
Abstract
Rechargeable sodium-ion batteries have recently attracted renewed interest as an alternative to Li-ion batteries for electric energy storage applications, because of the low cost and wide availability of sodium resources. Thus, the electrochemical energy storage community has been devoting increased attention to designing new cathode materials for sodium-ion batteries. Here we investigate P2- Na0.78Co1/2Mn1/3Ni1/6O2 as a cathode material for sodium ion batteries. The main focus is to understand the mechanism of the electrochemical performance of this material, especially differences observed in redox reactions at high potentials. Between 4.2 V and 4.5 V, the material delivers a reversible capacity which is studied in detail using advanced analytical techniques. In situ X-ray diffraction reveals the reversibility of the P2-type structure of the material. Combined soft X-ray absorption spectroscopy and resonant inelastic X-ray scattering demonstrates that Na deintercalation at high voltages is charge compensated by formation of localized electron holes on oxygen atoms.
Similar content being viewed by others


Introduction
Among the alternatives to lithium-based battery chemistries, the sodium-ion battery (SIB) technology has remained in the research focus due to its potential to decrease the cost1,2. Sodium and lithium both belong to the alkali metal group in the periodic table and thereby share common characteristics such as having the same valence state and they can be inserted or intercalated in layered oxides3,4. However, the larger ionic radius of sodium cation in comparison to lithium cations is accompanied with a lower energy density. Today, there is also a struggle to handle the instability of the crystal structures of typical cathode materials during sodiation/desodiation5.
Similarly, to the ongoing research on Li-ion batteries, there is a large variety of cathode materials being investigated for SIBs. In this context, sodium layered oxides (NaxMO2, (M = TM = transition metal)) have attracted significant attention thanks to their ease of synthesis, high capacity and good rate capability. Therefore, cathode materials such as NaxCoyMn1–yO26,7, NaxCoyTi1–yO28,9, and Na[FexMnyNiz]O210,11 have been extensively studied in literatures.
The NaxMO2 layered oxides are built up of sheets of edge sharing metal oxide octahedra (MO6), with Na ions located between these layers. According to Delmas’s nomenclature, sodium based layered materials can be categorized into two main groups: P2-type (0.6 < x < 0.7, where x = sodium stoichiometry) and O3-type (x ≈ 1). In these structures the sodium ions occupy the prismatic sites (P) or the octahedral sites (O), respectively12, and the number (n = 2 or 3) refers to the repeat period of the transition metals stacking. P2 and O3 type structures differ in their oxygen stacking sequences: ABBA for the P2 type and ABCABC for the O3 type.
The diffusion mechanism of Na+ in the P2 type is different than that in the O3 type. Although the O3 type structure has a high sodium content, the diffusion of the sodium from one octahedral site to another occurs through face-shared interstitial tetrahedral sites, which generate a comparatively slow diffusion13. The P2 type structure on the other hand has an open path for sodium ions, where they diffuse through an open square surrounded by four oxide ions3. However, the sodium deficiency in the P2 type structure leads to significant reduction of the delivered capacity when a sodium-free anode material, such as hard carbon, is used in a full-cell setup. Use of sacrificial salt to compensate the sodium deficiency has therefore been demonstrated to compensate for this shortcoming14,15.
Moreover, P2-type cathodes suffer from capacity fading when operated in a high voltage range, which in turn result in a shortened cycle life. It has been suggested that oxygen release as well as the cation migration associated with the oxygen activity at high voltages could be the reason behind this capacity loss16. At the same time, other researchers have shown that cathode materials with higher capacity can be realized by optimizing and controlling the oxygen activity, since charge can be stored in both transition metals and oxide ions17.
A large number of papers have discussed the oxygen activity in Li-excess cathode materials in lithium-ion batteries they believed that the introduction of alkali ions into the transition metal layers result in relatively ionic Li+-O2p-Li+ interactions, which places these O2p at the top of O-valence band and then promote oxygen redox activity in these materials18,19,20,21. Correspondingly, anionic redox activity in sodium ion batteries was rarely reported. However, it can also be expected. Using electron energy loss spectroscopy (EELS) and soft X-ray absorption spectroscopy (sXAS), Ma et al. demonstrated the existence of oxygen vacancies at the surface of Na0.78Ni0.23Mn0.69O2 at high voltages, which suggests the participation of oxygen anions in the charge compensation mechanism, thereby resulting in considerably higher capacity than the theoretical value22. Maitra et al. reported that Na2/3[Mg0.28Mn0.74]O2 exhibits oxygen redox activity, which displays that excess of alkali metal is not a requirement for oxygen redox activity. Furthermore, the presence of Mg2+ in the TM layers suppress the undesired oxygen loss23. This effect was further supported by the existence of both oxygen redox activity as well as absence of O2 release in alkali-deficient Na2/3Mn7/9Zn2/9O2, despite evidence of cation migration observed upon cycling. This was observed by Bai et al. who could see a tendency of Zn to migrate into the more favorable tetrahedral site, while these sites remains less favorable for Mn24.
The aim of this study is to understand the mechanism of redox activity of the P2-type cathode material Na0.78Co1/2Mn1/3Ni1/6O2. This composition was selected following extensive investigations performed by Doubaji et al.25, which showed a positive impact of cobalt substitution by Ni and Mn in terms of structural stability and electrochemical performance. The redox process at high voltages was, however, not clarified in these studies.
Herein, we investigate the redox mechanisms upon the charge/discharge process of a Na//Na0.78Co1/2Mn1/3Ni1/6O2 electrochemical cell by using several advanced analytical tools such as in situ X-Ray diffraction (XRD), X-ray absorption spectroscopy (XAS), and resonant inelastic X-ray scattering (RIXS), which elucidate the participation of the anions redox reactions to compensate the sodium deintercalation at high voltages.
Results and discussion
Characterization of the structure and morphology
The elemental composition of P2-NaxCo1/2Mn1/3Ni1/6O2 measured by ICP-AAS measurements showed the molar Na:Co:Mn:Ni ratio of 0.78:0.503:0.331:0.165. The ratio between the transition metals is thus consistent with expected stoichiometry, however, the high content of sodium observed is likely originating from the excess of sodium used during synthesis.
Figure 1a shows the XRD pattern of the pristine sample. All the intense diffraction peaks are indexed to a hexagonal lattice with space group P63/mmc, and additional small peaks can be indexed to a P3-type structure (space group: R3m). It is clear that the diffraction lines of the P2-type are much stronger than those of the P3-type, which is also illustrated from the Rietveld ratio (P2: 96 wt%, P3: 4 wt%). The cell parameters obtained (a = 2.83857 Å, c = 11.06986 Å) are in close proximity to those reported for P2-NaxCo2/3Mn2/9Ni1/9O2: a = 2.8274 Å, c = 11.0553 Å25. The different parameters obtained by Rietveld refinement for P2-type structure are presented in supplementary Table 1. A schematic illustration of the P2-type structure using the Vesta program for Na0.78Co1/2Mn1/3Ni1/6O2 is presented in the Fig. 1b, Cobalt, manganese and nickel ions are located in octahedral sites while Na ions are coordinated by oxygen in two trigonal prismatic sites: Nae, where Na is sharing edges with six surrounding (Co, Mn, Ni)O6 octahedra, and Naf, where Na shares faces with two neighbor (Co, Mn, Ni)O6 octahedra.

a Rietveld refined XRD patterns. b Schematic illustration of P2-structure. c SEM micrographs taken at different magnifications.
The SEM micrographs of Na0.78Co1/2Mn1/3Ni1/6O2 are shown in Fig. 1b. The sample exhibits hexagonal-shaped primary particles with an average size of 1–2 µm, which reflects the material’s hexagonal symmetry. It can also be seen that these primary particles undergo agglomeration to form secondary particles which are several micrometers.
Electrochemistry
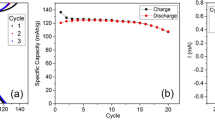
The electrochemical performance of Na0.78Co1/2Mn1/3Ni1/6O2 was tested in room temperature using sodium half-cells. The open circuit voltage (OCV) value was about 2.7 V (vs. Na+/Na) for all tested cells. Figure 2 displays galvanostatic cycling results of P2- Na0.78Co1/2Mn1/3Ni1/6O2 electrodes using two different cut-off potentials of 4.2 V and 4.5 V, respectively using a current density of 25 mA/g.

a, b Cycling performances in a potential window of 2–4.2 V. c, d Cycling performances in a potential window of 2–4.5 V.
Figure 2a shows that the first charge capacity is equal to 85 mAh g−1 corresponding to 0.334 Na+ when the cell is charged to the cut-off potential of 4.2 V. In the following discharge process, the capacity is about 118 mAh g−1, meaning that the Coulombic efficiency of the first cycle is above 100%. This means that the amount of Na in the cathode formulation has increased from 0.78 to 0.89 Na+ after the first cycle. This is due to the fact that P2-type materials contain sodium vacancies and when cycled vs. metallic sodium, a larger amount of sodium can be inserted into the structure. The electrochemical profiles of the subsequent 40 cycles almost completely overlap and the cathode material delivers capacities between 118 to 113 mAh g−1 with a retention of above 95% and a very promising Coulombic efficiency exceeding 99.3% (see Fig. 2b).
Figure 2c shows the galvanostatic charge/discharge voltage profiles when the high cut-off potential is increased to 4.5 V. The cathode material delivers a higher capacity compared to the cell with cut-off potential of 4.2 V; for the first charge and discharge the capacities of about 146 mAh g−1 and 143 mAh g−1, respectively, were achieved. These capacities correspond to removal of 0.58 Na+ and addition of 0.56 Na+ during the first charge and discharge, respectively. The extra capacity obtained is due to the appearance of a new and relatively flat high voltage plateau (above 4.2 V) which correspond to an additional capacity of 25 mAh g−1. The discharge capacities decreased after the first cycle, with a capacity retention of 88.3% and a coulombic efficiency of 98.8% after 40 cycles (Fig. 2d).
The flat plateau between 4.3 and 4.5 V indicates that a reaction occurred at high potential, which is fundamentally different from the charge plateau observed in narrow potential windows up to 4.2 V. This plateau is longer in the first charge, but it provides some reversible capacity from the second cycle. There are several mechanism that can explain the origin of this high voltage plateau: (i) phase transition such as from P2 to O226,27, or from P2 to OP428,29, or P2 to Z phase30 (ii) redox activity of oxygen anions22,31, or (iii) electrolyte decomposition on at the surface of electrode at high potentials32.
The latter is not the possible explanation for the extra capacity at high potentials since cyclic voltammetry and XPS spectra showed no indication of electrolyte decomposition. The C 1s, F 1s, and O 1s spectra of electrodes analyzed at different state of charge during the first cycle look very similar to the pristine electrode, implying absence of electrolyte degradation products (see supplementary Fig. 1). To understand whether the first two mechanism explain the origin of the plateau at high potentials, results from in situ XRD, XAS, and RIXS measurements of Na0.78Co1/2Mn1/3Ni1/6O2 electrodes are discussed below.
Structural evolution of P2-Na0.78Co1/2Mn1/3Ni1/6O2 during cycling
A phase transition is known to be associated with a volume change in the material structure, which contributes to structural irreversibility and leads to poor rate and cycling performances. The structural changes of P2-Na0.78Co1/2Mn1/3Ni1/6O2 during the first cycle in the voltage range of 2–4.5 V was monitored using in situ XRD. The voltage-time profile and selected in situ XRD patterns are plotted in Fig. 3. The overview of the diffraction patterns reveals that the major diffraction lines of the P2-type structure are maintained, which means that the material preserves its P2 structure during the entire electrochemical process.

a Voltage–time curve of the first cycle at 0.1C rate in the potential range of 2–4.5 V and the magnifications of 002, 004, 100 in situ XRD reflections during cycling. b The evolution of the hexagonal unit cell parameters during the first cycle obtained from Rietveld refinement. c extended view of the 104 and 106 reflections at high potentials.
Figure 3 shows that in the beginning of the charge process (00ℓ) and (10ℓ) reflections shift to lower angles, in contrast to (100) shifting to a higher angle (Fig. 3a). This indicates that the “c” lattice parameter increases, most likely due to the increase in the electrostatic repulsions between the oxygen layers upon sodium extraction. At the same time, the “a” lattice parameter which represents the M–M distance decreases. This is expected from the increase in the oxidation state of the transition metals (Co3+ → Co4+, Ni3+ → Ni4+), which is needed to maintain the neutrality of the material during sodium removal (Fig. 3b).
The XRD peaks become broader with lower intensity at high voltage, especially for the sample fully charged to 4.5 V as compared to at the beginning of the charge process. Simultaneously, the appearance and growth of a very weak peak between 17° and 18° can be seen, which is not observed in the in situ XRD pattern in the voltage range 2.0–4.2 V (see supplementary Fig. 2). These changes may be associated with the formation of O2 stacking faults (i.e., changes in the evolution of a and c lattice parameters). Indeed, the sodium ions in the P2-type structure occupy the prismatic sites between neighboring oxygen layers, and upon sodium extraction the MO2 slabs shift into the a–b plane to avoid oxygen–oxygen contacts. There are two possible directions for these shifts to form the O2-type structure. The stacking faults are formed because these two choices are selected randomly, which means that any long range ordered structure is lost33. The broadening of the (10ℓ) peaks (e.g., (104) and (106)) confirms the presence of these stacking faults in Na0.78Co1/2Mn1/3Ni1/6O2 (Fig. 3c)34.
The reverse process occurred during discharge: the well-defined and sharp P2 peaks were slowly recuperated upon sodium insertion, which means that the stacking faults formed previously during charge are eliminated during discharge. This is primarily owing to the incapability of the P2-type structure to accommodate stacking faults between adjacent MO2 layers, unlike the O2-type. Regardless of these variations during cycling, the material shows a good structural reversibility of the P2 type structure in the investigated voltage range.
XAS and resonant inelastic X-ray scattering measurements
The electronic structure of Co, Ni, Mn (L2,3-edge), and O (K-edge) during the first sodiation/desodiation was analyzed by a range of spectroscopic techniques, including soft XAS using different modes with different probing depths; total-electron yield (TEY) (surface sensitive), partial-fluorescence-yield (PFY), and inverse partial-fluorescence-yield (IPFY) (bulk sensitive)35, and RIXS measurements. The results provide information regarding the redox reactions taking place upon cycling as well as the hybridization states for metal-oxygen octahedral units. Figure 4 shows XAS spectra of nickel, cobalt and manganese L2,3-edges collected ex situ on electrodes at different state of charge.

a Co in PFY mode. b Co in TEY. c Mn in IPFY. d Mn in TEY. e Ni in PFY. f Ni in TEY mode. Insets: extended view of L3 peaks. (states of charges: OCV, charged to 4.2 V, charged to 4.5 V, discharged to 4.2 V, discharged to 4.5 V).
The Co L2,3-edge XAS corresponds to the transitions from the Co 2p3/2 and 2p1/2 core electrons, split by the spin–orbit interaction of the Co 2p core level, to the empty 3d orbitals. The profile and the peaks positions of the pristine spectrum obtained in both PFY and TEY modes (Fig. 4a, b) are very similar to those reported for LiCoO2 and thus clearly demonstrates that cobalt in the pristine material is in its trivalent state36. The spectra in PFY mode show that during charge, the main peak broadens and shifts slightly towards higher energy by 0.2 eV from the pristine to the fully charged material at 4.5 V, which suggests oxidation of Co3+ to Co4+. It has previously been claimed that the small peak shift is due to the low participation of cobalt in charge compensation process during charge37,38,39,40. The shift is reversible during discharge. However, in TEY mode the peaks of fully discharged electrode clearly shift toward lower energy, which is a much bigger shift compared to that in PFY mode, suggesting that the reduction of Co3+ to Co2+ mainly occurs at the surface of the material.
The Mn L2,3-edge spectra of the pristine electrode show a main peak at 643.4 eV and a sub-peak at 640.7 eV, which coincident with that of the Mn4+. No peak shift was observed during the charge process in either TEY or IPFY modes, indicating that Mn stays at tetravalent state and does not participate in the charge compensation during sodium deintercalation. The oxidation state of Mn seems unchanged during the discharge process in the IPFY mode, while the shift of the main peak for the fully discharged electrode is clearly seen in the TEY mode and reveal the reduction of Mn4+ at the surface.
Figures 4e, f show the Ni-L edge spectra taken in both PFY and TEY modes. Ni L2,3-edge XAS of the pristine electrode obtained in PFY mode shows that the Ni exist as Ni3+ in the bulk41. Nickel is oxidized to Ni4+ during sodium removal when charging the material up to 4.2 V, and no shift of the main peak was observed from 4.2 to 4.5 V. By discharging to 2 V, the spectrum almost returns to the same position of the pristine material but with different intensity ratio between L3high and L3low suggests the reduction to Ni2+. In contrast, the spectra in TEY mode shows that the Ni is mainly in its divalent state in the surface layer while it is oxidized during charging to 3+ and returns to its previous oxidation state during discharge. The transition metal oxidation gradient between the surface and the bulk of the material has been seen in previous studies and was then attributed to structural changes at the surface42,43,44.
The O K-edge XAS spectra of Na0.78Co1/2Mn1/3Ni1/6O2 electrodes at different state of charge are shown in Fig. 5a. The spectra can be divided into two regions. First, a broad feature at higher energy (≥535 eV) appears, which is associated with the electronic transitions from O 1s to O 2p-states that are hybridized with transition metal-4sp states. Second, the pre-edge peaks below 535 eV correspond to the transition from O 1s to O 2p-states that are hybridized with transition metal-3d states, which is discussed in the following. The peak around 534 eV might be associated with some carbonaceous components that exist on the surface of the material. The main contribution to the pre-edge peaks comes from transitions to Co3+ and Mn4+ states, as these are the dominant transition metal ions in the compound and accounts for 84% of the spectral transitions (see Supplementary Fig. 3 and Supplementary Note 1). Therefore, the pristine spectrum can be explained by a superposition of the O K-XAS peaks of LiCoO2 and MnO240,45. Figure 5b illustrates the changes in the area under the spectra between 520 and 534 eV of Fig. 5a and is a measure of the changes in the density of hole states just above the Fermi level.

a Oxygen K-edge XAS spectra collected at different states of charge in partial fluorescence yield mode. b Variation of the integrated intensity in the low-energy region (≤534 eV) for O K-edge SXAS. c RIXS on the O K-edge of Na0.78Co1/2Mn1/3Ni1/6O2 with an excitation energy of 531.5 eV.
When charging the material up to 4.2 V, the increase in the density of the empty states by 6.8% is caused by the oxidation of Ni3+ and Co3+ in Na0.78Co1/2Mn1/3Ni1/6O2, which creates more holes in the 2p–3d hybrid orbitals. A continuous increase in the density of the empty states is observed across the plateau (between 4.2 and 4.5 V) by an additional 7.27%. This increase cannot be explained by the oxidation of Ni, because the Ni L2,3-edge XAS spectra show no shift of the main peak in this region. However, a small shift was observed in the Co L2,3-edge XAS spectra above 4.2 V which corresponds to 4.28% (see Supplementary Fig. 4). Therefore, additional unoccupied states are associated with the removal of electrons from oxygen 2p orbitals, showing the small participation of anions in the redox activities in the charge compensation process in the material. To examine the nature of the hole states on oxygen, O K-edge RIXS spectra were collected as a function of state of charge (Fig. 5c).
RIXS allows us to detect the emission from the O 2p valence states below the Fermi level. There is a clear change in the RIXS spectra at 4.2 and 4.5 V (high voltage plateau) compared to the pristine spectrum. A considerable broadening at the main peak occurred and the elastic peak at the 531.5 eV is growing in intensity, which indicates the creation of hole states localized on the oxygen at high voltage17,20. However, if we compare the RIXs-spectrum of the OCV with the fully discharged electrode, a change in spectral weight is observed, suggesting lattice oxygen rearrangement during the first cycle.
Based on these results, we showed that electrons from oxygen atoms can participate in the charge compensation process during sodiation–desodiation and give rise to the excess of the capacity observed across the plateau at high voltage in Na0.78Co1/2Mn1/3Ni1/6O2 with a high proportion of cobalt cations.
In summary, the Na0.78Co1/2Mn1/3Ni1/6O2 cathode material can provide an extra reversible capacity of about 25 mAh/g between 4.2 and 4.5 V, in addition to reversible and stable capacity of about 118 mAh/g obtained in the voltage range between 2.0 and 4.2 V. The mechanism of redox reactions and the origin of the extra capacity is analyzed using variety of in-house and synchrotron-based analytical techniques. In situ XRD and XPS results excluded major crystal structure deformation or electrolyte degradation as the origin of extra capacity at high potential between 4.3 and 4.5 V. On the other hand, XAS and RIXs measurements revealed that upon charging up to 4.5 V the Mn valency remains at +4 state, while Co and Ni ions are oxidized to Co4+ and Ni4+. The continuous increase in the density of the empty states from 4.2 to 4.5 V indicated the removal of electrons from oxygen 2p orbitals implying contribution of anionic redox activities in charge process. The main findings of this study are schematically presented in Fig. 6.

Measurements showing the oxidation states of Ni, Mn, and Co in the bulk and surface.
Methods
Preparation of the samples
P2-Na0.78Co1/2Mn1/3Ni1/6O2 was prepared by a co-precipitation method. A solution made of Co(NO3)2·6H2O, Ni(NO3)2·6H2O and Mn(NO3)2·4H2O (1 M of the transition metals), was dripped into a NaOH solution (1 M) under rapid stirring. After 3 h stirring at room temperature, the precipitate formed was filtered, washed three times with distilled water and dried at 180 °C over night in air to remove residual water. The dried powder was mixed and grinded with Na2CO3 in an agate mortar for 45 min at room temperature. A 10 wt% excess of Na2CO3 was added to compensate for sodium volatility at high temperature. The obtained mixture was heated to 850 °C at a rate of 2 °C min−1 for 12 h in air, followed by slow cooling to room temperature and thereafter stored in an Ar-filled glove box.
Material characterization
The chemical composition of the NaxCo1/2Mn1/3Ni1/6O2 material was measured using inductively coupled plasma optical emission spectrometry (ICP-OES) (Agilent Technologies 5110). The structure of the synthesized material was characterized by XRD using a Bruker twin–twin [Cu Kα radiation, λ = 1.54056 Å] by measuring the diffraction angle (2θ) between 10° and 80° in a continuous scanning mode with a step size of 0.01°. XRD data were analyzed by the Rietveld method using FULL-PROF software. The morphology of the pristine material and the size distribution of the particles were measured by scanning electron microscopy using a Zeiss Leo 1550 scanning electron microscope.
Electrochemistry
Electrodes were fabricated by mixing 80 wt% of active material with 15 wt% carbon black and 5 wt% of polyvinylidene difluoride (PVDF) dissolved in N-methyl-2-pyrrolidone (NMP). The black slurry formed was coated on aluminum foil and was then dried at 80 °C for 4 h. Afterwards, electrodes were cut into 13 mm disks using a perforator and dried at 120 °C in a vacuum furnace inside the glovebox to remove any moisture or solvent contaminations. For cell assembly, the mass loading of the active material was approximately 2.1 mg cm−2, metallic sodium (Sigma-Aldrich) was used as both counter and reference electrodes. Whatman glass fiber membrane was used as the separator. The electrolyte was composed of 0.5 M of NaPF6 dissolved in a mixture of ethylene carbonate (EC) and diethyl carbonate (DEC) with a volume ratio of (1:1); 5 vol% of fluoroethylene carbonate (FEC) was added to improve the electrochemical performances. Cells were fabricated in CR2032 coin-type format and were galvanostatically cycled at room temperature using a current density of 25 mA/g. Two cutoff voltages of 2.0 and 4.2 V or 2.0 and 4.5 V, respectively, were used. All the potential values are hereafter reported vs. Na/Na+.
In situ X-ray diffraction
In situ analysis were performed on Stoe and Cie Gmbh Stadi X-ray powder diffractometer equipped with Ge monochromator (CuKα1) using “coffee bag” (polymer laminated aluminum pouch) cells. XRD patterns were continuously collected in reflection mode with a movable detector around the fixed sample, within the 2θ range of 10–80°. The data collection time for each XRD scan was 20 min.
X-ray photoelectron spectroscopy
Measurements were performed using a Scienta ESCA 300 instrument with the exciting radiation of Al Kα (hν = 1486.7 eV). The cells were disassembled in an argon-filled glove box, and the electrodes were thereafter washed with dimethyl carbonate (DMC) in order to remove excess electrolyte before the analysis. The samples were transferred into the introduction chamber of the XPS instrument using a special transfer cup to avoid any exposure to ambient air. The pressure in the analysis chamber was 1 × 10−8 mbar. The peak at 284.4 eV in the C 1s spectra was used as the reference to calibrate the binding energy scale. The spectra were analyzed using CasaXPS software.
XAS and resonant inelastic X-ray scattering
Soft XAS as well as resonant inelastic X-ray scattering (RIXS) measurements were performed, using beamline BL27SU of SPring-8, Japan46 XAS spectra were collected for the Mn L-edge, Co L-edge, Ni L-edge, and the O K-edge by collecting energy-resolved fluorescence x-rays (500–1000 eV) and by measuring the sample drain current simultaneously. Bulk representative XAS spectra for Co L-edges, Ni L-edges, and the O K-edges are obtained by using the partial fluorescence yield (PFY) mode while the Mn L-edge could only be adequately obtained by using the inverse partial fluorescence yield (IPFY) mode (derived by inverting the O K-edge PFY). The surface-related drain current produces XAS spectra equivalent to the total electron yield (TEY) mode. The (I)PFY mode XAS spectra were collected by using an energy resolving, liquid nitrogen cooled, solid state detector. The TEY mode XAS spectra and the incident intensity were measured by recording the sample drain current and focusing mirror current using pico-ammeters. (I)PFY mode XAS spectra are bulk sensitive with an average information depth of about 100 nm while TEY mode XAS spectra are more surface sensitive with an average information depth of about 10 nm. The monochromator band width was set to ~0.2 eV (O K-edge) and ~0.3 eV (transition metal L-edges), respectively. Ni L3-edge and O K-RIXS measurements were performed using a high-resolution, variable line space grating spectrometer. The combined instrumental (monochromator and spectrometer) resolution ranged between about 0.3 eV for O K-RIXS and 0.5 eV for Ni L-RIXS.
Data availability
All data used in this manuscript are available from the authors upon reasonable request.
References
Palomares, V. et al. Na-ion batteries, recent advances and present challenges to become low cost energy storage systems. Energy Environ. Sci. 5, 5884–5901 (2012).
Xu, G. L. et al. Challenges in developing electrodes, electrolytes and diagnostics tools to understand and advance sodium-ion batteries. Adv. Energy Mater. 8, 1–63 (2018).
Yabuuchi, N., Kubota, K., Dahbi, M. & Komaba, S. Research development on sodium ion batteries. Chem. Rev. 114, 11636–11682 (2014).
Kim, S. W., Seo, D. H., Ma, X., Ceder, G. & Kang, K. Electrode materials for rechargeable sodium-ion batteries: potential alternatives to current lithium-ion batteries. Adv. Energy Mater. 2, 710–721 (2012).
Slater, M. D., Kim, D., Lee, E. & Johnson, C. S. Sodium-ion batteries. Adv. Funct. Mater. 23, 947–958 (2013).
Chen, X. et al. Stable layered P3/P2 Na0.66Co0.5Mn0.5O2 cathode material for sodium ion batteries. J. Mater. Chem. A 3, 20708–20714 (2015).
Xu, X., Ji, S., Gao, R. & Liu, J. Facile synthesis of P2-type Na0.4Mn0.54Co0.46O2 as high capacity cathode material for sodium ion batteries. RSC. Adv. 5, 51454–51460 (2015).
Sabi, N. et al. Layered P2-Na2/3Co1/2Ti1/2O2 as a high-performance cathode material for sodium-ion batteries. J. Power Sources 342, 998–1005 (2017).
Sabi, N. et al. Effect of titanium substitution in a P2-Na2/3Co0.95Ti0.05O2 cathode material on the structural and electrochemical properties. ACS Appl. Mater. Interfaces 9, 37778–37785 (2017).
Kim, D. et al. Layered Na[Ni1/3Fe1/3Mn1/3]O2 cathodes for Na-ion battery application. Electrochem. Commun. 18, 66–69 (2012).
Yuan, D. D. et al. P2-type Na0.67Mn0.65Fe0.2Ni0.15O2 cathode material with high-capacity for sodium-ion battery. Electrochim. Acta 116, 300–305 (2014).
Delmas, C., Braconnier, J.-J., Fouassier, C. & Hagenmuller, P. Electrochemical intercalation of sodium in NaxCoO2 bronzes. Solid State Ion. 3/4, 165–169 (1981).
Kubota, K., Yabuuchi, N., Yoshida, H., Dahbi, M. & Komaba, S. Layered oxides as positive electrode materials for Na-ion batteries. MRS Bull. 39, 416–422 (2014).
Martinez De Ilarduya, J., Otaegui, L., López del Amo, J. M., Armand, M. & Singh, G. NaN3 addition, a strategy to overcome the problem of sodium deficiency in P2-Na0.67[Fe0.5Mn0.5]O2 cathode for sodium-ion battery. J. Power Sources 337, 197–203 (2017).
Zhang, B. et al. Insertion compounds and composites made by ball milling for advanced sodium-ion batteries. Nat. Commun. 7, 10308 (2016).
Assat, G. & Tarascon, J. Fundamental understanding and practical challenges of anionic redox activity in Li-ion batteries. Nat. Energy 3, 373–386 (2018).
Luo, K. et al. Anion redox chemistry in the cobalt free 3d transition metal oxide intercalation electrode Li[Li0.2Ni0.2Mn0.6]O2. J. Am. Chem. Soc. 138, 11211–11218 (2016).
La Mantia, F., Rosciano, F., Tran, N. & Novák, P. Direct evidence of oxygen evolution from Li1+x(Ni1/3Mn1/3Co1/3)1-xO2 at high potentials. J. Appl. Electrochem. 38, 893–896 (2008).
Seo, D. et al. The structural and chemical origin of the oxygen redox activity in layered and cation-disordered Li-excess cathode materials. Nat. Chem. 8, 692–697 (2016).
Luo, K. et al. Charge-compensation in 3d-transition-metal-oxide intercalation cathodes through the generation of localized electron holes on oxygen. Nat. Chem. 8, 684–691 (2016).
Yabuuchi, N. et al. Detailed studies of a high-capacity electrode material forechargeable batteries Li2MnO3−LiCo13Ni13Mn13O2. J. Am. Chem. Soc. 133, 4404–4419 (2011).
Ma, C. et al. Exploring oxygen activity in the high energy P2-Type Na0.78Ni0.23Mn0.69O2 cathode material for Na-ion batteries. J. Am. Chem. Soc. 139, 4835–4845 (2017).
Maitra, U. et al. Oxygen redox chemistry without excess alkali-metal ions in Na2/3[Mg0.28Mn0.72]O2. Nat. Chem. 10, 288–295 (2018).
Bai, X. et al. Anionic redox activity in a newly Zn-Doped sodium layered oxide P2-Na 2/3 Mn 1 − y Zn y O 2 (0 < y < 0. 23). Adv. Energy Mater. 2, 1–12 (2018).
Doubaji, S., Valvo, M., Saadoune, I., Dahbi, M. & Edström, K. Synthesis and characterization of a new layered cathode material for sodium ion batteries. J. Power Sources 266, 275–281 (2014).
Jung, Y. H., Christiansen, A. S., Johnsen, R. E., Norby, P. & Kim, D. K. In situ X-ray diffraction studies on structural changes of a P2 layered material during electrochemical desodiation/sodiation. Adv. Funct. Mater. 25, 3227–3237 (2015).
Wang, P. F. et al. Suppressing the P2–O2 phase transition of Na0.67Mn0.67Ni0.33O2 by magnesium substitution for improved sodium-ion batteries. Angew. Chem. Int. Ed. 55, 7445–7449 (2016).
Yabuuchi, N. et al. P2-type Nax[Fe1/2Mn1/2[O2 made from earth-abundant elements for rechargeable Na batteries. Nat. Mater 11, 512–517 (2012).
Clément, R. J. et al. Structurally stable Mg-doped P2-Na2/3Mn1-YMgyO2 sodium-ion battery cathodes with high rate performance: insights from electrochemical, NMR and diffraction studies. Energy Environ. Sci. 9, 3240–3251 (2016).
Talaie, E., Duffort, V., Smith, H. L., Fultz, B. & Nazar, L. F. Structure of the high voltage phase of layered P2-Na2/3−z[Mn1/2Fe1/2]O2 and the positive effect of Ni substitution on its stability. Energy Environ. Sci. 8, 2512–2523 (2015).
Yabuuchi, N. et al. New O2/P2-type Li-excess layered manganese oxides as promising multi-functional electrode materials for rechargeable Li/Na batteries. Adv. Energy Mater. 4, 1301453 (2014).
Theivaprakasam, S. et al. Understanding the behavior of LiCoO2 cathodes at extended potentials in ionic liquid - alkyl carbonate hybrid electrolytes. J. Phys. Chem. 29, 15630–15638 (2017).
Lu, Z. & Dahn, J. R. In situ X-ray diffraction study of P2-Na2/3[Ni1/3Mn 2/3]O 2. J. Electrochem. Soc. 148, A1225 (2001).
Xu, J. et al. Identifying the critical role of Li substitution in P2-Nax[LiyNiyMn1-y-z]O2 (0 < x, y, z < 1) intercalation cathode materials of high-energy Na-ion batteries. Chem. Mater. 26, 1260–1269 (2014).
Asakura, D. et al. Material/element-dependent fluorescence-yield modes on soft X-ray absorption spectroscopy of cathode materials for Li-ion batteries. AIP Adv. 6, 1–8 (2016).
Yoon, W. S. et al. Oxygen contribution on Li-ion intercalation-deintercalation in LiCoO2 investigated by O K-edge and Co L-edge X-ray absorption spectroscopy. J. Phys. Chem. B 106, 2526–2532 (2002).
Oishi, M., Fujimoto, T., Takanashi, Y., Orikasa, Y. & Kawamura, A. Material for Li-ion batteries analyzed by a combination of hard and soft X-ray absorption near edge structure. J. Power Sources 222, 45–51 (2013).
Oishi, M. et al. Direct observation of reversible charge compensation by oxygen ion in Li-rich manganese layered oxide positive electrode material. J. Power Sources 276, 89–94 (2015).
Chen, C. et al. Soft X-ray absorption spectroscopy studies on the chemically delithiated commercial LiCoO2 cathode material. J. Power Sources 174, 938–943 (2007).
Yoon, W. et al. Oxygen contribution on Li-ion intercalation-deintercalation in LiAlyCo1-yO2 investigated by O K-edge and Co L-edge X-ray absorption spectroscopy. J. Electrochem. Soc. 149, 1305–1309 (2002).
Qinghao, Li et al. Quantitative probe of the transition metal redox in battery electrodes through soft x-ray absorption spectroscopy. J. Phys. D 49, 413003 (2016).
Lin, F. et al. Profiling the nanoscale gradient in stoichiometric layered cathode particles for lithium-ion batteries. Energy Environ. Sci. 7, 3077 (2014).
Yoon, W., Yoon, K., Mcbreen, J., Fischer, D. A. & Yang, X. Electronic structural changes of the electrochemically Li-ion deintercalated LiNi0.8Co0.15Al0.05O2 cathode material by X-ray absorption spectroscopy. J. Power Sources 174, 1015–1020 (2007).
Yoon, W.-S. et al. Investigation of the charge compensation mechanism on the electrode system by combination of soft and hard X-ray absorption spectroscopy. J. Am. Chem. Soc. 127, 17479–17487 (2005).
Kobayashi, S., Kottegoda, I. R. M., Uchimoto, Y. & Wakihara, M. XANES and EXAFS analysis of nano-size manganese dioxide as a cathode material for lithium-ion batteries. J. Mater. Chem. 14, 1843–1848 (2004).
Tamenori, Y., Ohashi, H., Ishiguro, E. & Ishikawa, T. Performance of a grating monochromator at BL27SU beamline of SPring-8 in the higher energy region. Rev. Sci. Instrum. 73, 1588–1590 (2004).
Acknowledgements
This work has been supported by the Swedish Research Council (contract 2017-05466), and the strategic research area StandUp for Energy. Mr. Marco Carboni is gratefully acknowledged for his help with the XPS data analyzing. XAS and RIXs radiation experiments were performed at the BL27SU of SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2019A1459). The authors thank K. Tsuruta for technical assistance at the beamline.
Author information
Authors and Affiliations
Contributions
C.H. performed the synthesis, XRD, XPS and electrochemical tests. N.S. contributed to the analysis of in situ XRD results with the assistance from I.S. L.A.M. and L.C.D. contributed to the measurement of XAS and RIXS spectroscopy. C.H., I.S., R.Y., and M.D. participated in the XPS data discussion. C.H., R.Y., and L.C.D participated in the XAS and RIXS data discussion. C.H. analyzed the data and wrote the manuscript with the help of D.B. and K.E.. I.S. leads the project.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hakim, C., Sabi, N., Ma, L.A. et al. Understanding the redox process upon electrochemical cycling of the P2-Na0.78Co1/2Mn1/3Ni1/6O2 electrode material for sodium-ion batteries. Commun Chem 3, 9 (2020). https://doi.org/10.1038/s42004-020-0257-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42004-020-0257-6
This article is cited by
-
Surfactant-assisted synthesis of Na3V2(PO4)3 cathode material with a 3D carbon network for Na storage
Ionics (2023)
-
Layered NaxCoO2-based cathodes for advanced Na-ion batteries: review on challenges and advancements
Ionics (2021)
-
One-pot fabrication of pitch-derived soft carbon with hierarchical porous structure and rich sp2 carbon for sodium-ion battery
Journal of Materials Science: Materials in Electronics (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
