad, Hercules, CA, USA). RNA purity was evaluated by the A260/A280 ratio.
lncRNA microarray and computational analysis
For lncRNA expression profiling, the Human LncRNA Array (Arraystar, Rockville, MD, USA) which covered 18,534 lncRNAs was used. After hybridization and washing, the arrays were scanned by an Axon GenePix 4000B Microarray Scanner (Molecular Devices, Sunnyvale, CA, USA). Raw data were extracted and normalized by NimbleScan v2.5 software package (Roche NimbleGen, Madison, WI, USA).
Construction of the ceRNA network
The construction of ceRNA network included three steps (Fig. 6): (i) lncRNA screening: lncRNAs that were up- or down-regulated fold change ≥ 3.0 and P-value < 0.05 were first retained; then to enhance the data reliability, lncRNA whose sequences have not been recorded in ENCODE were removed; (ii) lncRNA-miRNA interactions were predicted by miRcode (http://www.mircode.org/); (iii) mRNAs that were targeted by miRNAs with experimental support were from TarBase (http://www.microrna.gr/tarbase).

A flowchart of ceRNA network construction.
(i) lncRNAs that are fold change ≥ 3.0 and P-value < 0.05 were retained; (ii) lncRNAs that have not been recorded in ENCODE were removed; (iii) miRNA-lncRNA interactions were predicted by miRcode; (iv) mRNAs that targeted by miRNAs were captured from TarBase; (v) ceRNA network construction.
Re-sampling analysis of ceRNA network
To get more robust results of ceRNA network, we assigned a P-value to each relationship by re-sampling analysis42. First, we re-sampled the set of above miRNAs from the pool of all human miRNAs in miRBase database and re-inferred the ceRNA relationships. Then we observed whether the previously predicted ceRNA relationships occurred in the newly inferred dataset. We repeated the re-sampling 10,000 times and counted the number of times that each ceRNA relationship occurred. The P-value was defined as the number of occurred times over 10,000. Finally, we obtained the ceRNA relationships using the cutoff of 0.01.
Regression analysis of ceRNAs' expression level
Several sets of gene expression data in cancers were analysed (data accessible at NCBI GEO database, accession GDS1667, Slebos et al, 2005; accession GDS4159, Vitari et al, 2011; accession GDS1665, He et al, 2005; accession GDS4275, Michaelis et al, 2011; accession GDS4168, Gutierrez et al, 2010; accession GDS4066, Spillman et al, 2011). Regression analysis and correlation coefficient were generated using SPSS Statistics v20.0 software package (IBM, Armonk, NY, USA).
Cells and culture conditions
Human gastric epithelial cell line, GES-1, was obtained from Cancer Institute and Hospital, Chinese Academy of Medical Sciences (Beijing, China). Human gastric cancer cell lines, AGS, MGC-803 and SGC-7901, were obtained from Shanghai Institutes for Biological Sciences, Chinese Academy of Sciences (Shanghai, China). Human embryonic kidney cell line, HEK 293T, was obtained from GeneChem Co., Ltd. (Shanghai, China). GES-1, AGS, MGC-803 and SGC-7901 were grown in RPMI Medium 1640 (Life Technologies) plus 10% fetal bovine serum (FBS). HEK 293T was grown in DMEM (Life Technologies) plus 10% FBS. All cell lines were grown at 37°C in a humidified atmosphere with 5% CO2. Cells were counted using a TC10 Automated Cell Counter (Bio-Rad).
Dual luciferase reporter assay
miR-106a-5p expression plasmid (GV268) was purchased from GeneChem Co., Ltd. (Shanghai, China). The wild-type and mutant DNA sequences of FER1L4 were custom synthesized by GeneChem Co., Ltd. and cloned into GV272 Firefly luciferase plasmid (GeneChem). The miR-106a-5p MRE of wild-type FER1L4 was 5′-GCACUU-3′, while the mutant sequence was 5′-UACAGG-3′. HEK 293T of 80% confluence in 24-well plates was transfected using Lipofectamine 2000 Reagent (Life Technologies) according to the manufacturer's protocol. Firefly luciferase plasmid and miR-106a-5p expression plasmid were co-transfected with pRL-TK Renilla luciferase vector (Promega, Madison, WI, USA) for normalization. 48 hours after transfection, luciferase activity was measured using Dual-Glo Luciferase Assay System (Promega). All experiments were performed in triplicate.
Transient transfection
For the transfection of siRNA, GES-1, AGS, MGC-803 and SGC-7901 (2 × 105) were seeded in 6-well plates. The following day they were transfected with 120 nM siRNAs using Lipofectamine 2000 Reagent (Life Technologies). FER1L4 siRNA was: 5′-CAGGACAGCUUCGAGUUAATT-3′ (sense) and 5′-UUAACUCGAAGCUGUCCUGTT-3′ (antisense). Negative control siRNA was: 5′-UUCUCCGAACGUGUCACGUTT-3′ (sense) and 5′-ACGUGACACGUUCGGAGAATT-3′ (antisense).
qRT-PCR analysis
qRT-PCR was performed using GoTaq 2-Step RT-qPCR System (Promega) in a Mx3005P QPCR System (Stratagene, La Jolla, CA, USA) according to the manufacturer's protocol. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as a control. Relative quantification of gene expression was performed with 2−ΔΔCt method43. FER1L4 primers: forward 5′-CCGTGTTGAGGTGCTGTTC-3′ and reverse 5′-GGCAAGTCCACTGTCAGATG-3′. RB1 primers: forward 5′-TGGCGTGCGCTCTTGAGGTT-3′ and reverse 5′-AGAGCCATGCAAGGGATTCCATGA-3′. GAPDH primers: forward 5′-AAGGTGAAGGTCGGAGTCAA-3′ and reverse 5′-AATGAAGGGGTCATTGATGG-3′. All experiments were performed in triplicate.
Statistical analysis
Data were presented as mean ± SD. Differences among groups were evaluated by two-tailed Student's t-test by using SPSS Statistics v20.0 software package (IBM). P < 0.05 was considered to be statistically significant.
References
Pasquinelli, A. E. MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat Rev Genet 13, 271–282 (2012).
Thomas, M., Lieberman, J. & Lal, A. Desperately seeking microRNA targets. Nat Struct Mol Biol 17, 1169–1174 (2010).
Zhang, H., Li, Y. & Lai, M. The microRNA network and tumor metastasis. Oncogene 29, 937–948 (2010).
Gennarino, V. A. et al. Identification of microRNA-regulated gene networks by expression analysis of target genes. Genome Res 22, 1163–1172 (2012).
Li, L. et al. 3'UTR shortening identifies high-risk cancers with targeted dysregulation of the ceRNA network. Sci Rep 4, 5406 (2014).
Salmena, L., Poliseno, L., Tay, Y., Kats, L. & Pandolfi, P. P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell 146, 353–358 (2011).
Ebert, M. S., Neilson, J. R. & Sharp, P. A. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods 4, 721–726 (2007).
Poliseno, L. et al. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 465, 1033–1038 (2010).
Wang, J. et al. CREB up-regulates long non-coding RNA, HULC expression through interaction with microRNA-372 in liver cancer. Nucleic Acids Res 38, 5366–5383 (2010).
Lee, D. Y. et al. Expression of versican 3'-untranslated region modulates endogenous microRNA functions. PLoS One 5, e13599 (2010).
Jeyapalan, Z. et al. Expression of CD44 3'-untranslated region regulates endogenous microRNA functions in tumorigenesis and angiogenesis. Nucleic Acids Res 39, 3026–3041 (2011).
Tay, Y. et al. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell 147, 344–357 (2011).
Karreth, F. A. et al. In vivo identification of tumor-suppressive PTEN ceRNAs in an oncogenic BRAF-induced mouse model of melanoma. Cell 147, 382–395 (2011).
Sumazin, P. et al. An extensive microRNA-mediated network of RNA-RNA interactions regulates established oncogenic pathways in glioblastoma. Cell 147, 370–381 (2011).
Cesana, M. et al. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 147, 358–369 (2011).
Chen, G. et al. LncRNADisease: a database for long-non-coding RNA-associated diseases. Nucleic Acids Res 41, D983–D986 (2013).
Li, Y. et al. HMDD v2.0: a database for experimentally supported human microRNA and disease associations. Nucleic Acids Res 42, D1070–D1074 (2014).
Yang, Z. et al. dbDEMC: a database of differentially expressed miRNAs in human cancers. BMC Genomics 11, S5 (2010).
Jeggari, A., Marks, D. S. & Larsson, E. miRcode: a map of putative microRNA target sites in the long non-coding transcriptome. Bioinformatics 28, 2062–2063 (2012).
Gu, S., Jin, L., Zhang, F., Sarnow, P. & Kay, M. A. Biological basis for restriction of microRNA targets to the 3' untranslated region in mammalian mRNAs. Nat Struct Mol Biol 16, 144–150 (2009).
Guo, J. et al. Differential expression of microRNA species in human gastric cancer versus non-tumorous tissues. J Gastroenterol Hepatol 24, 652–657 (2009).
McKiernan, P. J., Molloy, K., Cryan, S. A., McElvaney, N. G. & Greene, C. M. Long noncoding RNA are aberrantly expressed in vivo in the cystic fibrosis bronchial epithelium. Int J Biochem Cell Biol 52, 184–191 (2014).
Harrow, J. et al. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res 22, 1760–1774 (2012).
Vergoulis, T. et al. TarBase 6.0: capturing the exponential growth of miRNA targets with experimental support. Nucleic Acids Res 40, D222–D229 (2012).
Volinia, S. et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A 103, 2257–2261 (2006).
Ying, L. et al. Downregulated MEG3 activates autophagy and increases cell proliferation in bladder cancer. Mol Biosyst 9, 407–411 (2013).
Tripathi, V. et al. Long noncoding RNA MALAT1 controls cell cycle progression by regulating the expression of oncogenic transcription factor B-MYB. PLoS Genet 9, e1003368 (2013).
Wang, Y. et al. Endogenous miRNA sponge lincRNA-RoR regulates Oct4, Nanog and Sox2 in human embryonic stem cell self-renewal. Dev Cell 25, 69–80 (2013).
Kallen, A. N. et al. The imprinted H19 lncRNA antagonizes let-7 microRNAs. Mol Cell 52, 101–112 (2013).
Jalali, S., Bhartiya, D., Lalwani, M. K., Sivasubbu, S. & Scaria, V. Systematic transcriptome wide analysis of lncRNA-miRNA interactions. PLoS One 8, e53823 (2013).
Khachane, A. N. & Harrison, P. M. Mining mammalian transcript data for functional long non-coding RNAs. PLoS One 5, e10316 (2010).
Xiao, B. et al. Detection of miR-106a in gastric carcinoma and its clinical significance. Clin Chim Acta 400, 97–102 (2009).
Memczak, S. et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 495, 333–338 (2013).
Hansen, T. B. et al. Natural RNA circles function as efficient microRNA sponges. Nature 495, 384–388 (2013).
Karreth, F. A. & Pandolfi, P. P. ceRNA cross-talk in cancer: when ce-bling rivalries go awry. Cancer Discov 3, 1113–1121 (2013).
Tay, Y., Rinn, J. & Pandolfi, P. P. The multilayered complexity of ceRNA crosstalk and competition. Nature 505, 344–352 (2014).
Ala, U. et al. Integrated transcriptional and competitive endogenous RNA networks are cross-regulated in permissive molecular environments. Proc Natl Acad Sci U S A 110, 7154–7159 (2013).
Tang, G. et al. Construction of short tandem target mimic (STTM) to block the functions of plant and animal microRNAs. Methods 58, 118–125 (2012).
Liu, K., Yan, Z., Li, Y. & Sun, Z. Linc2GO: a human lincRNA function annotation resource based on ceRNA hypothesis. Bioinformatics 29, 2221–2222 (2013).
Li, J. H., Liu, S., Zhou, H., Qu, L. H. & Yang, J. H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res 42, D92–D97 (2014).
Almeida, M. I., Reis, R. M. & Calin, G. A. Decoy activity through microRNAs: the therapeutic implications. Expert Opin Biol Ther 12, 1153–1159 (2012).
Li, J. et al. Identification of high-quality cancer prognostic markers and metastasis network modules. Nat Commun 1,34 (2010).
Yu, X. et al. Growth inhibitory effects of three miR-129 family members on gastric cancer. Gene 532, 87–93 (2013).
Acknowledgements
This work was supported by the Zhejiang Provincial Natural Science Foundation of China (Grant No. LY14C060003), the Applied Research Project on Nonprofit Technology of Zhejiang Province (Grant No. 2014C33222), the National Natural Science Foundation of China (Grant No. 81171660 and 31301068), the Natural Science Foundation of Ningbo (Grant No. 2013A610207), the Scientific Innovation Team Project of Ningbo (Grant No. 2011B82014), the Outstanding (Postgraduate) Dissertation Growth Foundation of Ningbo University (Grant No. PY2012004) and the K. C. Wong Magna Fund in Ningbo University.
Author information
Authors and Affiliations
Contributions
T.X. and J.G. conceived and designed the experiments. T.X. Q.L. X.J. Y.S. B.X. performed the experiments. T.X. Q.L. X.J. B.X. Y.X. J.G. analyzed the data. T.X. J.G. wrote the paper.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International License. The images or other third party material in this article are included in the article's Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder in order to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/4.0/
About this article

Cite this article
Xia, T., Liao, Q., Jiang, X. et al. Long noncoding RNA associated-competing endogenous RNAs in gastric cancer. Sci Rep 4, 6088 (2014). https://doi.org/10.1038/srep06088
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep06088
This article is cited by
-
Comprehensive analysis on the regulation of differentially expressed of mRNA and ncRNA in different ovarian stages of ark shell Scapharca broughtonii
BMC Genomics (2023)
-
H19 encourages aerobic glycolysis and cell growth in gastric cancer cells through the axis of microRNA-19a-3p and phosphoglycerate kinase 1
Scientific Reports (2023)
-
DeepWalk based method to predict lncRNA-miRNA associations via lncRNA-miRNA-disease-protein-drug graph
BMC Bioinformatics (2022)
-
LncRNA HOTAIR facilitates proliferation and represses apoptosis of retinoblastoma cells through the miR-20b-5p/RRM2/PI3K/AKT axis
Orphanet Journal of Rare Diseases (2022)
-
hsa_circ_0000218/hsa-miR-139-3p/SOX4 regulatory feedback circuit influences the proliferation and apoptosis of gastric cancer cells
Cytotechnology (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
