
Abstract
Obesity affects gene expression and metabolism of white adipose tissue (WAT), which results in insulin resistance (IR) and type 2 diabetes. However, WAT is a heterogeneous organ containing many cell types that might respond differently to obesity-induced changes. We performed flow cytometry sorting and RNA expression profiling by microarray of major WAT cell types (adipocytes, CD45−/CD31−/CD34+ progenitors, CD45+/CD14+ monocytes/ macrophages, CD45+/CD14− leukocytes), which allowed us to identify genes enriched in specific cell fractions. Additionally, we included adipocytes and adipocyte progenitor cells obtained from lean and obese individuals. Taken together, we provide a detailed gene expression atlas of major human adipose tissue resident cell types for clinical/basic research and using this dataset provide lists of cell-type specific genes that are of interest for metabolic research.
Design Type(s) | parallel group design • cell type comparison design |
Measurement Type(s) | transcription profiling assay |
Technology Type(s) | microarray |
Factor Type(s) | Diagnosis • cell type |
Sample Characteristic(s) | Homo sapiens • white adipose tissue |
Machine-accessible metadata file describing the reported data (ISA-Tab format)
Similar content being viewed by others


Background and Summary
Obesity and its comorbidities are major health problems in modern society. Expansion of white adipose tissue (WAT) is often associated with fat cell hypertrophy (few, but large cells), low-grade chronic inflammation, changes in WAT-resident immune cell populations and altered secretion of proteins (adipokines) and lipids, all of which are important for the development of insulin resistance (IR), type 2 diabetes, hyperlipidemia and atherosclerosis1–3.
WAT is a heterogeneous organ composed of many cell types including adipocytes, adipocyte progenitor cells, endothelial cells, fibroblasts and various types of immune cells4–6. Less than 50% of the cells are the tissue specific fat cells. Obesity has a complex impact on WAT by affecting distinct cell populations differently. In obese WAT, macrophages acquire a pro-inflammatory phenotype7, the relative abundance of lymphocyte populations is changed8, and endothelial cells are activated9. In addition, WAT expansion causes changes in the progenitor cell population, and a change of caloric intake can affect differentiation/recruitment of new fat cells10,11.
WAT metabolic alterations caused by obesity are also reflected in gene expression, a measurement often used in clinical studies. mRNA profiling of WAT obtained from lean/obese individuals or before/after weight reduction is commonplace, e.g. refs 12–15. However, such an approach neither gives insights into the changes within specific cell types nor enables prediction of intracellular gene regulatory networks as obesity-affected genes might be differentially expressed in various WAT cell types. Only a few studies have tried to address these questions comparing gene expression in paired samples of SVF and adipocytes16 or analyzing expression in one particular cell fraction (magnetic-bead sorted macrophages/monocytes)17. Systemic comparison of transcriptomes in WAT cell types is lacking, but is of great interest for the field.
The aims of the current study were to determine transcriptomic profiles of the major cell types in human WAT and thus enable investigations of previously published obesity-regulated genes13, or other WAT-genes of interest, in relevant WAT cell populations. We also aimed to investigate how obesity affects gene expression and function of human adipocyte progenitor cells. The adipocyte progenitors are highly relevant for WAT morphology and metabolic phenotype as hyperplastic WAT (many, small fat cells) reflects efficient recruitment/differentiation of adipocyte progenitors and is associated with a favourable metabolic profile while hypertrophic WAT (few, large fat cells) is closely linked to a pernicious metabolic profile and IR18,19. The adipocytes have direct effects on whole body energy homeostasis by regulating lipid turnover and secretion of adipokines like adiponectin and leptin.
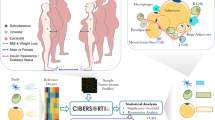
To address these questions in human WAT, we used flow cytometry sorting of the stroma vascular fraction (SVF) of WAT, performed transcriptional profiling in four major cell populations, and compared mRNA expression in adipocyte progenitor cells and adipocytes from lean and obese individuals (Fig. 1). Here we provide a full transcriptomic dataset for major WAT cell types as well as trancriptome of adipocyte progenitors and adipocytes obtained from lean and obese individuals. Analysis of fraction-enriched genes is also provided as an additional useful tool for the researchers in the field.

Outline of the project.
Methods
Human subjects and metabolic measurements
Subcutaneous (sc)WAT from 10 healthy obese and 10 non-obese healthy individuals undergoing cosmetic plastic surgery was collected (Table 1). Obesity-regulated genes have been defined in WAT from 56 non-obese/obese women13. All subjects were given written and oral information about the study before they provided their written informed consent. The study was approved by the regional committee on ethics at Karolinska Institutet.
Flow cytometry
scWAT SVF was isolated and cryopreserved, then stained and analyzed/collected by FACS analysis as described20. Mature adipocytes were prepared in parallel of SVF preparation as described21. The antibodies used are specified in Table 2. Progenitor cells (CD45−/CD34+/CD31−), endothelial cells (CD45−/CD34+/CD31+), monocytes/macrophages (CD45+/CD14+) and leukocytes (CD45+/CD14−) were collected for RNA purification. In addition, the occurrence of T-cell population (CD45+/CD3+/CD14−) was recorded.
RNA extraction
RNA from FACS-sorted cell fractions and SVF were extracted with RNeasy Micro Kit (Qiagen, Hilden, Germany) and from adipocytes with RNeasy Lipid tissue kit (Qiagen) in accordance with the manufacturer’s recommendations.
Microarray analysis
RNA expression was analyzed on Affymetrix GeneChip Human Transcriptome Array 2.0 (Affymetrix Inc., Santa Clara, CA) in accordance with the manufacturer’s instructions. Arrays were normalized (RMA, transcript cluster level) in the Expression Console (Affymetrix, Thermo Scientific).
Statistical analysis of microarray data
After RMA normalization in Affymetrix Expression console software all further analysis of microarray data was carried out in R statistical software (http://CRAN.R-project.org/).
Quality control
After RMA normalization, the array quality was assessed using the ArrayQualityMetrics package in R22.
Enrichment in adipocyte, adipocyte progenitor, macrophage and leukocyte fractions
We excluded array control transcripts before further analysis (by selecting only transcripts with affymetrix category ‘main’) from this step onward. Our data set contains adipocyte, adipocyte progenitor, macrophage and leukocyte microarrays from RNA from 6 non-obese female subjects. To identify genes enriched in one of the fractions versus all others we used the Bioconductor R-package LIMMA23. We made paired, pairwise comparisons of all fractions and selected probes for each fraction that had significantly (Benjamini-Hochberg corrected P-value<0.05) higher expression levels in that fraction compared to all others. We also calculated moderated F-statistics for all genes and filtered out any genes that were not significant (Benjamini-Hochberg adjusted P-value<0.05) according to that analysis. The R-code for this analysis can be found in Data Citation 1.
Differential expression in progenitors and adipocytes
Differential gene expression in progenitors and adipocytes from non-obese and obese patients was also determined using LIMMA23. Array probes without gene symbol annotation were filtered out before LIMMA analysis.
Data Records
All microarray data are accessible on GEO (GSE80654) (Data Citation 2) and analysis files are provided as supplemental tables (Data Citation 3, Data Citation 4, Data Citation 5 and Data Citation 6).
Description of files
File 1: Microarray data are decribed in Data Citation 2.
File 2: Tables of fraction-specific genes (Data Citation 3). RNA from non-obese paired adipocyte, leukocyte, macrophage/monocyte and adipocyte progenitor fractions (from six non-obese women) were analyzed and cell-fraction enriched genes were defined as described under methods and R-code (Data Citation 1 and Data Citation 3).
File 3: Pairwise comparison of adipose cell fractions: RNA from non-obese paired adipocyte, leukocyte, macrophage/monocyte and adipocyte progenitor were compared to each other and differentially expressed genes defined as described in methods and R-code (Data Citation 4).
File 4: Table with obesity-regulated genes in progenitors. Effect of obesity on RNA expression in adipocyte progenitor cells was analyzed using samples from 10 non-obese women, 7 obese women and 2 obese men (Data Citation 5).
File 5: Table with obesity-regulated genes in adipocytes. Effect of obesity on RNA expression in purified mature adipocytes was analyzed using samples from 8 lean women, 5 obese women and 1 obese man (Data Citation 6).
Technical Validation
Quality control of FACS sorting
Purity of each FACS-sorted fraction was evaluated by post-sort analysis. Mean purity of adipocyte progenitor cells was 97.7±1.68%; endothelial cells: 77.5±11.9%; monocytes/macrophages: 95.1±2.9%; leukocytes: 98.8±0.87%. Individual values of purity for each sample are shown in Table 1. Viability of cells was generally between 85% and 70% and was determined by 7-aminoactinomycin D (7-AAD)20 and by distribution on SSC/FSC scatters where alive and dead cells constituted distinct populations. Samples with lower viability than 65% were not used for the analysis. SVF viability for each sample is indicated in Table 1. Functional validation of FACS sorting quality was performed by inducing adipogenesis in vitro in all sorted fractions. Only progenitor cells and total SVF had capacity to differentiate. Purity of adipocyte preparation was examined in an earlier study and was found to be 99%24. A sorting scheme, flow cytometry plots, gating strategy and cell fraction frequencies are shown in Fig. 2. Differentiation ability of progenitor, monocyte/macrophage and leukocyte fractions are shown in Fig. 3.

(a) Representative FACS profiles and gating strategy with marked populations of cells used for analysis. (b) Relative frequency of cell populations in lean and obese SVF (*P<0.05, lean n=10, obese n=9). Non-obese and obese groups were compared by multiple T-test. Means and standard deviations are shown.

(a) Pictures of the cells one day after FACS-sorting/plating, before the induction of adipogenic differentiation. (b) Pictures of the cells at day 13 of differentiation. DNA dye Hoechst was used to visualize nuclei representing total amount of cells and neutral lipid dye Bodipy 493/503 was used to visualize lipid droplets.
Quality control of RNA integrity
To determine RNA quality, Agilent 2100 Bioanalyzer (Agilent Technologies Inc., Santa Clara, CA) was used. The integrity of RNA was calculated using RIN (RNA integrity number) algorithm, where higher numbers indicate higher quality, a maximum value being 10. Mean RIN value of the samples was 7.9 and lowest acceptable RIN in this study was 6.6. In adipocyte fraction, RIN values reflected well the amount of viable cells in the preparation.
Quality control of microarray profiling
RMA normalized data was quality controlled using the ArrayQualityMetrics package22 in R (Fig. 4c–g). Principal component analysis performed in R showed that samples grouped on cell fraction. The two immune cell fractions, leukocytes and macrophages/monocytes were separated by PC1/PC3 (Fig. 4b) and formed distinct clusters in array distance distribution pseudo-heat map (Fig. 4d). The array signal intensity indicated that two adipocyte samples (JL41_M_4 (non-obese) and JL51_M_17 (obese)) as outliers (Fig. 4c,f) but as neither MA-plots nor array distance analysis identified these samples as outliers (Fig. 4e,g), nor did the PCA analysis show these samples to be distant from others. Thus, we kept them in our downstream analyses. Quality of microarray profiling was also verified by RT-qPCR examining expression of known cell-type-enriched genes in all four major SVF fractions (Fig. 5a) and obesity-regulation of genes in progenitor cell fraction (Fig. 5b) (primers listed in Table 3).

(a) Color coding of samples throughout this figure. (b) Principal component analysis plot with PC1 versus PC2 and PC1 versus PC3 showing separation of the sorted fractions. (c) Box plot of array distance distributions after RMA normalization. (d) Heatmap of distance between arrays. Color scale covers the range of distances encountered between the arrays. (e,f,g) Outlier detection in array QualityMetrics based on the specified parameters. Only two possible outliers were detected, one adipocyte non-obese and one adipocyte obese sample (* in f). As this was based on signal intensity distribution alone and none of the other two outlier detection method called these (or other) samples, we decided to keep them in the analysis.

(a) Relative expression of selected known fraction-specific genes in four cell fractions from lean individuals (n=3–5). Groups are compared by multiple T-test and all fraction specific genes are significantly higher in the expected fraction (adjusted P<0.05). Means and standard deviations are shown. (b) Relative expression of selected genes in lean (n=8) and obese (n=7) women. Inclusion of 2 obese men in the obese group (original microarray cohort) does not affect significance for any of the tested genes. Groups are compared by T-test (**P<0.01). Mean and standard deviations are shown. n.d.—not detected.
Usage Notes
Genes enriched in specific WAT cell fractions
In clinical studies, it is often impossible for ethical reasons to obtain enough material to study the effect of different conditions/treatments in the individual cell types of WAT. However, when moving from gene associations to functional studies, the cell type that a gene is primarily expressed in is a crucial clue for designing experiments. Our data set provides a way to assess this, and we provide lists of genes enriched in each of the four cell types we sorted (Data Citation 3). Besides fraction-enriched gene lists, tables include mean expression of the gene in a fraction where it is enriched, log fold change (logFC) compared to each of other fractions and adjusted P values (adj.P.Val). Minimal logFC and maximal adj.P.Val against other fractions are included as separate columns to enable easy sorting of the data. A table where 100 highest-ranked genes from each fraction (based on highest logFC_min and lowest adj.P.val_max) is also provided (Data Citation 3). We also provide pairwise comparisons between all fractions so that researchers can quickly check the magnitude of the differential expression for a specific gene (Data Citation 4). The results are summarized in Venn diagrams (Fig. 6a–d).

Genes differentially expressed in adipocyte progenitors (a), adipocytes (b), macrophages/monocytes (c), leukocytes (d). Amount of genes enriched in the indicated fraction compared to the other three is shown in the middle of the graphs.
Our enrichment analysis is well in line with previously reported data. For example the well known ‘markers’ Adiponectin (ADIPOQ), Leptin (LEP) and Perilipin-1 (PLIN1) were among the top enriched adipocyte genes, CD3G and CD69 were enriched in leukocytes, MMP2 and COL1A2-in adipocyte progenitors. In the monocyte/macrophage fraction we found 23 out of 24 earlier reported WAT macrophage-specific genes17 among the most enriched. Only HLA-DRA from the previous study was not defined as macrophage/monocyte-enriched, which goes well with it’s reported expression in all types of antigen-presenting cells, such as B-lympocytes, dendritic cells and others25. There are also lesser known fraction-enriched genes, of particular interest may be the non-coding genes, that to date have not been well characterized.
Splicing and non-coding transcripts
The Human transcriptome 2.0 arrays contain exon level information and can be used to analyze splicing using e.g., the affymetrix software ‘Transcriptome analysis console’ that is available for free download on Affymetrix/ThermoFisher Scientific’s webpage https://www.thermofisher.com/se/en/home/life-science/microarray-analysis/microarray-data-analysis/genechip-array-annotation-files.html. This analysis can be useful for determining e.g., differential splicing between cell types, or the expression of a specific splice variant in a cell type.
Furthermore, the HTA2.0 array contains probes for many non-protein coding transcripts, which many other older arrays do not. Thus, this data set can be of specific importance for researchers in e.g., the lncRNA field. Annotation to all included probes can be obtained from Affymetrix/Thermo Scientific’s webpage as indicated above.
Effects of obesity on scWAT adipocyte progenitor cells
To investigate how gene expression in human adipose progenitors is affected by obesity, we performed microarray analysis on this cell fraction in 10 non-obese and 9 obese individuals. We were primarily interested in annotated genes so we filtered out all probesets without an associated gene symbol before the start of the analysis. When global gene expression in non-obese and obese WAT progenitors was compared, all multiple hypothesis corrected P-values were >0.05, probably due to small cohort size and the still large amount of transcripts tested. However, even if false discovery rate is rather high in this data set alone, it may still be used for hypothesis generation, especially when combined with other data and perhaps also with cut-offs on e.g., gene expression fold change. To see whether such an approach had any merit we selected genes that had an unadjusted P-value <0.05 and ≥50% up-/downregulation. This gave 35 genes (Data Citation 5), including DKK126, CD3627 and BMP228, known to be involved in regulation of adipogenesis and progenitor proliferation. To add more value to biological significance of our findings, we have selected genes that are regulated by obesity in progenitors and highly enriched in this fraction (15 out of 34 genes). We assumed that in intact adipose tissue, most of the expression detected from these genes is coming from progenitor cells. Therefore, we investigated obesity-regulation of these 15 genes in the earlier published cohort reporting obesity-regulated genes in intact WAT13. Out of 15 genes, 13 were found in this dataset and 9 of them were regulated by obesity with 5% FDR. This suggests that even this small dataset of obesity regulated-progenitor genes can be used for hypothesis generation before deeper functional studies when combined with other data sets or in larger transcriptomic studies.
Additional information
How to cite this article: Ehrlund, A. et al. The cell-type specific transcriptome in human adipose tissue and influence of obesity on adipocyte progenitors. Sci. Data 4:170164 doi: 10.1038/sdata.2017.164 (2017).
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
References
Arner, P. Human fat cell lipolysis: biochemistry, regulation and clinical role. Best Pract Res Clin Endocrinol Metab 19, 471–482 (2005).
Guilherme, A., Virbasius, J. V., Puri, V. & Czech, M. P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 9, 367–377 (2008).
Lee, B. C. & Lee, J. Cellular and molecular players in adipose tissue inflammation in the development of obesity-induced insulin resistance. Biochim Biophys Acta. 1842, 446–462 (2014).
Sengenes, C. et al. Chemotaxis and differentiation of human adipose tissue CD34+/CD31− progenitor cells: role of stromal derived factor-1 released by adipose tissue capillary endothelial cells. Stem Cells. 25, 2269–2276 (2007).
Villaret, A. et al. Adipose tissue endothelial cells from obese human subjects: differences among depots in angiogenic, metabolic, and inflammatory gene expression and cellular senescence. Diabetes 59, 2755–2763 (2010).
Schipper, H. S., Prakken, B., Kalkhoven, E. & Boes, M. Adipose tissue-resident immune cells: key players in immunometabolism. Trends Endocrinol Metab. 23, 407–415 (2012).
Wentworth, J. M. et al. Pro-inflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes 59, 1648–1656 (2010).
Travers, R. L., Motta, A. C., Betts, J. A., Bouloumie, A. & Thompson, D. The impact of adiposity on adipose tissue-resident lymphocyte activation in humans. Int J Obes (Lond) 39, 762–769 (2015).
Sengenes, C., Miranville, A., Lolmede, K., Curat, C. A. & Bouloumie, A. The role of endothelial cells in inflamed adipose tissue. J Intern Med. 262, 415–421 (2007).
Rossmeislova, L. et al. Weight loss improves the adipogenic capacity of human preadipocytes and modulates their secretory profile. Diabetes 62, 1990–1995 (2013).
Esteve, D. et al. Human White and Brite Adipogenesis is Supported by MSCA1 and is Impaired by Immune Cells. Stem Cells. 33, 1277–1291 (2015).
Viguerie, N. et al. Adipose tissue gene expression in obese subjects during low-fat and high-fat hypocaloric diets. Diabetologia 48, 123–131 (2005).
Arner, E. et al. Adipose tissue microRNAs as regulators of CCL2 production in human obesity. Diabetes 61, 1986–1993 (2012).
Dahlman, I. et al. Downregulation of electron transport chain genes in visceral adipose tissue in type 2 diabetes independent of obesity and possibly involving tumor necrosis factor-alpha. Diabetes 55, 1792–1799 (2006).
Capel, F. et al. Macrophages and adipocytes in human obesity: adipose tissue gene expression and insulin sensitivity during calorie restriction and weight stabilization. Diabetes 58, 1558–1567 (2009).
Cancello, R. et al. Reduction of macrophage infiltration and chemoattractant gene expression changes in white adipose tissue of morbidly obese subjects after surgery-induced weight loss. Diabetes 54, 2277–2286 (2005).
Klimcakova, E. et al. Macrophage gene expression is related to obesity and the metabolic syndrome in human subcutaneous fat as well as in visceral fat. Diabetologia 54, 876–887 (2011).
Arner, E., Ryden, M. & Arner, P. Tumor necrosis factor alpha and regulation of adipose tissue. N Engl J Med. 362, 1151–1153 (2010).
Gustafson, B., Hammarstedt, A., Hedjazifar, S. & Smith, U. Restricted adipogenesis in hypertrophic obesity: the role of WISP2, WNT, and BMP4. Diabetes 62, 2997–3004 (2013).
Acosta, J. R. et al. Increased fat cell size: a major phenotype of subcutaneous white adipose tissue in non-obese individuals with type 2 diabetes. Diabetologia 59, 560–570 (2015).
Pettersson, A. M. et al. LXR is a negative regulator of glucose uptake in human adipocytes. Diabetologia 56, 2044–2054 (2013).
Kauffmann, A., Gentleman, R. & Huber, W. arrayQualityMetrics--a bioconductor package for quality assessment of microarray data. Bioinformatics 25, 415–416 (2009).
Wettenhall, J. M. & Smyth, G. K. limmaGUI: a graphical user interface for linear modeling of microarray data. Bioinformatics 20, 3705–3706 (2004).
Ryden, M. et al. Transplanted Bone Marrow-Derived Cells Contribute to Human Adipogenesis. Cell Metab. 22, 408–417 (2015).
Giorda, E. et al. Modular usage of the HLA-DRA promoter in extra-hematopoietic and hematopoietic cell types of transgenic mice. FEBS J. 272, 3214–3226 (2005).
Gustafson, B. & Smith, U. The WNT inhibitor Dickkopf 1 and bone morphogenetic protein 4 rescue adipogenesis in hypertrophic obesity in humans. Diabetes 61, 1217–1224 (2012).
Christiaens, V., Van Hul, M., Lijnen, H. R. & Scroyen, I. CD36 promotes adipocyte differentiation and adipogenesis. Biochim Biophys Acta. 1820, 949–956 (2012).
Lee, S. Y. et al. BMP2 increases adipogenic differentiation in the presence of dexamethasone, which is inhibited by the treatment of TNF-alpha in human adipose tissue-derived stromal cells. Cell Physiol Biochem. 34, 1339–1350 (2014).
Data Citations
Ehrlund, A., & Laurencikiene, J. Figshare https://doi.org/10.6084/m9.figshare.4910372 (2017)
Gene Expression Omnibus GSE80654 (2017)
Ehrlund, A. Figshare https://doi.org/10.6084/m9.figshare.5277727 (2017)
Ehrlund, A. Figshare https://doi.org/10.6084/m9.figshare.4910381 (2017)
Ehrlund, A. Figshare https://doi.org/10.6084/m9.figshare.4929638 (2017)
Ehrlund, A. Figshare https://doi.org/10.6084/m9.figshare.5277658 (2017)
Acknowledgements
The technical assistance of Gaby Åström, Eva Sjölin, Elisabeth Dungner, Kerstin Wåhlén, Yvonne Widlund and Katarina Hertel (Dept. of Medicine Huddinge, Karolinska Institutet, Sweden) is greatly appreciated. Cell sorting was performed at MedH Flow Cytometry Facility, supported by a grant from Karolinska Institutet. We also would like to thank the core facility at Novum, BEA, Bioinformatics and Expression Analysis, which is supported by the board of research at the Karolinska Institute and the research committee at the Karolinska hospital and the Karolinska High Throughput Center, funded by SciLifeLab. This work was supported by the Swedish Research Council, Novo Nordisk Foundation including the Tripartite Immuno-metabolism Consortium (TrIC), Grant Number NNF15CC0018486, CIMED and the Diabetes Research Program at Karolinska Institutet.
Author information
Authors and Affiliations
Contributions
J.L., A.E. and P.A. designed a study, I.D., J.R.A. and J.L. designed flow cytometry protocols, J.L., A.E., C.B., I.D., J.R.A., P.H., P.A. researched and analyzed data, J.L. and A.E. wrote the manuscript, J.L., A.E,. C.B., I.D., J.R.A., P.H., P.A. contributed to the discussion, revised critically the manuscript and approved the final version. J.L. is the guarantor of this work and takes full responsibility for the full content.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
ISA-Tab metadata
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/ The Creative Commons Public Domain Dedication waiver http://creativecommons.org/publicdomain/zero/1.0/ applies to the metadata files made available in this article.
About this article

Cite this article
Ehrlund, A., Acosta, J., Björk, C. et al. The cell-type specific transcriptome in human adipose tissue and influence of obesity on adipocyte progenitors. Sci Data 4, 170164 (2017). https://doi.org/10.1038/sdata.2017.164
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/sdata.2017.164
This article is cited by
-
IL-1β promotes adipogenesis by directly targeting adipocyte precursors
Nature Communications (2024)
-
Adipose stem cells are sexually dimorphic cells with dual roles as preadipocytes and resident fibroblasts
Nature Communications (2024)
-
TM4SF19-mediated control of lysosomal activity in macrophages contributes to obesity-induced inflammation and metabolic dysfunction
Nature Communications (2024)
-
Shared genetic loci for body fat storage and adipocyte lipolysis in humans
Scientific Reports (2022)
-
A Novel Hypothesis and Characterization to Isolate Microvascular Endothelial Cells Simultaneously with Adipose-Derived Stem Cells from the Human Adipose-Derived Stromal Vascular Fraction
Tissue Engineering and Regenerative Medicine (2021)
