
Abstract
The promoter landscape of several non-human model organisms is far from complete. As a part of FANTOM5 data collection, we generated 13 profiles of transcription initiation activities in dog and rat aortic smooth muscle cells, mesenchymal stem cells and hepatocytes by employing CAGE (Cap Analysis of Gene Expression) technology combined with single molecule sequencing. Our analyses show that the CAGE profiles recapitulate known transcription start sites (TSSs) consistently, in addition to uncover novel TSSs. Our dataset can be thus used with high confidence to support gene annotation in dog and rat species. We identified 28,497 and 23,147 CAGE peaks, or promoter regions, for rat and dog respectively, and associated them to known genes. This approach could be seen as a standard method for improvement of existing gene models, as well as discovery of novel genes. Given that the FANTOM5 data collection includes dog and rat matched cell types in human and mouse as well, this data would also be useful for cross-species studies.
Design Type(s) | species comparison design • organism part comparison design • cell type comparison design |
Measurement Type(s) | DNA-templated transcription, initiation |
Technology Type(s) | cap analysis of gene expression |
Factor Type(s) | Species • animal body part • cell type |
Sample Characteristic(s) | Canis lupus familiaris • Rattus norvegicus • aortic smooth muscle cell • hepatocyte • mesenchymal stem cell |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others


Background & Summary
The recent years have seen a renewed interest in non-human model organisms, mainly thanks to the advances in DNA sequencing technologies. Even though next generation sequencing made the most disparate genomes available1,2, building correct gene models either by de-novo or alignment-based assembly, remains difficult. That is mainly due to the inability to accurately map very long transcripts (i.e., >10 kb)3–6.
Defining gene models, though, is one of the main steps in the characterization of an organism’s DNA repertoire. Comparison of genome sequences across multiple organisms is one powerful approach to uncover evolutionary gain/loss and constraints of genetically inherited information, thus shedding a light not only on evolution but also on disease7. Animal model organisms, as a matter of fact, have been broadly used in research under the assumption that understanding the systems in these organisms would translate into understanding the same systems in human. This is of particular importance when human experimentation is unfeasible or unethical. However, all genomic studies rely on accurate gene models in order to locate functional elements in genomes and assess the impact of conservation. The lack of correct and comprehensive genome annotations impedes further progresses in the field.
Among the technologies that allow identification of transcripts, RNA-seq and Cap Analysis of Gene Expression (CAGE)8 paired with sequencing are the most widely used9,10. They both reveal the presence and the amount of RNAs in a given biological sample at a given state, but their final readout is different. RNA-seq facilitates the determination of intron/exon boundaries and alternative splicing, even though with some limitations in defining precise 5′ and 3′ termini. CAGE, although it cannot generate the entire structure of RNAs, can explain promoter usage and architecture due to its ability to map capped 5′-ends of transcripts with high accuracy. Moreover, CAGE can detect promoters of capped ncRNAs, making it a useful tool to study antisense transcription, or to identify actively transcribed enhancer RNAs11, thus providing immediate insights on gene regulation. As shown by recent findings on human long noncoding RNAs (lncRNAs)12, complementing CAGE- and RNA-seq approaches can contribute significantly to improve the definition of gene models.
The functional annotation of the mammalian genome (FANTOM) project aimed at identifying functional elements of genomes9,13–15. Within FANTOM5, the fifth collaborative effort of the fifteen-year-long project, CAGE was applied to nearly 2,000 human and 1,000 mouse primary cell, tissue and cellular states of differentiation and stimulation types, to generate promoter atlases of unprecedented coverage, by using the same technology and the same platform16,17. The outcomes of this large-scale effort include: gene expression quantification across all samples, promoters at a high resolution, bi-directionally transcribed enhancers, transcription factor activities, and co-expression networks of genes. All the data, raw and processed, were further integrated into a public web resource18 for the benefit of the scientific community (http://fantom.gsc.riken.jp/). Partner web browsers and other web tools are also accessible from our gateway site19–24.
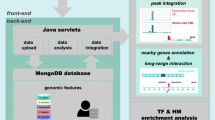
The human (Homo sapiens) and mouse (Mus musculus) genomes have been extensively studied and have well-characterized transcriptomes13,14,16,25. Other mammalian genomes, however, have not benefited from this level of in-depth analyses. Therefore, in order to improve the definition of gene models for the less-studied organisms, the FANTOM consortium generated CAGE data for several other species, like dog (Canis lupus familiaris) and rat (Rattus norvegicus). Experimentally defined transcriptome in these species will provide a baseline for cross-species studies of functional genomics and medical researches using disease models. From each species, we profiled 13 samples representative of three primary cell types matching human, mouse and chicken (Gallus gallus) primary cells. We identified CAGE peaks and associated them to the existing gene models as a part of our quality assessment; this also highlighted the utility of CAGE to refine incorrectly characterized gene models. Although the number of collected samples is meagre and not representative of all cell types, the same cell types are profiled with the same technology across 5 species, providing opportunities to understand similar cellular systems and functions across multiple organisms, and to study cell-type-specific promoters architecture and evolution. The full sets of CAGE promoters are available both via DDBJ data repository (mapping results) and via the FANTOM web resource (expression and genomic visualization), which can be accessible at http://fantom.gsc.riken.jp. A schematic of the entire workflow is summarised in Fig. 1.

The steps of sample collection, CAGE data production, post-processing and further analyses are shown as arrows with their results indicated by squares.
Methods
Sample collection and RNA extraction
Samples used in this study were collected in order to allow comparison of promoter sets and transcriptional regulatory models across several organisms. They were chosen in order to represent varied cells, such as derived from different germ layers, and cells with self-renewal potential. For both beagle dog (Canis lupus familiaris) and rat (Rattus norvegicus), a total of 13 matched samples each were used. One universal RNA tissue sample and 12 RNA primary cell samples from three biological replicates (6 aortic smooth muscle cell, 3 hepatocyte and 3 mesenchymal stem cell) were purchased. Specifically, universal RNA tissue for both dog and rat was obtained from Biochain (Newark, CA, USA); aortic smooth muscle and mesenchymal cells of dog and of all 12 rat primary cell samples from Cell Applications (San Diego, CA, USA); dog hepatocytes from Celsis (Chicago, IL, USA) and BD Gentest (Franklin Lakes, NJ, USA). The rat samples purchased were derived from Sprague Dawley strain, except two aortic smooth muscle cell samples that were derived from Lewis strain rats. We obtained two sets of aortic smooth muscle cells for both dog and rat, depending on which medium they are kept in; one set is kept to proliferate in a myoblast-like state, while the other set is made differentiate towards myotubes (where the suffix ‘diff’ is used to tell them apart). Total RNA was extracted using miRNeasy kit (QUIAGEN Valencia, CA, USA), following manufacturer’s instructions. Age or developmental stage information for neither dog nor rat samples could be obtained. A detailed list of all samples used in this study is available in Supplementary Table 1.
Library preparation and sequencing
CAGE libraries were prepared for single molecule sequencing as described previously26. Two protocols were applied for the preparation of dog samples. The standard library preparation method used 5 ug of total RNA, whereas a low quantity protocol was used for a sample of 1 ug or less total RNA (dog aortic smooth muscle cells donor1). CAGE libraries were subsequently sequenced on HeliScope sequencers27, following the manufacturer’s instructions.
Mapping and data processing
Sequenced libraries were first filtered for reads aligning to ribosomal DNA with up to two mismatches and for artifacts. We used our own developed tool Tag-dust28 to remove artifactual sequences. Retained reads were aligned to canFam3 (Sept 2011, Broad canFam3.1/canFam3) and rn6 (July 2014, RGSC 6.0/rn6) reference genomes by delve (see Code availability), which leverages on a hidden markov model (HMM) to assign probability scores to all mapped positions in the genome. It then uses all the probability scores obtained to calculate the most likely alignment. Mapping of each library was achieved by performing the following commands:
‘delve seed -l 12 -s 8 -o SEEDED_FILE.SAM -t 8 FILTERED.FA GENOME’ and ‘delve align -u 1 -o ALIGNED.BAM -t 8 SEEDED_FILE.SAM GENOME’
SEEDED_FILE.SAM is the output seeded file from FILTERED.FA, ALIGNED.BAM is the final alignment and GENOME is the genome assembly used to map the reads to.
The first command is used to seed the alignment, with a seed length of 12 bases (-l option), step size 8 (-s) and with max 8 threads (-t). The second command is used to build the HMM model and assign the scores to the aligned reads. Option -u ensures that only one alignment is reported (with the highest score).
Expression quantification was performed as described previously16. Simply, frequencies of the observed transcription start sites (TSSs) at a single base resolution were extracted from the alignments by using a combination of samtools29 and bedtools30, such that all mapped CAGE reads aligning at the same 5′ position accounted for the expression of the TSS at that position. We retained only aligned reads with 99% accuracy (mapping quality q>20) and with sequence identity above 85%. In practice, each alignment, for both dog and rat, was processed as follows:
‘samtools view -q 20 -F 768 -u ALIGNED.BAM|bamToBed -i stdin| awk 'BEGIN{OFS="\t"}{if($6=="+"){print $1,$2,$5}}'| sort -k1,1 -k2,2n| groupby -i stdin -g 1,2 -c 3 -o count| awk 'BEGIN{OFS="\t"}{print $1,$2,$2+1, $1":"$2".."$2+1",+",$3,"+"}'
The commands ensure that the reads are filtered for any other bad alignments (using the flag value 768) that may still pass the mapping quality threshold, then converted to a bed file. TSS-level expression is then obtained by grouping, and counting, the reads with the same start position (done for plus and minus strand orientation separately, here shown only for ‘+’ strand). The total number of uniquely mapped reads for each sample is given in Supplementary Table 1. Aligned reads (BAM format) and 1-base resolution frequencies of transcription initiation (BED format) are publicly available via DDBJ (see Data Records, Data Citations 1 and 2).
Identification of promoter regions
CAGE peaks were defined by using a Decomposition Peak Identification (DPI) method as described previously16. Briefly, regions of continuous, composite signal were identified genome-wide, were subsequently decomposed across samples in order to discriminate the signal coming from distinct samples, and finally peaks were called based on distance and a minimum read counts metrics. Peaks were obtained by running the software with default setting, taking as input the BED files of 5′-end frequencies produced as described above. Two sets of peaks were generated: a ‘permissive’ set of promoters with minimum 3 read counts in a single position in at least one sample, and a ‘robust’ set with minimum 1 tpm and 10 read counts in a single position in at least one sample. This ensured that the sets of peaks for dog and rat were comparable to those generated for human and mouse16. Since HeliScopeCAGE is an amplification-free protocol based on single-molecule sequencing, individual reads represent independent evidence of capped 5′-ends. In defining the peak sets, read counts corresponding to the number of observations were primarily employed as a threshold, and the expression level (1 tpm threshold) was added to ensure the robust set included only peaks with a certain minimum level of expression.
The peaks were annotated based on Ensembl transcripts, Augustus, GeneScan, GeneID, RefSeq and EST gene models obtained from UCSC genome browser22 (download date 06/2016). As an annotation rule we followed what was done previously16, that is a CAGE peak overlapping a 1 kb region centred on the gene’s 5′ end on the same strand orientation is associated to that gene.
Projections of human promoters to dog and rat
The conversion of the promoters’ genomic coordinates from human to dog and rat was performed using liftOver tool (see Code availability). Default parameters with pairwise alignment chain files ‘hg19ToCanFam3.over.chain.gz’ and ‘hg19ToRn6.over.chain.gz’ were used to convert human coordinates into rat and dog (downloaded from the UCSC site http://hgdownload.cse.ucsc.edu/goldenPath/hg19/liftOver/). In total, 129,287 (64%) and 111,218 (55%) human CAGE promoters could be projected to dog and rat, respectively. In order to make sure that we considered likely conserved promoters only, we required that a projected promoter be within 50 bp both upstream and downstream of the promoter in the destination genome, following a similar rationale as applied in Young et al.31. Past works on promoter architecture and evolution across species showed that conservation decreases beyond a 70 nt distance32. Moreover, since the set of human CAGE peaks was obtained from profiling thousands of samples, we filtered the set to only use those likely conserved peaks that were expressed in the corresponding cell types to rat and dog. A summary of the number of projected promoters is given in Table 1.
RNA-seq data processing
Publicly available RNA-seq data were downloaded for dog (Data Citations 3 to 5) and rat (Data Citation 6). The rat (Fisher 344 strain) data sets comprise tissues sampled from 11 different organs (muscle, spleen, brain, uterus, testis, kidney, heart, thymus, lung, liver, and adrenal gland), of both sexes and at varying development stages (2 to 100 weeks old). We arbitrarily chose three female and three male samples for each tissue origin, with the exceptions of uterus and testis. RNA-seq samples for dog represented heart (normal and diseased), pituitary, adrenal cortex, and lymph node tissues (normal and B-cell lymphoma). Ages of the dog samples used in the heart disease study varied from 3 to 17 years, and pituitary and adrenal samples were collected from adult dogs; we couldn’t find age information for the lymphoma study samples. A summary of the data sets used is given in Supplementary Table 2 for rat and Supplementary Table 3 for dog.
Data sets were reprocessed using HISAT2, an improved version of HISAT tool33, to align them to the canFam3 and rn6 reference genomes.
Additional data processing
Genomic distribution of the CAGE peaks and screening for TATA-box motifs was performed via HOMER34 (v4.8.1) using the ‘annotatePeaks.pl’ function with standard parameters, except for the TATA motif annotation where a region 500 bp upstream and 200 bp downstream around the peak was scanned. The HOMER default reference gene set for genomic distribution annotations is RefSeq, and the default promoter region window is from 1,000 bp upstream to 500 bp downstream of a gene TSS:
‘annotatePeaks.pl PEAK_FILE.BED GENOME -size -500,200 -m data/knownTFs/motifs/tata.motif -multi -CpG -noann>tata_annotation.txt’
‘annotatePeaks.pl PEAK_FILE.BED GENOME -size given -go DEST_DIR -genomeOntology DEST_DIR -annStats stats_record.txt>annotation.txt’
Student’s t-test was performed on the two sets of CpG- and TATA-associated promoters using the R language function t.test() with default parameters. Clustering of samples was performed using R bioconductor edgeR package35. Specifically, normalization of expression values was calculated with the function ‘calcNormFactors()’ and the plot was visualized with the function ‘plotMDS()’. Figures were all generated using R, version 3.1.3.
Genomic views
All our data is visualized via our original genome browser ZENBU36. Pre-configured views for rat and dog sets can be accessed at these URLs:
http://fantom.gsc.riken.jp/zenbu/gLyphs/#config=3eolUxzyN1nm167YHayYLC (rat)
http://fantom.gsc.riken.jp/zenbu/gLyphs/#config=ddICFtA-H1upfbn6qjjFID (dog)
Code availability
Mapping was performed by our own algorithm delve, version 0.95, described in Djebali et al.37 specifically developed for handling data derived from HeliScope single molecule sequencing. The software is available at this URL (fantom.gsc.riken.jp/5/suppl/delve/).
The DPI peak calling was performed using our own software, detailed in Forrest et al.16. It can be freely downloaded at this URL (https://github.com/hkawaji/dpi1/).
The liftOver tool was downloaded from the UCSC software repository38 (http://hgdownload.soe.ucsc.edu/admin/exe/).
The samtools (v1.3) and bedtools (v2.23.0) packages29,30 were downloaded from (https://sourceforge.net/projects/samtools/ and https://github.com/arq5x/bedtools2).
The HISAT package33 (v2.0.5) was downloaded from (https://github.com/infphilo/hisat).
The HOMER34 motif discovery tool (v4.9) can be found at the following URL (http://homer.salk.edu/homer/download.html).
Data Records
The data resulting from sequencing (fastq format), mapping (BAM format) and TSS profiling (BED format) can be found in DDBJ data repository (Data Citations 1 and 2). The same datasets, together with the identified CAGE peaks sets and association to gene models can also be found within the FANTOM data repository (http://fantom.gsc.riken.jp/5/datafiles/latest). The folder ‘basic’ contains the primary mapping data (BAM format) and TSSs (BED format). The folder ‘extra’ contains the subfolder ‘CAGE_peaks’ for the coordinates of the identified peaks (BED format), peak-based expression tables for raw and normalized counts (tabular text format) and associations to gene models (tabular text format). A detailed description of the samples used in this study, such as cell type, strain, species, tissue of origin, is also given in Supplementary Table 1.
Technical Validation
Primary cell data show high reproducibility across replicates
Biological replicates of each cell type were tested in order to assess the reproducibility of the experiments. Results show agreement within all three replicates for all cell types (Table 2), as seen, for instance, for two of the rat aortic smooth muscle (AoSM) cells replicates where Spearman correlation is 0.97 (Fig. 2a). The same level of reproducibility is evident also in the dog primary cell replicates (Fig. 2b), although we obtained less amount of RNA for one of the dog AoSM cell replicate. That forced us to adopt a variation of the standard library preparation protocol (see Methods), which resulted in a lower total read count and consequently in a slightly lower correlation (0.83, Spearman).

Scatter plots and correlation values of normalized expression values between AoSMC samples (replicate 1 versus replicate 2 on the left and replicate 1 differentiated versus non differentiated in the center), and MDS plots highlighting the separation across cell types are shown for rat (a) and dog (b).
The correlation between the differentiated and genuine AoSM cells is still relatively high, whereas it decreases when AoSM cells are compared to phenotypically distinct cell types such as hepatocytes, which means that the CAGE expression alone is already enough to distinguish between those two states. Clustering analysis via multi-dimensional scaling (see Methods) shows that samples group based on cell type (Fig. 2).
Characterization of the dog and rat CAGE promoters
All samples were sequenced on HeliScope single molecule sequencer27. While the number of mapped reads across samples is comparable in general, many factors can affect it, like RNA amount, RNA quality, protocol efficiency or sequencing errors. The total number of mapped reads for the rat samples varied between 1 M (mesenchymal stem cell donor 2 and 3) and 7 M (AoSM cell), while the universal RNA tissue sample topped 10 M uniquely mapped reads. Uniquely mapped reads for dog samples were, as stated above, generally lower than for rat but with some exceptions, varying between 500,000 of mesenchymal stem cells and 10 M of hepatocytes (Supplementary Table 1). We observed a consistent lower number of uniquely mapped reads for the mesenchymal stem cells in both species, compared to the other cell types. At any rate, the percentage of mapped reads at putative TSSs is more or less constant, around 70% (Fig. 3a), suggesting that the majority of the CAGE signal is concentrated around specific genomic locations (i.e., CAGE peaks) identifying genuine transcription initiation rather than being scattered everywhere.

(a) Percentage of mapped reads at promoters identified by DPI for each sample. Labels description: AoSMC=aortic smooth muscle cell; AoSMCdiff=differentiated aortic smooth muscle cell; MESbm=mesenchymal stem cell from bone marrow; Hep=hepatocyte; UniTis=Universal tissue; (b) histograms of CAGE peaks lengths; (c) enrichment of TATA motifs near CAGE peaks; (d) graphs showing TATA-rich versus CpG-rich peaks. TATA-only bound CAGE peaks tend to be sharp whereas CpG-only peaks are generally broader; (e) percentage of genes that can be associated to a CAGE peak for each of the inspected known models; (f) distribution of the distances of CAGE peaks from their closest gene TSS. Colours: orange denotes rat and blue dog, except for (d), where colour-code is specified in the legend.
We calculated the CAGE based expression levels in each sample separately, and then aggregated the signal into CAGE peaks representing transcription initiation events, or promoters (see Methods). The total number of CAGE peaks is comparable across species: we obtained 28,497 and 23,147 ‘robust’, and 92,031 and 85,324 ‘permissive’ promoters for rat and dog, respectively. Unless explicitly stated, all the analyses presented in this work are based on the robust set of promoters only.
One of the main points of the DPI method in identifying peaks is the ability to decompose big regions of CAGE signal into smaller, non overlapping segments with expression. In order to verify such ability, we calculated the distribution of CAGE peaks sizes and confirmed that they were a) short and b) were consistent between species. Distributions of peak sizes show that they tend to be less than 150 bp long, with the majority of them being 10 to 30 bp (Fig. 3b). By comparison, human and mouse promoters can be longer, up to 300 and 200 bp, respectively16, but the majority of them are around 20–25 nucleotides long, resembling the distributions in rat and dog datasets.
Finally, we evaluated the ratio of CpG- and TATA-rich regions. Promoters are generally classified as either sharp, TATA-rich, shorter regions that harbour most of the expression within a few nucleotides, or broad, CG-rich ones where the expression is distributed on a larger region with sub-peaks of sharp expression32,39. Moreover, TATA-rich promoters tend to be associated to housekeeping genes16,32. CpG islands are one of the most prominent indications of promoters32, and indeed we found 57% (16,327) and 60% (13,863) of rat and dog CAGE peaks, respectively, overlapping a CpG. Since there is no publicly available dataset for TATA-rich regions in dog and rat, we used HOMER motif finding tool34 to check for enrichment of TBP (TATA binding protein) sites around our promoters. An appreciable number of CAGE peaks harboured TATA-box motifs in the region 500 bp upstream and 200 bp downstream of the peak region (32.7% (9,338) and 29% (6,747) in rat and dog respectively), with a clear preference for the region 30–35 bp upstream of a TSS (Fig. 3c). We next subdivided the CAGE peaks into three categories, TATA-only (3,775 dog and 4,763 rat), CpG-only (10,891 and 11,752 dog and rat respectively) and both, and compared their sizes. Although it is not as evident as in human promoters, we could observe that TATA-only promoters tend to be shorter than the CpG-only ones (Student’s t-test, P-value<2.2e-16 in both species) (Fig. 3d). These results are in line with what is known for other model organisms13,16.
CAGE can contribute to refine the promoter landscape of poorly characterized genomes
In order to have a rough estimate of what genomic features the CAGE promoters are closest to, we took the dog and rat peaks and annotated them by using HOMER’s annotation tool (see Methods). We found that 58% (16,625) of rat CAGE peaks are located near TSSs of known genes (Table 3). In the case of dog CAGE peaks, the picture that emerged was different, with only 1,394 peaks annotated as promoters of known RefSeq genes, while the majority of them were annotated as CpG-island (Table 3), highlighting the fragmentary coverage of the dog genome. In fact, the total number of RefSeq genes for dog is as low as about 2,300. As we would like to employ CAGE as an accessory means to improve resolution, coverage and accuracy of the existing gene models, we first downloaded all available gene models, either manually curated or predicted, from UCSC genome browser22 for both dog and rat, and associated them to the CAGE peaks (see Methods) to see how many we could recover.
For the rat CAGE peaks, we could link 19,254 of them (68%) to the TSS of a known Ensembl transcript and 17,188 (60%) to that of a known RefSeq gene (Fig. 3e). Distributions of the distances between all CAGE-defined TSSs and the genes’ TSSs show that CAGE technology captures loci of genuine transcription initiation (Fig. 3f). For 2,289 (8%) CAGE peaks we found no association to any of the gene references. Of those, however, 1,761 (77%) were overlapping with one or more of CpG islands (harbouring the potential for novel TSSs), repeats, gene bodies (i.e., introns or exons more than 500 bp downstream of the TSS). The remaining 520 un-annotated peaks are either intergenic, potential bidirectional enhancers11, or are located more than 500 bp upstream of a known gene TSS.
In the case of the dog dataset we could associate 10,542 (46%) of the CAGE peaks to Ensembl transcripts, and only 1,350 (6%) to known RefSeq genes (Fig. 3e). The large difference is likely due to gene coverage, as only ~2,200 genes are annotated by RefSeq, which adopts strict experimental validation criteria40, against the ~39,000 transcripts (corresponding to about 29,000 genes) reported in Ensembl. On the other hand, the proportion of Ensembl transcripts covered by a CAGE peak in dog is as low as 21% (Table 4), not much lower than what observed in rat (27%). This, and the fact that CAGE peaks tend to align at the TSS of known genes (Fig. 3f) together suggest likely incorrect or incomplete gene annotations. The number of un-annotated peaks for the dog data set was 4,550 (19%), with 873 of them found far from other known features. The peaks-to-genes associations are summarised in Table 4, while the total numbers of CAGE peaks associated to zero or more gene models are listed in Table 5.
To check whether we could increase the gene annotation rate, we converted the coordinates all human robust CAGE promoters identified in a previous study16 to both dog and rat genomes via the liftOver tool (see Methods). These human projections would thus serve to confirm the already annotated peaks and reduce the number of the un-annotated ones, in a similar manner to a guilt-by-association exercise. We chose the human CAGE promoters set primarily because it is the most accurate, comprehensive and characterized, but also because a lift-over of full transcripts to other species is error-prone due to length, splicing, or directionality of genes. For all peaks, we adopted a distance rule requiring the human projected peak be within 50 bp of a dog or rat peak in order to consider the alignments reliable (see Methods). Overall, 79% of dog and 67% of rat CAGE peaks were aligned with a projected human peak expressed in the same cell types (Table 1). Regarding the un-annotated peaks, we required in addition that their aligned human peak be annotated and within 10 kb of a dog or rat orthologous gene to be eligible for annotation. We used Ensembl genes for this survey. Following this principle, we isolated 205 and 801 unique rat and dog peaks respectively, and named them Rescued CAGE Peaks (RCP) (Data Citation 7).
As an additional support, we also used publicly available RNA-seq data for dog and rat (Data Citation 3 to 6), as close to the cell types we profiled as possible, and used them to check whether the RCP are located at the 5′-end of the RNA-seq model as well. Even though it is not a general rule, we’ve noticed several cases where if the RCP are near the dominant human peak (most expressed, noted as ‘p1@gene’) they exhibit higher expression, the RNA-seq model supporting the gene is better defined and chances of them representing a better TSS for the gene are higher.
One example is LOXL3 gene, which was not associated to any CAGE peak in dog because the TSS of its corresponding Ensembl transcript (ENSCAFT00000013310.3) is more than 1 kb away; however, there are two lifted-over human LOXL3 peaks within 50 bases of the un-annotated dog CAGE peaks (Fig. 4a). RNA-seq data shows that the 5′ end of LOXL3 gene is indeed further upstream than the current defined model, judging by the drop of signal at the start of the CAGE peak. As a comparison, we checked the rat genome, where Loxl3 gene is associated to a CAGE peak, instead, and exhibits a similar RNA-seq profile defining the 5′ end together with the same human LOXL3 peaks (Fig. 4b).

Screen shots of (a) LOXL3 gene in dog with RCPs supported by RNA-seq and human lift-over promoters, and (b) Loxl3 gene in rat annotated with CAGE peaks, also supported by RNA-seq and human promoters.
We tried to find out whether those RCP or their associated genes had some features in common. We observed that all RCP in both dog and rat were relatively short, with a median size of 11 bp and tend to be expressed at low levels (Data Citation 7). We next looked at the RCP nearby genes, noticing that, for the vast majority, they are very big, with a median length of 80 kb in rat and 65 kb in dog (Data Citation 7). Since Ensembl genes are predicted, many of them may represent mere open reading frames (ORFs), and in that case the reported TSS would not represent the real one, but just the beginning of the ORF; this scenario could explain why several peaks could not be associated to genes in the first place. We checked this by calculating the distance between the genes’ transcription and coding sequence start, which was zero for 48% (301/622) and 34% (68/201) of dog and rat RCP associated genes (Data Citation 7). In comparison, of the genes associated to a CAGE peak only 12.5% of them in rat are ORFs, while for dog, where the number of curated genes (i.e., RefSeq) is overall lower, the proportion is 52%. Alternatively, the observation that most genes were associated to multiple RCP found within their body could signify the existence of previously unknown isoforms13,16,32 (i.e., Ncor2 gene in rat, http://fantom.gsc.riken.jp/zenbu/gLyphs/#config=3eolUxzyN1nm167YHayYLC;loc=rn6::chr12:36831460..37074148+). Relative to the type of the RCP associated genes, we found that 25% (36/147) of those in rat and 32% (155/478) in dog are known TFs in human; 8 TF genes (AHR, AHRR, HOXB6, JARID2, KDM2A, NCOR2, TSC22D2, ZC3H4) are listed in both RCP sets.
This simple survey showed that, although a systematic validation of all RCP should be pursued, CAGE profiling can be considered as a useful tool to refine existing gene models, even with a limited number of cell types at disposal.
Usage Notes
The FANTOM web portal (fantom.gsc.riken.jp) provides a starting point for data download and exploration. Genomic coordinates and frequencies of TSSs at a single base-pair resolution can be found in fantom.gsc.riken.jp/5/datafiles/latest/basic/, whereas the CAGE peaks, their expression across samples and their gene annotation can be found in fantom.gsc.riken.jp/5/datafiles/latest/extra/. Transcription initiation signals can also be used to identify transcribed enhancers11; however, it has to be noted that the signal from enhancers are quite weak in general. Given the quite limited number of samples, it was not possible to identify enhancer profiles in the two species dog and rat.
Visual inspection of CAGE peaks positions and expression patterns, in relation to gene models and other genomic elements is possible via ZENBU genome browser (fantom.gsc.riken.jp/zenbu/), offering a quick and interactive overview of a genome’s complex structure, as well as via a FANTOM5 UCSC track hub (https://genome.ucsc.edu/cgi-bin/hgHubConnect?hubUrl=http%3A%2F%2Ffantom.gsc.riken.jp%2F5%2Fdatahub%2Fhub.txt). Promoter-level expression based on the CAGE peaks can be easily obtained by using the Table Extraction Tool (TET, fantom.gsc.riken.jp/5/tet/). Finally, CAGE peak centric page and sample centric ones are accessible in SSTAR semantic browser (fantom.gsc.riken/5/sstar/).
All data available through the web portal are public and thus free to use.
Additional Information
How to cite this article: Lizio, M. et al. Monitoring transcription initiation activities in rat and dog. Sci. Data 4:170173 doi: 10.1038/sdata.2017.173 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Tomato Genome, C. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485, 635–641 (2012).
Zeng, X. et al. The draft genome of Tibetan hulless barley reveals adaptive patterns to the high stressful Tibetan Plateau. Proc Natl Acad Sci USA 112, 1095–1100 (2015).
Conesa, A. et al. A survey of best practices for RNA-seq data analysis. Genome Biol 17, 13 (2016).
Engstrom, P. G. et al. Systematic evaluation of spliced alignment programs for RNA-seq data. Nat Methods 10, 1185–1191 (2013).
Fang, Z. & Cui, X. Design and validation issues in RNA-seq experiments. Brief Bioinform 12, 280–287 (2011).
Robert, C. & Watson, M. Errors in RNA-Seq quantification affect genes of relevance to human disease. Genome Biol 16, 177 (2015).
Alfoldi, J. & Lindblad-Toh, K. Comparative genomics as a tool to understand evolution and disease. Genome Res 23, 1063–1068 (2013).
Takahashi, H., Kato, S., Murata, M. & Carninci, P. CAGE (cap analysis of gene expression): a protocol for the detection of promoter and transcriptional networks. Methods Mol Biol 786, 181–200 (2012).
de Hoon, M., Shin, J. W. & Carninci, P. Paradigm shifts in genomics through the FANTOM projects. Mamm Genome 26, 391–402 (2015).
Wang, Z., Gerstein, M. & Snyder, M. RNA-Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 10, 57–63 (2009).
Andersson, R. et al. An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461 (2014).
Hon, C. C. et al. An atlas of human long non-coding RNAs with accurate 5' ends. Nature 543, 199–204 (2017).
Carninci, P. et al. The transcriptional landscape of the mammalian genome. Science 309, 1559–1563 (2005).
Consortium, F. et al. The transcriptional network that controls growth arrest and differentiation in a human myeloid leukemia cell line. Nat Genet 41, 553–562 (2009).
Ravasi, T. et al. An atlas of combinatorial transcriptional regulation in mouse and man. Cell 140, 744–752 (2010).
Forrest, A. R. et al. A promoter-level mammalian expression atlas. Nature 507, 462–470 (2014).
Arner, E. et al. Transcribed enhancers lead waves of coordinated transcription in transitioning mammalian cells. Science 347, 1010–1014 (2015).
Lizio, M. et al. Gateways to the FANTOM5 promoter level mammalian expression atlas. Genome Biol 16, 22 (2015).
Arenillas, D. J. et al. CAGEd-oPOSSUM: motif enrichment analysis from CAGE-derived TSSs. Bioinformatics 32, 2858–2860 (2016).
Ienasescu, H. et al. On-the-fly selection of cell-specific enhancers, genes, miRNAs and proteins across the human body using SlideBase. Database (Oxford) 2016, baw144 (2016).
Medvedeva, Y. A. et al. EpiFactors: a comprehensive database of human epigenetic factors and complexes. Database (Oxford) 2015, bav067 (2015).
Rosenbloom, K. R. et al. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res 43, D670–D681 (2015).
Theocharidis, A., van Dongen, S., Enright, A. J. & Freeman, T. C. Network visualization and analysis of gene expression data using BioLayout Express(3D). Nat Protoc 4, 1535–1550 (2009).
Abugessaisa, I. et al. FANTOM5 transcriptome catalog of cellular states based on Semantic MediaWiki. Database (Oxford) 2016, baw105 (2016).
Okazaki, Y. et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature 420, 563–573 (2002).
Kanamori-Katayama, M. et al. Unamplified cap analysis of gene expression on a single-molecule sequencer. Genome Res 21, 1150–1159 (2011).
Thompson, J. F. & Steinmann, K. E. Single molecule sequencing with a HeliScope genetic analysis system. Curr Protoc Mol Biol Chapter 7, Unit7 10 (2010).
Lassmann, T., Hayashizaki, Y. & Daub, C. O. TagDust--a program to eliminate artifacts from next generation sequencing data. Bioinformatics 25, 2839–2840 (2009).
Li, H. et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Quinlan, A. R. & Hall, I. M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010).
Young, R. S. et al. The frequent evolutionary birth and death of functional promoters in mouse and human. Genome Res 25, 1546–1557 (2015).
Carninci, P. et al. Genome-wide analysis of mammalian promoter architecture and evolution. Nat Genet 38, 626–635 (2006).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12, 357–360 (2015).
Heinz, S. et al. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589 (2010).
Dai, Z. et al. edgeR: a versatile tool for the analysis of shRNA-seq and CRISPR-Cas9 genetic screens. F1000Res 3, 95 (2014).
Severin, J. et al. Interactive visualization and analysis of large-scale sequencing datasets using ZENBU. Nat Biotechnol 32, 217–219 (2014).
Djebali, S. et al. Landscape of transcription in human cells. Nature 489, 101–108 (2012).
Tyner, C. et al. The UCSC Genome Browser database: 2017 update. Nucleic Acids Res 45, D626–D634 (2017).
Lenhard, B., Sandelin, A. & Carninci, P. Metazoan promoters: emerging characteristics and insights into transcriptional regulation. Nat Rev Genet 13, 233–245 (2012).
O'Leary, N. A. et al. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44, D733–D745 (2016).
Data Citations
DNA Data Bank of Japan DRA004814 (2016)
DNA Data Bank of Japan DRA004813 (2016)
NCBI Sequence Read Archive SRP055477 (2015)
NCBI Sequence Read Archive SRP051588 (2014)
NCBI Sequence Read Archive SRP016141 (2013)
NCBI Sequence Read Archive SRP037986 (2014)
Lizio, M. figshare http://doi.org/10.6084/m9.figshare.c.3907471 (2017)
Acknowledgements
FANTOM5 project was funded by the following grants: Research Grant for RIKEN Omics Science Center from MEXT to Yoshihide Hayashizaki; Grant of the Innovative Cell Biology by Innovative Technology (Cell Innovation Program) from MEXT to Yoshihide Hayashizaki; Research Grant from MEXT to the RIKEN Center for Life Science Technologies; Research Grant to RIKEN Preventive Medicine and Diagnosis Innovation Program from MEXT to Yoshihide Hayashizaki. We also thank all members of the FANTOM5 consortium for contribution of samples and GeNAS sequencing facility for data production.
Author information
Authors and Affiliations
Contributions
ARRF designed the experiments. M.O., S.W., M.I. generated the CAGE data. M.L., A.K.M., A.H., T.L., J.S., J.H., I.A., T.K., H.C.C., and H.K. processed the data. P.C., Y.H., A.R.R.F., H.K. managed the project. M.L. and H.K. wrote the manuscript. All the authors approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Lizio, M., Mukarram, A., Ohno, M. et al. Monitoring transcription initiation activities in rat and dog. Sci Data 4, 170173 (2017). https://doi.org/10.1038/sdata.2017.173
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2017.173
