
Abstract
Traditional means for scoring the effects of anti-cancer drugs on the growth and survival of cell lines is based on relative cell number in drug-treated and control samples and is seriously confounded by unequal division rates arising from natural biological variation and differences in culture conditions. This problem can be overcome by computing drug sensitivity on a per-division basis. The normalized growth rate inhibition (GR) approach yields per-division metrics for drug potency (GR50) and efficacy (GRmax) that are analogous to the more familiar IC50 and Emax values. In this work, we report GR-based, proliferation-corrected, drug sensitivity metrics for ~4,700 pairs of breast cancer cell lines and perturbagens. Such data are broadly useful in understanding the molecular basis of therapeutic response and resistance. Here, we use them to investigate the relationship between different measures of drug sensitivity and conclude that drug potency and efficacy exhibit high variation that is only weakly correlated. To facilitate further use of these data, computed GR curves and metrics can be browsed interactively at http://www.GRbrowser.org/.
Design Type(s) | cross over design • database creation objective |
Measurement Type(s) | negative regulation of growth rate |
Technology Type(s) | cell counting |
Factor Type(s) | cell line • perturbagen |
Sample Characteristic(s) | breast cancer cell line |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others
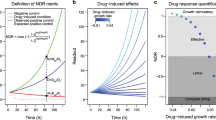


Background & Summary
The prioritization of compounds for lead discovery, identification of pharmacogenomic associations, and study of cellular machinery requires reproducible and accurate data on drug response in cultured cell lines. In the case of anti-cancer drugs, which act by slowing cell proliferation or increasing the rate of cell death, the traditional approach is to expose cells to a drug over a range of concentrations, measure the number of cells at the end of a fixed period of time (typically 3 days) and then compare that number to the number of cells in a vehicle-only control. The ratios are usually fitted to a sigmoidal curve from which it is possible to compute IC50, the drug concentration at which cell count is half the control value, which is a traditional measure of potency, and Emax, the maximum effect at the highest concentration tested, which is a measure of efficacy. Although in use for many decades, this calculation is sensitive to the number of cell divisions taking place over the course of the assay, which differ when cell division rates vary. As a consequence, it is possible for IC50 values for a single drug in a single cell line to vary 100-fold or more simply as a consequence of exogenously imposed changes in the rate of division1,2. Natural cell division rates vary from one cell line to the next, often in a systematic manner depending on tissue of origin2,3, media composition, culture conditions, and plating density1. Because division rate is a powerful confounder in the calculation of IC50 and Emax values, and because it is often poorly controlled, we speculate that it is a contributor to the observed irreproducibility of drug dose-response data2–5.
The normalized growth rate inhibition (GR) method recently introduced by Hafner et al.1,2 corrects for variation in division rates by estimating the magnitude of drug response on a per-division basis. The GR value in the presence of drug at concentration c is GR(c)=2k(c)/k(0)−1 where k(c) is the growth rate of drug-treated cells and k(0) is the growth rate of untreated control cells. Growth rates can be estimated using time course data or, as in this paper, the number of cells prior to and at a fixed time after the addition of drug. Metrics of drug potency, GR50, and efficacy, GRmax, can then be calculated from the dose-response curve. The sign of the GR value is related directly to response phenotype: negative values reflect cytotoxicity, a value of 0 corresponds to complete cystostasis, and positive values reflect partial growth inhibition. GR calculations are described in detail at http://www.GRbrowser.org/, and a detailed method for performing the calculations with web-based software and an R pipeline is also available6,7.
The datasets presented here make use of drug dose-response data that have been described previously8,9 as well as substantial new data and comprise, after filtering low-quality data points, GR-based dose-response data for 71 breast cancer cell lines from all three major subtypes of breast cancer (HER2 amplified, hormone receptor positive, and triple negative) and 107 perturbagens (104 small molecule drugs and 3 therapeutic antibodies). We found that low growth was a good criterion for filtering out data with high relative error, as judged by standard error of the mean (SEM) for GR values, presumably because: (a) a very low division rate (fewer than one division per 72 h) is an indicator that the experimental setup is not optimized for the growth of a particular cell line; (b) fixed error in Cell Titer-Glo (CTG) assays makes measurement of smaller differences in cell number between control and experimental samples unreliable; and (c) partial cell cycle synchronization introduced during plating of cells with division times longer than the assay duration violates the assumption in GR calculations that growth is homogenous and constant10. As a quality control filter, we therefore eliminated from further consideration all samples in which the vehicle-only controls underwent too few divisions. Despite the need to filter out some data, the 4,700 unique cell line/perturbagen pairs described in this paper represent one of the largest collections of growth-corrected drug-response measurements currently available.
Methods
Cell culture and treatment
The original dataset [DS0, Data Citation 1] comprised the effects of 139 perturbagens (small molecules and therapeutic antibodies) on 73 breast cancer cell lines, some in multiple cell growth conditions, determined by plating cells at a density such that they remained in logarithmic growth during the assay11. This required adjusting passage and plating conditions for each line.
Cells were allowed to attach overnight onto 96-well plates before being treated in technical triplicate for 72 h with nine doses of each perturbagen in a 1:5 serial dilution. The concentration range was optimized on six cell lines, and, for most cell line/perturbagen pairs, the highest concentration tested fell between 33 μM and 167 μM.
Cell count
Cell count was estimated using the Cell Titer-Glo (CTG) Luminescent Cell Viability Assay (Promega) after three days in drug (the endpoint of the assay). Cell count was also measured in untreated plates at the time of treatment. The raw CTG assay readout values were considered proportional to the number of viable cells and used as surrogates for cell count values, which is a widely used assumption with some caveats12. A background CTG value was obtained from wells containing culture medium but no cells (values between 35 and 250). Dataset DS0 [Data Citation 1] comprises all raw CTG values.
Cell count normalization and GR values
Dataset DS1 [Data Citation 2] comprises data for the 107 perturbagens (104 small molecules and 3 therapeutic antibodies) and 71 cell lines that could be unambiguously identified (i.e. could be unequivocally matched to records in public databases such as PubChem or ATCC), representing 87% of the original dataset selected for further analysis (see Data Records). First, background CTG value was subtracted from the raw CTG values (which fell between 20 and 10,000) in drug-treated wells. Background-corrected values below 1 or negative, which represented <0.1% of the data, were set to 1 to eliminate artefacts from near-background CTG values. Then, we calculated the following values:
-
–x(c), a robust average of three technical replicates for the background-subtracted CTG value following drug treatment at concentration c; x(c)=mean({xi∈x | abs(log10(xi)–log10(mean(x)))<1}) where x is the vector of treated values.
-
–xctrl, the robust average background-subtracted CTG value in DMSO-treated control wells from the same plate; xctrl=mean({xi∈x | abs(log10(xi)–log10(mean(x)))<1.5)}) where x are all DMSO-treated control values.
-
–x0, the background-subtracted median value from the untreated samples (one per cell line and biological replicate) measured at the time of treatment.
Dataset DS1 [Data Citation 2] comprises the GR value for each treated condition calculated as follows1:
Dose-response curves and GR metrics
GR values for a specific drug across multiple concentrations were fitted to a sigmoid curve with the equation using the MATLAB fitting function. More details about the procedure can be found in Hafner et al.6. Given the data points and fitted equation, we extracted the response parameters:
-
GR50: the primary metric of drug potency; the concentration of drug c at which GR(c)=0.5. If the value for GRinf (see below) is above 0.5, GR50 cannot be defined and we set its value to +∞. If GRinf is below 0.5 but the inferred GR50 value is half an order of magnitude (3.16-fold) above the highest tested concentration, we set the GR50 value to +∞ to avoid artefacts due to extrapolation (this affected 0.6% of conditions). In plots of GR50 values, GR50 values were capped at 100 μM for visualization purposes.
-
GRmax: the primary metric of drug efficacy; the GR value at the highest tested dose of the drug. GRmax lies between –1 and 1; negative values correspond to a cytotoxic response (i.e. cell death), a value of 0 corresponds to a fully cytostatic response (no increase or decrease in cell number), and positive values less than one correspond to partial growth inhibition. GRmax can only be compared across drugs or cell lines when the highest tested doses are the same.
-
GRAOC: the integrated effect of the drug across a range of concentrations as estimated from the ‘area over the curve’. A value of 0 means no effect of the drug across the full dose-response range. GRAOC can only be compared across drugs or cell lines when the dose range is the same.
-
GRinf: a measure of drug efficacy extrapolated to an infinitely high drug concentration as determined from the asymptote of the dose-response curve; . For dose-response curves that reach a plateau under experimental conditions, the value of GRinf is similar to GRmax.
-
hGR: the Hill coefficient of the fitted curve; it reflects the steepness of the dose-response curve. We constrained its value between 0.1 and 5.
-
GEC50: an analogy to EC50 corresponding to the concentration of drug at half-maximal effect. GEC50 is relevant for drugs that have poor potency and that do not reach a GR value below 0.5. In the fitting procedure, GEC50 is constrained to lie within two orders of magnitude of the highest and lowest tested drug concentrations.
We excluded from further consideration cell line/perturbagen pairs in which fewer than 0.3 cell divisions (a 23% increase in cell number) were recorded for the no-treatment control over the course of a 3-day assay (this affected 8.5% of the data, the majority of which were associated with 6 specific cell lines). This was done because GR values quantify the effects of drugs on cell growth. If untreated control cultures are not growing, the response to drug is generally weak, and GR values become highly variable and biologically meaningless. In principle, it is possible to quantify the effects of highly cytotoxic drugs under these conditions using traditional Emax values, but fewer than 1% of the responses in the overall dataset were sufficiently cytotoxic that Emax values would not have been biased by the low division rate. Following filtering, we obtained 4,788 unique cell line/perturbagen pairs, the majority of which were recorded in biological duplicate for a total of 8,882 cell line/perturbagen pairs, each tested at nine doses in technical triplicate. Before averaging biological replicates when available, we further filtered the data to exclude measurements for which division time was greater than 80 h (corresponding to 0.9 division over the course of a 72 h assay). We included this cutoff based on preliminary data showing that GR values obtained from slow-growing cell lines had high uncertainty (see Technical Validation section below). The final dataset comprised 4,650 cell line/perturbagen pairs corresponding to a median of 88 perturbagens per cell line [DS2, Data Citation 2].
Code availability
GR calculation, curve fitting, and extraction of metrics was performed using scripts published in Hafner et al.1,6 and found on the GitHub repo at https://github.com/datarail/gr_metrics. Other analysis was performed using MATLAB standard functions.
Data Records
This manuscript describes 4 new datasets (DS1, DS2, DS3, DS4) that are publically available as a single data package in the Dryad Digital Repository (Data Citation 2) alongside a fifth file bundle (DS0) consisting of the reference files used to process the file containing the raw dose-response data described in Heiser et al.8 and Daemen et al.9 and additional data generated subsequently (Data Citation 1). DS1 reports relative cell counts estimated from the available CTG values as well as computed GR values and nominal division rates for all cell line/perturbagen pairs from the raw dataset for which the cell line and perturbagen reagents used could be unambiguously identified (representing 87% of the original dataset, DS0) and for which the cell line demonstrated sufficient growth over the course of the assay to exceed our cutoffs (see Methods). DS2 reports the calculated GR metrics for the same set of cell line/perturbagen pairs. DS3 and DS4 report the median, upper quartile, and lower quartile GR metrics per perturbagen and per perturbagen class, respectively, across all cell lines in DS2. DS0 consists of a downloadable zip file containing multiple reference files necessary for annotating the raw data file with reagent identifiers and perturbagen concentration details, reference file metadata in the form of data column lookup tables (LUTs), experimental metadata including a detailed protocol, and a ReadMe file. DS1, DS2, and DS3 each consist of a downloadable zip file containing a data file, data file metadata in the form of a data column LUT, experimental metadata including a detailed protocol and reagent metadata tables, and a ReadMe file. DS4 consists of a downloadable zip file containing a data file, data file metadata in the form of a data column LUT, experimental metadata including a detailed protocol, and a ReadMe file. All files are provided in their original format and, where relevant, also in a non-proprietary, preservation-friendly format (e.g. .csv, .txt). All dose-response curves and metrics corresponding to DS2 also are available for interactive viewing at http://www.GRbrowser.org.
Technical Validation
The GR method used to perform calculations in this paper has previously been described1, and the code has been validated on multiple datasets. Use and installation instructions, as well as bug fixes, are available at our GitHub repo (https://github.com/datarail/gr_metrics). Methods for plating cells, adding drugs, and performing CTG assays have also been described in Heiser et al.8 and elsewhere. We have recently published an in-depth protocol10 for performing drug dose-response experiments in cell lines that includes advances in methodology not available at the time the current data were collected. We recommend that future studies follow these more recent guidelines.
One of the key biological premises implicit in computing GR metrics is that cells in untreated wells are proliferating in a uniform, exponential, and continuous manner. In principle, this should be true for all established cell lines, but in a large-scale drug-response experiment it is inevitable that some variability will be encountered. For this reason, we discarded any plate in which control cells underwent fewer than 0.3 cell divisions. When we examined cell line/perturbagen pairs in which cells had undergone more than 0.9 divisions in the absence of drug, we observed a substantially lower standard error of the mean (SEM; Fig. 1). The 95th-percentile SEM was 0.35 for all cell line pairs tested and dropped to ~0.30 when we removed the 27% of the conditions that represent the slowest growing conditions (fewer than 0.9 divisions). We suspect that this arises because ratios between x(c), xctrl, and x0 are more sensitive to fixed measurement noise in the case of slow growing cultures. Low division number can arise because some cells are inherently slow growing, but sporadic examples of slow growth are more likely to reflect defects in the plate, excess evaporation of the medium, or pipetting error. Regardless, eliminating data for these slow growing conditions improved the SEM of the GR values and metrics.

Each plot shows the median, 75th-, 90th-, 95th- and 99th-percentiles for SEM of the GR values (top left), goodness of the sigmoidal fit (top right), GEC50 values (bottom left), and GR50 values (bottom right).
The SEM of the GR values was below 0.1 for 75% of the data in dataset DS1, but 10% of the conditions in DS1 remained substantially variable across replicates (SEM > 0.2). Among the GR values with SEM > 0.2, we observed little bias by cell line. We therefore concluded that the data were impacted primarily by sporadic rather than systematic error (see Methods). With respect to estimation of drug potency, the SEM for GR50 across all perturbagens and cell lines was low (SEM below half an order of magnitude in drug concentration for 95% of the cell line/perturbagen pairs), but this estimate is biased by the many cases in which cells are resistant for which GR50 is set at infinity. For the subset of cell line/perturbagen pairs in which a substantial biological response is observed (GR50 <10 μM), 77% of the data have SEM values below half an order of magnitude. This is consistent with the GEC50 values, whose SEM is below an order of magnitude for 75% of the cell line/perturbagen pairs. Overall, the sigmoidal fit was good (r2>0.75) for 90% of the cell line/perturbagen pairs that show a substantial biological response (GRmax<0.5). Taken together, these results show that, for this dataset, differences in GR value above 0.2 or in GR50 values above half an order of magnitude should reflect substantial differences in sensitivity.
Usage Notes
Uses of our data include identifying cell lines or subtypes that are particularly sensitive or resistant to a class of drugs (or the converse) for follow-up investigation. Using GR metrics for systematic pharmacogenomics studies has two advantages relative to standard approaches2: (1) it ensures that enrichment analysis is not biased by differences in division rate, and (2) it makes it possible to distinguish genes associated with drug-induced cytotoxicity (negative GRmax values) from cytostasis (GRmax values between 0 and 1). In the current study, we first compared potency, as measured by GR50 values, and efficacy, as measured by GRmax values, by drug class across cell lines (see Table 1 (available online only) for annotations; Fig. 2 and DS3 [Data Citation 2]). This showed that drugs targeting tubulin and the proteasome are among the most potent (low GR50), whereas HDAC inhibitors and DNA cross-linkers have the highest efficacy (low GRmax). Many drugs induced a cytotoxic response at the highest concentrations (negative GRmax values), but most MAPK inhibitors and the majority of the ErbB, PI3K, and RTK inhibitors had positive GRmax values, showing that such drugs are partially cytostatic in most breast cancer cell lines. When results were sorted by clinical subtype, we found, for example, that tubulin-binding drugs were generally less efficacious in non-malignant (NM) and hormone-receptor-positive cell lines (HR+) than in HER2-amplified (HER2amp) and triple-negative (TNBC) cells; such drugs are used most widely in the treatment of TNBC.

(a) Distribution of the GR50 values (x-axis) versus GRmax values (y-axis) by drug for all analyzed cell lines (averaged across biological replicates). (b,c) Distribution of the cell line/perturbagen pairs by drug class based on the response classes defined by efficacy (positive or negative GRmax value) and potency (GR50 value below or above 1 μM) for all cell lines (b) or separated by clinical subtype (c).
Contemporary analysis of tool compounds and therapeutic drugs focuses almost entirely on comparing differences in drug potency, with relatively little attention paid to efficacy as measured by maximum drug effect. We have previously shown, however, that both IC50 and Emax values for the Heiser et al.8 dataset are highly variable across drugs and cell lines13. When we repeated this analysis using the expanded dataset and GR-based drug metrics described in this work, we came to the same conclusion, namely that values for GR50, GRmax, GRAOC, and hGR are variable across cell lines and drugs (Fig. 3). For some drugs, predominantly MAPK and ErbB inhibitors, GR50 and GRmax values were correlated, but further analysis showed that this arose simply because a few cell lines were drug sensitive whereas the majority were resistant. When we examined the subset of cell line/perturbagen pairs that resulted in cytotoxicity (GRmax<0), we observed no correlation between GR50 and GRmax (Spearman’s p > 0.05).

(a) GR50 values. (b) GRmax values. (c) GRAOC. (d) Hill coefficient (hGR).
Aggregating drugs by class (based on nominal targets–see Table 1 ((available online only) for these annotations; Fig. 4 and DS4 [Data Citation 2]) also revealed systematic variation in efficacy, potency, and Hill coefficient by drug class. For example, GR50 values for tubulin-binding drugs are very similar across cell lines; variation in response primarily involves GRmax values, many of which are negative (Figs 3a,b and 4a,b). mTOR and PI3K inhibitors as a class have very shallow dose-response curves characterized by low Hill coefficients (hGR; Figs 3d and 4d) and positive GRmax values, reflecting partial growth inhibition. Area over the dose-response curve (GRAOC; Figs 3c and 4c) can also be used to assess drug potency and efficacy in a single metric, but we have shown that GRAOC values have lower information content than GR50 and GRmax values taken jointly2 because the latter two are biologically different. We therefore propose that future studies of cellular factors, genetic changes, and epigenomic variants controlling the responsiveness of cells to anti-cancer drugs consider associations with GR50, GRmax, and hGR independently.

(a) GR50 values. (b) GRmax values. (c) GRAOC. (d) Hill coefficient (hGR).
Additional information
How to cite this article: Hafner, M. et al. Quantification of sensitivity and resistance of breast cancer cell lines to anti-cancer drugs using GR metrics. Sci. Data 4:170166 doi: 10.1038/sdata.2017.166 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Hafner, M., Niepel, M., Chung, M. & Sorger, P. K. Growth rate inhibition metrics correct for confounders in measuring sensitivity to cancer drugs. Nat. Methods 13, 521–527 (2016).
Hafner, M., Niepel, M. & Sorger, P. K. Alternative drug sensitivity metrics improve preclinical cancer pharmacogenomics. Nat. Biotechnol. 35, 500–502 (2017).
Haverty, P. M. et al. Reproducible pharmacogenomic profiling of cancer cell line panels. Nature 533, 333–337 (2016).
Haibe-kains, B. et al. Inconsistency in large pharmacogenomic studies. Nature 504, 389–393 (2013).
Cancer Cell Line Encyclopedia Consortium & Genomics of Drug Sensitivity in Cancer Consortium. Pharmacogenomic agreement between two cancer cell line data sets. Nature 528, 84–87 (2015).
Hafner, M., Niepel, M., Subramanian, K. & Sorger, P. K. Designing Drug-Response Experiments and Quantifying their Results. Curr. Protoc. Chem. Biol. 9, 96–116 (2017).
Clark, N. A. et al. GRcalculator: an online tool for calculating and mining dose-response data. BMC Cancer, 17, 698 (2017).
Heiser, L. M. et al. Subtype and pathway specific responses to anticancer compounds in breast cancer. Proc. Natl. Acad. Sci. USA. 109, 2724–2729 (2012).
Daemen, A. et al. Modeling precision treatment of breast cancer. Genome Biol. 14, R110 (2013).
Niepel, M., Hafner, M., Chung, M. & Sorger, P. K. Measuring Cancer Drug Sensitivity and Resistance in Cultured Cells. Curr. Protoc. Chem. Biol. 9, 55–74 (2017).
Kuo, W.-L. et al. A systems analysis of the chemosensitivity of breast cancer cells to the polyamine analogue PG-11047. BMC Med. 7, 77 (2009).
Chan, G. K. Y., Kleinheinz, T. L., Peterson, D. & Moffat, J. G. A Simple High-Content Cell Cycle Assay Reveals Frequent Discrepancies between Cell Number and ATP and MTS Proliferation Assays. PLoS ONE 8, e63583 (2013).
Fallahi-Sichani, M., Honarnejad, S., Heiser, L. M., Gray, J. W. & Sorger, P. K. Metrics other than potency reveal systematic variation in responses to cancer drugs. Nat. Chem. Biol. 9, 708–714 (2013).
Data Citations
Heiser, L. M., Wang, N. J., Korkola, J. E., & Gray, J. G. SAGE Bionetworks http://dx.doi.org/10.7303/syn8094063.1 (2017)
Hafner, M. Dryad Digital Repository http://dx.doi.org/10.5061/dryad.03n60 (2017)
Acknowledgements
This work was supported by NIH grant U54 HG008100 (J.W.G.), NCI grant U54 CA 112970 (J.W.G.), Stand Up To Cancer–AACR Dream Team Translational Cancer Research grant SU2C-AACR-DT0409 (J.W.G. and P.K.S.), Susan G. Komen Foundation grant SAC110012 (J.W.G.), NIH LINCS grant U54-HL127365 (P.K.S.), and the Prospect Creek Foundation (J.W.G. and L.M.H.). L.M.H. was also supported by the Jayne Koskinas Ted Giovanis Foundation for Health and Policy and the Breast Cancer Research Foundation, private foundations committed to critical funding of cancer research. The opinions, findings, conclusions or recommendations expressed in this material are those of the author(s) and not necessarily those of the Jayne Koskinas Ted Giovanis Foundation for Health and Policy or the Breast Cancer Research Foundation or their respective directors, officers, or staffs.
Author information
Authors and Affiliations
Contributions
L.M.H., N.J.W. and J.E.K. generated the raw data that were used in this manuscript. M.H. analyzed the data. E.H.W. curated the data. J.W.G. and P.K.S. supervised the study. All authors wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Hafner, M., Heiser, L., Williams, E. et al. Quantification of sensitivity and resistance of breast cancer cell lines to anti-cancer drugs using GR metrics. Sci Data 4, 170166 (2017). https://doi.org/10.1038/sdata.2017.166
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2017.166
