
Abstract
Due to its complex and multifaceted nature, developing effective treatments for epilepsy is still a major challenge. To deal with this complexity we introduce the concept of degeneracy to the field of epilepsy research: the ability of disparate elements to cause an analogous function or malfunction. Here, we review examples of epilepsy-related degeneracy at multiple levels of brain organisation, ranging from the cellular to the network and systems level. Based on these insights, we outline new multiscale and population modelling approaches to disentangle the complex web of interactions underlying epilepsy and to design personalised multitarget therapies.
Similar content being viewed by others

Introduction
Degeneracy, “the ability of elements that are structurally different to perform the same function or yield the same output”1, is a general principle, present in most complex adaptive biological systems1,2. Degeneracy (sometimes also called ’non-uniqueness’3) should be distinguished from redundancy, which results from multiple identical elements performing the same function1,4,5,6, but see also ref. 7. In contrast to redundant components, degenerate components may generate dissimilar outputs in different contexts2,8. Therefore, although degeneracy is sometimes called also ‘partial redundancy’9, we follow the terminology of Edelman and Gally1 and distinguish degeneracy from redundancy.
The ubiquity of degeneracy is rooted in the advantages it entails for organisms’ evolvability9,10 and robustness11,12,13. Degeneracy allows organisms to satisfy the two seemingly contradictory goals of preserving already evolved functions and concurrently searching for and evolving new functions10. In this way degeneracy is linked to both robustness and innovation in evolution1,10.
It is becoming increasingly clear that the brain also exhibits degeneracy1,4,14,15,16,17, with similar physiological states arising from a multitude of different subcellular, cellular and synaptic mechanisms18,19, for recent reviews see refs. 6,20,21. However, not only similar physiological but also similar pathological brain states may arise from structurally disparate mechanisms6,15,22,23. Therefore, the existence of degeneracy in the brain has important implications for understanding and treating complex brain disorders such as epilepsy.
Despite considerable progress in epilepsy research, reviewed by24,25,26, its complex multicausal and variable nature27,28,29,30 has made it difficult to disentangle the underlying mechanisms. This has hindered the development of effective therapies. Apparently contradictory observations still fuel deep controversies about the origin of epilepsy. In this review, we argue that competing hypotheses can be reconciled by taking into account the concept of degeneracy. Moreover, we emphasize that degeneracy can help explain why multiple minor changes can sometimes induce pathological phenotypes, while at other times their effects cancel, preserving healthy/physiological circuit states.
Our aim is to review examples for epilepsy-related degeneracy at three different levels of brain organization: the cellular, network and systems level. We propose that epilepsy is a group of multiscale disorders with a potential involvement of degenerate mechanisms at each level (see Fig. 1).

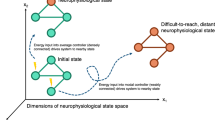
a In this article we exemplify degeneracy in epilepsy across the cellular, network and system level. Levels and sub-components are highlighted. This list reflects the organisation and scope of the article and is not intended to be exhaustive. There are likely to be other levels of organisation that express degeneracy in the context of epilepsy. b The concept of degeneracy, e.g. different changes leading to a similar outcome, can be visualized by a many-to-one relationship between the parametric and the functional space. Diverse changes in the parametric space can lead to a similar pathological outcome in the functional space. Green/violet spheres represent the healthy/pathological case in functional space and in the corresponding parametric space. Black thin arrows symbolise the many-to-one relationship, violet arrows the corresponding pathological transition. c Epilepsy is often multicausal: Pathological changes at the cellular, network and system levels can interact across multiple brain regions. d Thus, similar pathology indices in multiple animals can be caused by degenerate modifications of multiple properties across various levels. Some elements of a and c were adapted from287, published under CC BY license http://creativecommons.org/licenses/by/4.0/.
Degeneracy in epilepsy at the cellular level
Intrinsic properties and ion channel degeneracy
It is now well established that indistinguishable single-cell and network activity can result from a variety of parameter combinations of different ion channels18,31,32. This phenomenon has been termed ion channel degeneracy33,34; for recent reviews see refs. 21,23. Ion channel degeneracy means that “distinct channel types overlap in their biophysical properties and can thus contribute collectively to specific physiological phenotypes”23.
There have been extensive studies of epilepsy-relevant changes in ion channels35,36,37. However, their role in the context of ion channel degeneracy and epilepsy has not yet been studied extensively. Neuronal hyperexcitability and associated epilepsy often arise as a result of combinatorial effects of multiple ion channel mutations. This has been demonstrated by an important sequencing study by Klassen et al., examining over 200 ion channel genes in epilepsy patients and healthy controls38. The study found a highly complex pattern of gene changes in single individuals. Even multiple nonsynonymous mutations in known monogenic risk genes for epilepsy were frequently identified in healthy individuals38 (see Fig. 2a). This suggests that, due to ion channel degeneracy, different channel types are able to partially compensate each other’s defects23. When a mutation affects a function of a given ion channel, other channels may rescue normal excitability23 (see Fig. 2b). This applies both to loss-of-function as well as gain-of-function changes in ion channels. Accordingly, individuals with epilepsy typically have more than one mutation in human epilepsy genes38.

a Genetic analysis reveals that both healthy and pathological individuals commonly carry multiple mutations in the 17 known ion channel risk genes for familial human epilepsy. Data reproduced from ref. 38. b To illustrate why the combined effect of multiple mutations may sometimes, but not always, be detrimental, we create a hypothetical scenario with three voltage-gated ion channels, which are all equally effective. Let us assume, that a healthy state requires that the peak of the combined activation remains in a certain voltage band, green zone. In contrast, if the peak of the combined activation is shifted to a lower or higher voltage a pathological situation may occur, violet zone. In the upper row, all three channels are present. Due to their symmetric activation profiles, the combined activation will be strongest at the center, dashed line, and thus remains in the healthy zone. If both channel 2 and channel 3 are lost, middle row, peak activation shifts to the left and pathology ensues. In contrast, if channel 1 and channel 3 are deleted, lower row, the healthy state is maintained. Modified from ref. 21. c The combined effect of multiple mutations depends on their specific trajectory in the abstract functional space: Multiple mutations can be neutral if the function simply remains in the healthy zone, upper left green trajectory, or if a detrimental mutation is compensated by a second mutation, right green trajectory. Mutations can be pathological, if they together cause the functional state to leave the healthy zone, violet trajectory.
Similarly, clinical genetic testing in patients with epilepsies that are considered to be genetic (e.g., childhood absence epilepsy or childhood epilepsy with centrotemporal spikes) often fails to detect mutations in known disease-causing genes, despite an intensive search for monogenic causes in affected families. This suggests polygenic inheritance of these classic genetic epilepsies. The difficulty in finding the contributing genes in these patients is probably related to the multicausal nature of their epilepsy.
The results of Klassen et al.38 indicate that epilepsies are more often caused by an accumulation of mutations in multiple ion channels than by mutation in one particular ion channel39.
This is supported also by computational modelling showing that combinations of small changes in the voltage activation of two or three ion channels can lead to network hyperexcitability40. The degeneracy of ion channels may also explain why in some individuals a lower number of mutations or hits leads to a pathological epilepsy phenotype, while other individuals with a higher number of hits appear unaffected23. The net effect of combined mutations will depend, first, on the initial, pre-mutation distance of a given individual (i.e. a given set of ion channel parameters) from the pathological hyperexcitable region of the possible functional space. Second, the net effect will depend on the specific trajectory in functional space determined by single or multiple mutations (Fig. 2c; see also Fig. 4). Multiple mutations in ion channels can sometimes cancel each other’s effects, paradoxically preserving physiological firing behavior of affected neurons23. In line with this, the degeneracy between ion channel types leads to high-dimensional parameter spaces, which support robustness21,23,41,42.
There is indirect evidence that degeneracy of dendritic ion channels is involved in epilepsy. The way in which mutations in ion channels affect the excitability of neurons is complex. This is because excitability depends not only on ion channels in the axon or the soma, but also on dendritic channels. The interaction between dendritic and somatic channels can change the way neurons respond to stimuli37,43,44,45,46,47,48. Mutations in some dendritic ion channels, such as A-type potassium and HCN channels, have been associated with increased excitability; however, their effects vary depending on cell type and brain region35,49,50,51,52,53,54,55,56,57,58,59. Nevertheless, experimental evidence clearly points to a significant involvement of dendritic ion channel alterations in acquired or genetic epilepsy35,37,51,57,58 or in epilepsy-related comorbidities such as memory impairment60.
In parallel, there is a second, epilepsy-unrelated but complementary line of direct evidence for the manifestation of ion-channel degeneracy in the emergence of characteristic somato-dendritic properties that shape normal dendritic and somatic excitability of neurons. In several mammalian principal neurons, multiple intraneuronal functional maps61 co-exist on the same electrotonically non-compact morphology. The question is therefore whether these multiple spatial constraints on function, imposed on the same neuronal morphology, would limit the expression of degeneracy in these neurons. However, several lines of evidence, spanning different neuronal subtypes, demonstrate the emergence of similar somatodendritic functional properties despite widespread heterogeneity in the underlying ion-channel expression profiles and gating properties20,21,48,62,63,64,65,66,67. Thus, there are independent lines of evidence for different ion channels mediating epilepsy-associated changes to somato-dendritic excitability, and for how different combinations of ion channels might yield similar somato-dendritic functional maps. However, the convergence of these two lines of research directly investigating degeneracy in the manifestation of pathological excitability states of neuronal dendrites has been much less explored.
Nevertheless, although epilepsy research has not directly and explicitly addressed ion channel degeneracy, there is an increasing amount of indirect and implicit experimental evidence.
For example, a recent study has suggested that neural circuits express degeneracy and robustness in circuit excitability due to heterogeneous components68. The authors have shown that cortical neurons extracted from epileptic tissue respond more homogeneously to external stimulation than cells from healthy controls68. This is due to both a reduced variability in the distance between resting membrane potential and spiking threshold and a reduced dynamic range of the FI response curve. Surprisingly, neurons from epileptogenic tissue required more input to generate a low number of spikes and showed a reduced increase in firing rate for additional input. Thus, counter-intuitively the principal neurons from epileptogenic tissue were less excitable than neurons from control tissue. This suggests that hyperexcitability of individual neurons may not be necessary to induce the hyperexcitable seizure state. Compare, for example, a right-shifted FI curve in Cunha et al.69 with a left-shifted one in Whitebirch et al.70. Importantly, computer simulations of Rich et al.68 indicated that the reduced variability in excitability could make neuronal networks more susceptible to generating synchronous epilepsy-like discharges. While it is not clear whether the reduced variability here is cause or effect of the epilepsy, these observations fit well with the concept of degeneracy. By definition, degeneracy requires heterogeneous components. Thus, reduced variability limits the ability of the system to adapt and maintain healthy function71 and may underlie an epileptogenic transition.
Further, neuronal recordings in animal models and in the tissue of epileptic patients have generated additional data on the diversity and degeneracy of ion channel changes associated with epilepsy36,37. This is the case for different cell types. For example, hippocampal CA1 pyramidal cells show changes in multiple ion channel types, including upregulation of T-type calcium channels50,72,73, downregulation of A-type potassium channels51, and HCN channels53. These changes are thought to lead to increased excitability36 in the form of enhanced firing and bursting74. In addition, gain-of-function mutations in the sodium channels have been identified as a cause of hyperexcitability in CA1 pyramidal neurons75.
Similarly, changes in multiple ion channel types of neocortical pyramidal cells have been observed during epilepsy and some of them identified as a cause of pathologically increased excitability. These include epilepsy-associated changes in KCNQ2 potassium channels76,77, BK potassium channels78, HCN cation channels79,80,81, and Nav1.6 sodium channels82,83.
Viewed together, these studies suggest that there are multiple different routes towards hyperexcitability and its prevention or reversal in hippocampal and neocortical pyramidal neurons. Ion channel degeneracy implies that the effect of changes in one channel type will depend on the biophysical context, i.e. on the activation other intrinsic or synaptic channels in a given neuron. Indeed, for example, depending on their synaptic and intrinsic context, sodium84, A-type potassium33,85, SK37,86 and HCN channels87,88,89,90 can contribute to a suppression or an enhancement of neuronal spiking. Therefore, predicting how a specific channel alteration affects neuronal excitability will require detailed models and experimental analyses of ion channel degeneracy. We would like to encourage future studies on the role of ion channel degeneracy in epilepsy. We believe it is a promising direction for epilepsy research. In pain research, for example, recent studies have directly shown that different configurations of ion channels can lead to the hyperexcitability that underlies chronic pain15,91,92.
Morphological properties
In response to epileptogenic changes in the circuitry, neurons change the structure of their dendritic trees. Altered dendritic branching has been reported in different animal models of epilepsy93,94,95,96. The impact of such epilepsy-related dendritic remodelling on neuronal function is poorly understood.
Computational and experimental studies have shown that morphological changes of dendrites are able to affect neuronal excitability97,98,99. Even if one keeps electrotonic properties in neuronal models unchanged, spiking behavior of neurons changes strongly depending on their size and topology97,98,100,101,102. This is the case for spiking behavior driven by somatic current injections.
In contrast, the situation may be different, if spikes are triggered by distributed synaptic inputs instead of somatic current injections. In such synaptic stimulation scenarios, spike rates, but not spike patterns, are largely independent of dendritic size and topology, provided synaptic density is preserved103. This has been generalized to different cell types as a universal dendritic constancy principle103. In a degeneracy-like manner, different morphological shapes help make firing rates, but not patterns, more similar.
Likewise, with active dendritic trees, diversity in neuronal morphologies has been found to be a sloppy parameter, that is, a parameter with little influence104, in models of hippocampal CA1 pyramidal cells driven by distributed synapses65. CA1 pyramidal cell models remained functionally similar despite the cell-to-cell variability in dendrites because the variability was compensated by ion channel degeneracy and synaptic democracy65. Synaptic democracy describes a compensatory mechanism, which ensures the somatic impact remains the same irrespective of the synapse’ location on the dendritic tree105. On the other hand, in contrast to spike rate, spiking pattern, for instance the presence of bursting in healthy98,103 or epileptic cells74, depends strongly on dendrite morphology and intrinsic as well as synaptic ion channels.
Overall, these studies imply that the net impact of dendritic changes in epileptic tissue depends on the interplay between morphological, synaptic and nonsynaptic ion channel changes. Indeed, this has been supported by computational modelling of epilepsy-related dendrite changes in adult-born dentate granule cells. The models have shown that isolated morphological changes, observed in epileptic animals, made neurons less excitable106. However, when placed in a network context with pathologically altered excitatory inputs, for example in the form of synaptic sprouting and synapse loss, altered morphologies either did not change network hyperexcitability or enhanced it107. Taken together, the degeneracy of morphological and biophysical parameters makes it unlikely that morphological changes of dendrites can monocausally explain epileptic hyperexcitability or its reversal.
Degeneracy in epilepsy at the network level
Degenerate synaptic and intrinsic properties
Degeneracy can be found not only at the single cell level but also at the network level of synaptically connected neurons. Analogous to multiple configurations of intrinsic ion channels leading to indistinguishable electrical behavior of single neurons108, multiple configurations of synaptic properties can support indistinguishable electrical behavior of neuronal networks18,31,85,109,110. In addition, circuit degeneracy is enhanced by the fact that intrinsic and synaptic channels can compensate for each other18,31,111. This further increases the parameter space of indistinguishable network activity. Hence both extrinsic (synaptic) as well as intrinsic properties belong to important degenerate parameters of the circuit112. Therefore, in principle, synaptic mechanisms can compensate for the impaired function of intrinsic ion channels and vice versa31,111, thus increasing the robustness of the nervous system23 by counterbalancing changes in overall synaptic drive and intrinsic excitability113.
Degeneracy of synaptic and intrinsic properties is directly relevant in the context of epilepsy. For example, degenerate synaptic and intrinsic properties are important for the spatially and temporally sparse firing of dentate granule cells34,42. The sparsely active dentate gyrus is considered a protective gate for the spread of epilepsy-related hyperexcitability in the hippocampus114,115,116. The degeneracy hypothesis predicts that effective protection of the dentate gate is supported by degenerate intrinsic and synaptic mechanisms mediating default but also inducible homeostatic maintenance of firing behavior of dentate granule cells. If one set of protective mechanisms was impaired, the other set would compensate and keep the gate intact.
There exists ample experimental evidence for degenerate intrinsic and synaptic mechanisms to protect the dentate gate117. It is well established that even strong excitatory input from the entorhinal cortex leads only to limited activity in dentate granule cells. Their relative inertia to perforant path input is partly due to specific intrinsic biophysical properties such as hyperpolarized resting potential, spike rate adaptation and a low expression of active channels in dendrites117,118. Moreover, dentate granule cells have been reported to upregulate their leak (Kir, Kv1.1) and HCN channels (but see also below the discussion of the complexity of HCN channel changes) and thereby further decrease their excitability in temporal lobe epilepsy (TLE)119,120,121,122,123. This can be seen as an antiepileptic compensatory reaction36 to proepileptic changes such as the enhancement of main excitatory perforant path synapses of dentate granule cells124. This suggests that a compensatory recruitment of three different ion channels can support robust maintenance of firing rate homeostasis, which would otherwise be impaired by synaptic pathology in epileptic tissue. This might partially explain the robustness of dentate granule cells against excitotoxic cell death in hippocampal sclerosis, which is associated with TLE36. Intriguingly, ion channels that get upregulated in dentate granule cells in epilepsy might be suitable candidates for pharmacological or genetic antiepileptic treatment36,125.
In addition to intrinsic channels, extrinsic network mechanisms also contribute to the protection of the dentate gate. For example synaptic, phasic as well as extrasynaptic, tonic GABAergic inhibition is known to be exceptionally strong in the dentate gyrus126. Moreover, even synaptic inhibition itself is supported by structurally diverse mechanisms. Specifically, the existence of a stunning diversity in GABAergic interneurons of the dentate gyrus127,128,129,130,131 suggests significant degeneracy in maintaining synaptic dendritic and somatic inhibition controlling the recruitment of dentate granule cells132. And if, despite its degeneracy and robustness, synaptic inhibition becomes impaired, tonic extrasynaptic inhibition can still provide some protection. The evidence shows namely that extrasynaptic inhibition remains preserved or even becomes enhanced in some animal models of epilepsy133,134. Hence, the increase in extrasynaptic inhibition in case of impaired synaptic inhibition suggests that degenerate mechanisms protect the dentate gate at the network level.
Degenerate protection mechanisms allow for multiple routes to failure. Because multiple mechanisms protect the dentate gate, there are different ways to make it fail. This is reflected in the variety of animal models of epilepsy, ranging from kindling or status-epilepticus models induced by pilocarpine, kainate or electrical stimulation117,135 to traumatic brain injury (TBI) models136. Stereotypical changes in the dentate gyrus network in all these different animal models of TLE have been well characterized137. They include loss of mossy cells and hilar GABAergic neurons, sprouting of inhibitory synapses and changes in GABA currents and their reversal potentials, increased recurrent excitation (due to mossy fiber sprouting), enhanced adult neurogenesis and astrocytic gliosis117,135,136,138,139,140.
However, despite similarities between distinct animal models of TLE, there are also differences that are still not well understood. For example, intrinsic properties of granule cells are largely unaltered in TBI models136 in contrast to intrahippocampal kainate injection models121. Also changes in dendritic and somatic GABAergic synaptic inhibition are complex and difficult to interpret139. This is further complicated by depolarizing shifts in GABA reversal potentials caused by chloride dysregulation, reviewed in ref. 141. We believe that future research guided by the degeneracy framework might help reconcile some controversies regarding seemingly conflicting findings of intact142, reduced143,144, enhanced145 or first reduced and later enhanced146 GABAergic inhibition in TLE. The concept of degeneracy could help elucidate how changes in inhibition in early and later stages of epileptogenesis interact with changes in excitation (including mossy fibre sprouting), cell loss and neurogenesis. The challenge is to determine, which of these changes are adaptive or maladaptive117.
In early epileptogenesis induced by TBI (first days to weeks upon injury), an integrated picture starts emerging for synaptic and cellular changes in the dentate circuitry136. Interestingly, this picture seems to include degeneracy at the molecular, cellular and network levels. For example, convergent mTOR, TLR4 and VEGF receptor signaling has been found to be involved in synaptic changes, that is, enhanced excitatory AMPA currents, altered inhibition, chloride pump changes and mossy fiber sprouting, as well as in cellular changes, that is, cell loss, neurogenesis and astrogliosis (see also a recent review on neuroinflammatory cellular components of TBI-induced epileptogenesis in ref. 147). At the network level, these synaptic and cellular changes drive jointly dentate gate disruption and contribute to epileptogenesis136. The details still need to be clarified but multiscale network models of the dentate gyrus emerge as a promising tool to reveal the conditions and limits under which these synaptic and cellular changes are adaptive or maladaptive24,148,149,150. Their combination with multiscale experimental approaches will help resolve controversies concerning the question to what extent do altered excitation124 and inhibition139, neurogenesis151,152, mossy fiber sprouting153,154,155 or mossy cell loss156,157,158 contribute to dentate hyperexcitability and epileptogenesis.
The framework of degeneracy may help reconcile the ‘impaired inhibition’ with the ‘enhanced excitation’ hypotheses not only in TLE but also for example in Dravet syndrome. A Dravet-like epileptic phenotype can arise as a consequence of a Nav1.1 sodium channel deletion in GABAergic interneurons. Such an interneuron-specific Nav1.1 deletion reduces GABAergic inhibition due to impaired firing of GABAergic interneurons and is sufficient to cause Dravet-like seizures159,160,161,162; but see also a surprising robustness of CA1 pyramidal cells in ref. 163. However, in line with the concept of multiple degenerate routes toward epilepsy, the Dravet-like phenotype can also result from enhanced excitability of excitatory neurons164,165 or enhanced synaptic excitation166 in spite of mostly intact GABAergic inhibition.
Consequently, the efficiency of personalized therapy for Dravet syndrome may depend to some extent on the underlying circuit changes, which may be cell type specific59. In patients with deficient sodium channels in GABAergic inhibitory neurons, a pharmacological block of sodium channels would be ineffective or even counterproductive37,167, whereas in patients with hyperactive sodium channels in excitatory neurons it may be an effective therapy. Indeed, clinicians usually avoid giving sodium channel blockers to Dravet patients with a loss of function mutation in the SCN1A sodium channel subunit. This is also the case for those with a loss-of-function mutation in the SCN2A subunit, whereas patients with a gain-of-function mutation in the SCN2A subunit usually benefit greatly from sodium channel blockers168,169; see also ref. 59 for the cell type dependence of the loss/gain-of-function concepts. Similarly, in TLE patients with chloride dysregulation, leading to excitatory GABA reversal potentials, enhancement of GABAergic signaling (e.g., by benzodiazepines) might worsen rather then suppress seizures, despite pathologically diminished GABA-A conductances170, see also ref. 171. Thus, by drawing our attention to multiple causal links and their context, degeneracy can account for therapy failures and suggest new therapy approaches.
Degeneracy may help to explain counter-intuitive observations about inhibition and excitation not only in epileptogenesis, but also in ictogenesis. For example, a GABAergic initiation hypothesis has recently been proposed172 to account for increased interneuronal activity prior to seizure onset173,174,175. This is an example of another potential route to seizures outside the typical context of excitation-inhibition imbalance. It also illustrates that although hyperexcitability of principal neurons may be a key feature of seizure-prone circuits, it is not the only pathological change leading to seizures and epilepsy. As noted above, individual excitatory neurons may not be hyperexcitable as judged from their FI curves68 and population responses may be preceded by strong inhibition. Thus, single-neuron hyperexcitability should be distinguished from circuit hyperexcitability underlying seizures. Nevertheless, on average, excitatory and inhibitory cell activity is higher during a seizure than in the interictal state175.
Similar seizure dynamics despite diverse biophysical mechanisms
There is an astounding variety of causes and features of epileptic seizures. However, surprisingly, computational analysis has revealed that seizure dynamics display certain invariant properties. Dynamical systems analysis of transitions occurring at seizure onset and offset showed that most seizures can be classified into one of 16 dynamotypes176,177,178. These dynamotypes, composed as a combination of 4 onset and 4 offset bifurcations, characterise seizure dynamics based on measurable local field potentials, with certain dynamotypes occurring more frequently and some occurring in combination178. The existence of multiple dynamotypes is a prime example of the degenerate nature of seizure dynamics.
Unexpectedly, similar dynamics were found in different brain regions and in three different species including human, zebrafish and mouse176. A combination of experiments and dynamical modelling in the study by Jirsa et al.176 revealed that seizure dynamics share general fundamental properties179, described in more detail below. However, this does not mean that biophysical mechanisms of seizure generation are identical across distinct brain regions and species. On the contrary, it is likely that multiple different molecular, cellular and circuit mechanisms are capable of generating the observed seizure dynamics. Therefore, the authors have proposed that “there exists a wide array of possible biophysical mechanisms for seizure genesis, while preserving central invariant properties”176. This proposal is in agreement with the concept of degeneracy in epilepsy.
The corresponding dynamical model, called Epileptor, captures invariant dynamical properties of seizure events using only five state variables176, for a critical discussion see ref. 180. A slow variable, the permittivity variable181, determines the emergence of seizure onsets and offsets. Several different biophysical mechanisms could underlie this slow variable such as accumulation of extracellular potassium182,183,184 or changes in energy metabolism176. Jirsa et al.176 experimentally showed that potassium accumulation and metabolic changes (oxygen and ATP consumption) were indeed correlated with the slow onset of seizure-like events. However, other slow processes, operating on a similar time scale, e.g. short-term synaptic depression185 or concentration changes of other ion species such as chloride184 might also contribute to the biophysical implementation of the slow permittivity variable.
This is the case also for the remaining four faster variables. Based on electrophysiological recordings, the authors have linked the first state variable of the Epileptor to excitatory glutamatergic synaptic activity and the second state variable to inhibitory GABAergic activity176. Nevertheless, they showed in their own experiments that invariant dynamics of seizure-like events remain preserved even after dramatic changes of conditions such as disrupting synaptic release. This is another example showing that the Epileptor state variables, which underlie the invariant properties of simulated seizures, can be instantiated by different underlying physiological mechanisms. In agreement with this interpretation and fully in the spirit of degeneracy, the authors concluded that the above mentioned physiological correlates of the five state variables (potassium, ATP, glutamatergic and GABAergic activity) “may be only valid for (…) very specific experimental conditions” and emphasized that many other parameter configurations and trajectories could lead to the conserved system dynamics and behaviour176, see also ref. 171).
Similar LFP and EEG discharges despite diversity of underlying mechanisms
An important example for degeneracy in epilepsy at the border between macro- and micro-circuit scales is related to extracellular electrical recordings, including local field potentials (LFP), electrocorticogram (ECoG), and electroencephalogram (EEG). These extracellular recordings constitute a crucial tool to discern physiological and pathological patterns of network activity at different spatial scales186,187,188,189,190,191,192,193,194.
With specific reference to LFPs, it is clear from the analyses above that epileptic disorders are associated with changes in ion channel densities and synchrony or correlation in activity patterns. LFP and its frequency-dependent characteristics are critically reliant on single-neuron properties and on input correlations190,194,195,196,197,198. Together these imply that changes in LFP recordings are expected with epileptic disorders, which have indeed been shown across different brain regions174,199,200,201,202,203,204,205,206,207. Importantly, these observations also imply that the interpretations and analyses of extracellular recordings, their frequency-dependent properties, and spatial spread must account for pathological changes in ion channel distributions and input synchrony.
There are several lines of support for degeneracy in the generation of similar LFP and EEG patterns20,171,194,208,209,210. For instance, although EEG can be macroscopically similar across different subjects, the firing patterns and the ion-channel compositions of the distinct neurons can still be very different. In addition, LFP oscillations in the theta frequency range, which are observed in the hippocampus during exploratory behavior, have been linked both to intrinsic intra-hippocampal activation211,212,213,214,215,216 as well as to the activation of several afferent areas including entorhinal cortex217 and the medial septal-diagonal band of Broca211,212,214,216. Similar observations have been reported about the differential origins of gamma oscillations as well216,218,219. Thus, physiological LFP oscillations might emerge due to intra-regional but also inter-regional circuit mechanisms.
Degeneracy suggests that not only physiological patterns of activity such as theta and gamma oscillations but also pathological patterns might emerge due to multiple combinations of disparate mechanisms. For instance, hypersynchronous seizure-associated oscillations can arise from altered activity in several brain regions due to changes in disparate network components such as excitatory and inhibitory transmission or cell-specific intrinsic properties20,171,194,208,209,210.
Epileptologists are becoming increasingly aware of the fact that similar macroscopic activity (as measured e.g., by EEG) can be brought about by different underlying microscopic activities. As suggested in a recent review on micro-macro relationships in seizure networks, there are possible, although not yet experimentally shown, scenarios in which similar EEG in different patients could emerge from different activities of distinct hippocampal cell types208. If validated in experiments, this would be an important lesson teaching us that “ostensibly similar epilepsy expression at the macroscopic scale can originate from a variety of mechanisms at the microscopic scale”208. In a similar vein, the authors of a recent study on “divergent paths to seizure-like events” concluded that “ostensibly similar pathological discharges can arise from different sources”171.
The expression of such degeneracy, and the consequent macro-micro disconnect has critical implications for how extracellular recordings are interpreted and how therapeutic targets are designed171,208,210,220. Failure to recognize degeneracy or the manifestation of the many-to-one mappings between neural circuit components and extracellular recordings might result in negative side effects of drugs that are ineffectual as well208,220. Thus, analyses and interpretation of extracellular potentials must carefully account for the manifestation of considerable heterogeneity in the parametric space of neural circuits expressing degeneracy in the generation of these potentials. Basic research at the microcircuit and cellular scales is essential to elucidate the complex one-to-many mapping between similar electrographic recordings of seizures and distinct local network mechanisms171,208,210,220.
Together, we emphasize the need to recognize degeneracy in the emergence of disrupted extracellular signatures associated with epilepsy and the divergent paths to similar seizure events. These observations caution against extrapolation of similarities in extracellular signatures at the macroscopic scale to imply similarities of sources and mechanisms at the microscopic scale, involving disparate combinations of cellular and synaptic components171,208,209,210,220.
Degeneracy in epileptogenesis at the systems level
Similarly to the network and single-cell levels, also at the systems level, many interacting components are known to contribute to the development of epilepsy. Here we highlight the immune system, the blood brain barrier and their interactions with neural circuits.
Data from clinical and animal model studies show that similar epilepsy phenotypes can be associated with distinct underlying neuronal and inflammatory mechanisms. For example, seizures are accompanied by neuronal death in some221 but not in other222 animal models of epilepsy. This has led to controversies and questions whether seizures cause neuronal loss, and whether neuronal death is necessary and/or sufficient for triggering epileptogenesis223. Current evidence indicates that traumatic brain injury and associated neuronal death224 are capable of triggering epilepsy development in humans225 and model animals226. However, other experimental evidence suggests that epileptogenesis is possible also without apparent signs of neuronal loss when triggered by BBB breakdown222. Thus, neuronal loss seems sufficient but not necessary for triggering epileptogenesis. This can be understood by looking at epileptogenesis in the context of degeneracy.
One origin of degeneracy in epileptogenesis (Fig. 3) lies in the complexity of neuroimmunity227 and its crosstalk to multiple processes in the central nervous system228. Recent studies emphasize the key role of the immune system in the development of epilepsy147,228,229,230. This is unsurprising given that nervous and innate immune systems have close developmental trajectories with extensive mechanistic overlap231.

a Complex interactions at the system level illustrate why multiple distinct pathomechanisms may be sufficient but not necessary for epileptogenesis. b Experiments and simulations243 show that different causes, such as blood-brain barrier (BBB) disruption or neuroinflammation, can lead to a similar epileptic outcome.
The interconnectedness of nervous and immune systems can be illustrated with functions of microglia, the major player of brain innate immunity232,233. This cell type is involved not only in neuroinflammation, but also in inhibiting neuronal excitability in an activity-dependent fashion234. Furthermore, microglia regulate the secretion of, among others, IL-1 and TNF that are known to modulate neuronal excitability and seizure threshold228,229,235,236. Moreover, microglia are only one among a growing list of neuroimmunity-associated players involved in epileptogenesis.
Glial activity and the associated spectrum of neuroinflammatory reactions have been shown to be in close interplay with blood-brain barrier (BBB) permeability237, neuronal activity234,235, neuronal loss223 and network reorganization 238. Additional complexity is introduced by the fact that neuroimmune responses can be triggered not only by neurological injury, but also by downstream pathological changes which are characteristic of epilepsy and the ictal activity itself. For instance, enhanced BBB permeability was shown to be induced by epileptic seizures239,240. And vice versa, the spillover of brain-born substances, following BBB leakage, can cause secondary neuroinflammation241. In this way, seizures may develop via multiple mechanisms including BBB disruption, neuroinflammation and/or neuronal loss and network remodelling (Fig. 3).
We have recently created a phenomenological model of these major epileptogenic processes and their interactions at realistic time scales (Fig. 3)242. We have described the neuro-immune crosstalk in the context of neurological injury using a dynamical systems approach. In agreement with experimental data from three animal models221,222,243,244,245, simulations showed that neuronal loss can be sufficient but is not necessary to drive epileptogenesis. Overall, computational modelling supports the concept that in the brain with its degenerate mechanisms, multiple different pathological mechanisms may contribute to epileptogenesis requiring different kinds of interventions for successful treatment242. However, there are still many unanswered questions related to cell loss. For example, the loss of certain interneuron subtypes246 and the associated reduced inhibition and epilepsy247 has not yet been fully understood and compared with the loss of principal neurons in the context of epileptogenesis.
Multiscale and population modelling of degenerate circuits and epilepsy
Degeneracy is a multiscale phenomenon present at multiple levels of brain structure. Degeneracy implies that different processes on a lower level can lead to a certain phenomenon on a higher level. If phenomena on different scales interact, multiscale modelling becomes necessary. Therefore, deeper understanding of degenerate excitable systems requires multiscale computational approaches248. Creating models of neural circuits that can bridge two or more scales is a challenging task because of the nested hierarchy of molecular, cellular and supracellular networks with many feedforward and feedback loops between the scales. However, in well studied systems, multiscale models can provide new insights, which are relevant for epilepsy in the context of degeneracy.
A simulation study of the thalamo-cortical network in childhood absence epilepsy249 is an intriguing example of multiscale modelling connecting the ion channel, cellular and microcircuit scales. The model showed that enhancing T-type calcium channel activation or reducing inhibitory GABA-A synaptic channel activation—in isolation as well as in combination—converted physiological network activity to seizure-like discharges. The results were in agreement with clinical observations of multiple different mutations in GABA-A receptors and T-type calcium channels in human patients and animals. The model predicted that these different mutations can lead to the same phenotype of absence epilepsy. In this way, the simulations have linked individual genetic variability in patients, simulated as the variability in the parameters of GABA-A and T-type calcium channels, to childhood absence epilepsy. Moreover, the model suggested plausible explanations for the failure or success of pharmacological medications targeting GABA-A and/or T-type calcium channels. Importantly, modelling predicted the necessity of multitarget therapy, simultaneously enhancing GABA-A transmission and suppressing T-type calcium channel activation, for patients with mutations in both ion channels (see also Fig. 4). Furthermore, these simulations highlight the need for personalized epilepsy therapy in patients with different genetic backgrounds. Notably, in line with the degeneracy and multicausal pathogenesis, the alterations in GABA-A and T channels do not lead to monogenic epilepsies, and therefore clinical testing does not typically reveal abnormalities in these genes in patients with childhood absence epilepsy.

Epileptic hyperexcitability or restoration of normal excitability can be achieved by individual or combined changes in ion channels. The transition between normal excitation (green) and pathological hyperexcitability (violet) occurs when a tipping line is crossed. This transition can be induced for example by variation of sodium (horizontal axis) and potassium (vertical axis) conductance. Decrease in potassium conductance (a), increase in sodium conductance (c), or combining both (b) induces a transition from normal to pathological firing behavior of neurons. Increasing both conductances (d) maintains normal excitability. Conversely, reversal of epileptic hyperexcitability can be achieved by increasing potassium conductance (e), decreasing sodium conductance (f), or applying both changes simultaneously (g). Combined modification (g) moves the system farther away from the dangerous tipping point than isolated modification (e) or (f). For this reason, drugs might treat certain forms of epilepsy better if they modulate two or more types of ion channels simultaneously. This illustrates the potential advantage of multi-target therapy. Modified from refs. 15,261.
Another recent multiscale simulation study, although not focusing on epilepsy, has provided novel insights on degeneracy in the context of pathological perturbations linked to hyperexcitability associated with chronic pain109. Using network simulations of the spinal dorsal horn, the authors have shown that under physiological conditions, similar circuit activity can emerge in different models with disparate configurations of synaptic properties. However, following identical pathological perturbations, such as a reduction of inhibition or cell type diversity, these models displayed heterogeneous circuit responses. This is in agreement with previous work showing that perturbations in degenerate, superficially similar circuits can reveal hidden variability in synaptic and intrinsic properties leading to heterogeneous responses8,41,250,251. Such modelling insights are highly relevant also for epilepsy. Different individuals with similar physiological circuit output may exhibit different resilience to ictogenic and epileptogenic perturbations, due to hidden variability of circuit parameters. Likewise, for the same reason, different patients with similar hyperexcitable circuit output may exhibit different susceptibility to different therapies.
In degenerate systems, instead of a single model, a large population of models with different parameter combinations is able to generate similar behavior. Consequently, in addition to multiscale modelling, degeneracy requires population modelling of neurons and neural circuits19, also called ensemble or database modelling20,34,48,62,65,110,252,253. One downside of population modelling is that it is unclear which models/parameter combinations from the large theoretically possible parameter space exist in real brains. However, it is plausible to assume that evolution has removed suboptimal models from the parameter space254. Indeed, evolutionary optimization principles might be useful to greatly simplify the parameter space by restricting it to the models that are Pareto optimal for the evolutionary trade-offs between multiple biologically plausible objectives255,256. A phenotype (cell, circuit, organism) or its model is said to be Pareto optimal if there is no other phenotype/model that improves any of its biological objectives without worsening at least one other objective257. Pareto optimality for the trade-off between energy efficiency and functional effectiveness has been suggested as a guiding principle for modelling neurons and neural circuits258,259 and for reducing their degenerate parameter space (including ion channel space) to low-dimensional manifolds259. Hence Pareto theory might be used to improve population modelling of healthy but also epileptic neurons and neuronal circuits.
The upside of using populations of (single scale or multiscale) models is the ability to predict novel multicausal treatment options. Currently many computer models of epilepsy treatment simulate only monocausal pharmacological effects on one type of ion channels. Using populations of conductance-based models of neurons and their circuits, implementing synaptic and intrinsic channel variability would enable predictions for a combination of multiple therapeutic targets, that is, multitarget therapy, in silico. The use of such population-based in silico models could contribute to the discovery of new antiepileptic multitarget drug cocktails260, see Fig. 4. A recent simulation study provided a successful example for using population neuronal models as a tool for designing new multitarget drugs, which rescue pathologically increased excitability in Huntington’s disease261. The authors called such hypothetical medicaments that optimally modulate multiple targets holistic virtual drugs. A similar approach could be adopted in epilepsy research for finding therapeutically efficient sets of perturbations of multiple ion channels that would switch hyperexcitable neuronal phenotypes to control phenotypes with normal excitability.
Towards population models of human neurons and circuits in epilepsy
Recent studies of human neurons and neural circuits have begun to reveal unique properties of the human brain at the cell and circuit level. For example, human pyramidal neurons appear to exhibit enhanced dendritic compartmentalisation262 and distinct h-channel kinetics and expression263,264,265. In addition, dendrites of human pyramidal cells show complex calcium-mediated action potentials266, a high threshold for NMDA spikes267 and faster backpropagation of action potentials as well as forward propagation of excitatory postsynaptic potentials268. The uniqueness or generality of these recently characterized properties of human neurons with respect to their animal counterparts needs to be investigated in more detail269,270. Nevertheless, it is plausible to assume that the cell-to-cell variability and degeneracy discovered in animal neurons21 is also present in human neurons and their circuits.
For instance, the diversity in human h-channels (HCN channels)264,265, see also ref. 271, might be linked to the degeneracy of electrical resonance properties20 that are potentially important for theta oscillations in the cortex265. The influence of a change in HCN channel expression or kinetics on network excitability is complex due to the intricate role that HCN-mediated current (Ih) displays within a neuron47,87,88,89,272, on the one hand depolarizing resting membrane potential273, on the other hand decreasing input resistance and selectively diminishing dendritic excitability57,274. Degeneracy implies that the effects of ion channel changes may depend on the context, namely by the properties of other intrinsic and synaptic mechanisms. Therefore, compensatory or maladaptive changes in other mechanisms might explain why both loss-of-function as well as gain-of-function in Ih can lead to epilepsy90,275.
Epilepsy research has begun to benefit from the increased availability of living human brain samples. Transcriptomic, morphological and electrophysiological data from resected human epileptic tissue started providing insights into the complex interplay between altered ion channels and morphological changes. In a recent study, such human multimodal data were obtained from living hippocampal granule cells276. Combined with populations of realistic cell models and network modelling, this work has provided a first step towards a better understanding of morphological and ion channel changes associated with hyperexcitability of the dentate gyrus circuitry.
Degeneracy and its consequences for the pathophysiology and therapy of epilepsy
From a disease-etiology as well as a therapeutic standpoint, expression of degeneracy in pathological hyperexcitability states has two critical implications: (i) The source of hyperexcitability could manifest animal-to-animal and circuit-to-circuit variability, thus precluding the targeting of individual ion-channels or receptors across all animals and circuits. (ii) As hyperexcitability could emerge through disparate routes, it is equally possible that reversal of hyperexcitability could be achieved through disparate routes15,260 (Fig. 4). The use of inhibitory receptor agonists as anticonvulsants, in many cases irrespective of the mechanistic origins of synchronous firing, constitutes an example of the latter. Thus, it is not always necessary that the reversal of neuronal hyperexcitability is achieved by reversing the mechanisms that resulted in hyperexcitability. Such reversal could be achieved through other routes without probing the mechanistic origins behind the pathological characteristic. On the other hand, under some conditions, knowledge of the precise mechanistic route towards hyperexcitability in an individual patient may be necessary for the choice of effective personalized treatment. For example, therapeutic effects of benzodiazepines and sodium channel blockers can be strongly dependent on the context of specific network pathology (see above discussion of benzodiazepines and sodium channel blockers in the section on network level degeneracy).
The recognition of degeneracy in epileptic pathology could enable simultaneous use of multiple disparate components and routes to reverse hyperexcitability. Hence there is no need for sticking to one specific drug target. In fact, to repair a failed degenerate system, it is often not enough to restore a single target mechanism (Fig. 4). One reason for this is that, due to degeneracy, many different pathologically altered mechanisms are sufficient, but usually none of them is by itself necessary for pathological malfunction6. In addition, a degenerate nervous system sometimes displays compensatory adaptations, which undermine therapeutic interventions focusing on a single target15,92. Therefore, multitarget strategies in pharmacology or in neuromodulatory stimulation might be more promising than monotarget strategies (although carrying a higher risk for side effects). In fact, many already approved antiseizure drugs affect multiple targets277,278. Degeneracy offers rationale also for another currently considered option of multitarget pharmacology, namely using combinations of drugs with different single targets instead of single multitarget drugs15,278 or even using combinations of multitarget drugs. However, an intense basic and clinical research is still needed to find drug combinations with synergistic (supraadditive) therapeutic effects and low (infraadditive) toxicity278,279,280,281. Similarly, future clinical research is needed to provide evidence for beneficial effects of neurostimulation targeting multiple brain areas. Such evidence is currently sparse282.
As there are many potential drug targets, the choice of specific drugs could be allowed to take advantage of circuit-specific differences in target expression profiles. For instance, if hyperexcitability is specific to a neuronal subtype in a given brain region, this could be reversed by identifying mechanisms that are abundant in that neuronal subtype but not others. However, due to degeneracy, it is sometimes impossible to determine which neuron subtype or ion channel subtype is more crucial than others since it might depend on other, e.g., synaptic parameters23,109.
Degeneracy of the brain creates opportunities but also challenges for precision or personalized medicine, which tries to develop therapies targeted to the specific etiology and pathophysiology of individual patients. One challenge in basic and clinical research of degeneracy is measuring multiple hyperexcitability-relevant parameters in the same individual (animal or human). Ideally, in a denegerate circuit, one would need to know most or all contributing components/mechanisms of hyperexcitability including the knowledge of most relevant hub mechanisms with highest “cruciality score” of their involvement6. In other words, reductionist monocausal research strategies focusing on isolated components are only partially suitable for studying complex degenerate systems6,283. Moreover, the conceptual framework of degeneracy with its emphasis on multitarget and multicausal thinking has consequences not only for therapy development but also for basic research and its perturbation and lesion strategies. For example, multitarget lesion experiments6 or multi-knockout studies might be necessary to evoke dysfunction in degenerate neural or molecular networks. Measuring and perturbing multiple parameters and mechanisms simultaneously is challenging but can be facilitated by computational approaches such as multiscale and population modelling (see above).
Epilepsy and the evolutionary costs and benefits of degeneracy
How is it possible that degeneracy makes the excitability of neural circuits robust and yet susceptible to epilepsy? We would like to argue that the evolution of the complex nervous system has led to the emergence of multiple protective mechanisms against the hyperexcitability of neural circuits but at the same time to multiple potential pathways towards the failure of this protection. In a healthy state, multiple compensatory mechanisms may act jointly with the potential to compensate for each other’s failure, thereby supporting the robustness of physiological neural excitability. The compensation may be immediate (“default” or “constitutive” compensation) or it can be recruited on demand with a time delay as a form of “inducible” homeostatic plasticity (“homeostatic compensation”).
Notably, degeneracy in homeostatic plasticity mechanisms has been demonstrated in visual cortex284 and elsewhere20,285,286,287. The authors of visual cortex studies have explicitly stated that “multiple, partially redundant forms of homeostatic plasticity may ensure that network compensation can be achieved in response to a wide range of sensory perturbations”284. As we mentioned before, partial redundancy is a term that is sometimes used in the literature instead of degeneracy. We believe that similar research in the context of epilepsy will reveal degeneracy in homeostatic plasticity mechanisms protecting the hippocampus and other brain regions against hyperexcitability and epileptogenesis. Support for this idea comes also from computational models288 and control theory289, which indicate that degenerate homeostatic mechanisms provide functional benefits. For example, an effective control of neuronal firing rate, including both its mean and variance, can be achieved through the degeneracy of cooperative synaptic and intrinsic homeostatic plasticity289.
Degeneracy in biological systems is closely linked to their complexity in terms of the number of mutually interacting individual components and mechanisms1,290. The greater the number of interacting mechanisms, the more complex, flexible and robust the system becomes. At the same time, the higher are also the system’s energy costs linked to the extent of functional redundancy of identical components6. Since energy and material resources are limited, complex biological organisms display universal trade-offs between (1) functional performance, its (2) robustness and (3) flexibility and (4) energy costs258,291. Degeneracy facilitates the robustness and flexibility of functional behavior10,13. In contrast, pure redundancy increases the energy costs6. Through degeneracy evolution seems to have optimized biological systems for these multi-objective trade-offs254,255,257. A biological system tries to maximize functionality, flexibility and robustness but minimize energy expenditure292,293. Therefore, it is plausible that degenerate living systems evolved to become Pareto optimal for these multiple objectives. Thus, one would expect biological systems, including nervous systems258,259,294,295, to exist close to an optimal compromise between low energy costs and high functionality (as reflected in functional effectiveness, and its robustness and flexibility). In line with these ideas, one recent study proposed that “degeneracy affords a flexibility that offsets the cost of redundancy”296. Another recent work indicated that heterogeneity of synaptic parameters decreases the number of synapses needed for the processing of visual inputs, resulting in a cost benefit297. And, intriguingly, the results of Yang et al.295 suggested that the achievement of multiple biological goals (such as specific firing rates and energy efficiency) may only be possible with a sufficiently large diversity of interacting mechanisms (e.g., ion channels; see also71,298).
The conceptual link between degeneracy and evolutionary optimization opens many interesting questions. For example, counterintuitively, expressing multiple complementary protective mechanisms may paradoxically help reduce the overall cost of protection. This could be the case if activation of multiple protective mechanisms allows for optimal cost sharing among them299. That would open the possibility for every mechanism being expressed at the lower (and hence cheaper) end of its dynamical range299. In this way, the function (e.g., the homeostasis of normal excitability) would be preserved at a lower cost of its protection. Currently such optimal cost sharing is only a hypothesis that needs to be tested in experiments and simulations. On the other hand, it is also possible that the relatively high vulnerability of the nervous system with respect to hyperexcitability is a high evolutionary price that we have to pay for the enormous computational capabilities of our energy-efficient brains292,293, which seem to operate close to criticality that maximises information-processing300, but see also ref. 301.
Another largely unexplored area is the potential trade-off between immune defense and homeostasis of neural excitability. Immune defense of the brain is costly and under threatening conditions it might get activated at the expense of neural homeostasis299. Brain injuries (e.g., trauma, infection) may shift the balance and energy expenditure towards immune defense302. Future research may clarify whether epileptogenesis can be understood as a consequence of chronically enhanced immune defence (with its collateral damage) at the expense of suppressed excitability homeostasis.
Summary
Developing effective treatments for epilepsy remains a challenge. The complex and multifaceted nature of this disease continues to fuel controversies about its origins. In this perspective article, we argued that conflicting hypotheses could be reconciled by considering the degeneracy of the brain, which manifests itself in multiple routes leading to similar function or dysfunction. We exemplified degeneracy at three different levels, ranging from the cellular to the network and systems level. First, at the cellular level, we described the relevance of ion channel degeneracy for epilepsy and discussed its interplay with dendritic morphology. Second, at the network level, we provided examples of degenerate synaptic and intrinsic neuronal properties which support the robustness of neuronal networks but may also lead to diverse responses upon ictogenic and epileptogenic perturbations. Third, at the system level, we provided examples for degeneracy in the intricate interactions between the immune and nervous systems. Finally, we showed that computational approaches including multiscale and so called population (or ensemble/database) models of neurons and neural circuits might help disentangle the complex web of physiological and pathological adaptations. Such models may contribute to identifying the best personalized multitarget strategies for directing the system towards a physiological state.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
References
Edelman, G. M. & Gally, J. A. Degeneracy and complexity in biological systems. Proc. Natl Acad. Sci. 98, 13763–13768 (2001).
Tononi, G., Sporns, O. & Edelman, G. M. Measures of degeneracy and redundancy in biological networks. Proc. Natl Acad. Sci. 96, 3257–3262 (1999).
Prinz, A. A. Degeneracy rules! J. Physiol. 595, 2409 (2017).
Mason, P., Domínguez D, J., Winter, B. & Grignolio, A. Hidden in plain view: degeneracy in complex systems. Biosystems 128, 1–8 (2015).
Man, M., Zhang, Y., Ma, G., Friston, K. & Liu, S. Quantification of degeneracy in hodgkin–huxley neurons on newman–watts small world network. J. Theor. Biol. 402, 62–74 (2016).
Kamaleddin, M. A. Degeneracy in the nervous system: from neuronal excitability to neural coding. BioEssays 44, 2100148 (2022).
Mizusaki, B. E. & O’Donnell, C. Neural circuit function redundancy in brain disorders. Curr. Opin. Neurobiol. 70, 74–80 (2021).
Morozova, E., Newstein, P. & Marder, E. Reciprocally inhibitory circuits operating with distinct mechanisms are differently robust to perturbation and modulation. Elife 11, e74363 (2022).
Whitacre, J. & Bender, A. Degeneracy: a design principle for achieving robustness and evolvability. J. Theor. Biol. 263, 143–153 (2010).
Whitacre, J. M. Degeneracy: a link between evolvability, robustness and complexity in biological systems. Theor. Biol. Med. Model. 7, 1–17 (2010).
Wagner, A. Distributed robustness versus redundancy as causes of mutational robustness. Bioessays 27, 176–88 (2005).
Mellen, N. M. Belt-and-suspenders as a biological design principle. Integr. Respir. Control. 605, 99–103 (2008).
Whitacre, J. M. Biological robustness: paradigms, mechanisms, and systems principles. Front. Genet. 3, 67 (2012).
Cropper, E. C., Dacks, A. M. & Weiss, K. R. Consequences of degeneracy in network function. Curr. Opin. Neurobiol. 41, 62–67 (2016).
Ratté, S. & Prescott, S. A. Afferent hyperexcitability in neuropathic pain and the inconvenient truth about its degeneracy. Curr. Opin. Neurobiol. 36, 31–37 (2016).
Seifert, L., Komar, J., Araújo, D. & Davids, K. Neurobiological degeneracy: a key property for functional adaptations of perception and action to constraints. Neurosci. Biobehav. Rev. 69, 159–165 (2016).
Marder, E., Gutierrez, G. J. & Nusbaum, M. P. Complicating connectomes: electrical coupling creates parallel pathways and degenerate circuit mechanisms. Dev. Neurobiol. 77, 597–609 (2017).
Prinz, A. A., Bucher, D. & Marder, E. Similar network activity from disparate circuit parameters. Nat. Neurosci. 7, 1345–1352 (2004).
Marder, E. & Taylor, A. L. Multiple models to capture the variability in biological neurons and networks. Nat. Neurosci. 14, 133–138 (2011).
Rathour, R. K. & Narayanan, R. Degeneracy in hippocampal physiology and plasticity. Hippocampus 29, 980–1022 (2019).
Goaillard, J.-M. & Marder, E. Ion channel degeneracy, variability, and covariation in neuron and circuit resilience. Annu. Rev. Neurosci. 44, 335–357 (2021).
Neymotin, S. A., Dura-Bernal, S., Lakatos, P., Sanger, T. D. & Lytton, W. W. Multitarget multiscale simulation for pharmacological treatment of dystonia in motor cortex. Front. Pharmacol. 7, 157 (2016).
O’Leary, T. Homeostasis, failure of homeostasis and degenerate ion channel regulation. Curr. Opin. Physiol. 2, 129–138 (2018).
Bui, A., Kim, H. K., Maroso, M. & Soltesz, I. Microcircuits in epilepsy: heterogeneity and hub cells in network synchronization. Cold Spring Harb. Perspect. Med. 5, a022855 (2015).
Symonds, J. D., Zuberi, S. M. & Johnson, M. R. Advances in epilepsy gene discovery and implications for epilepsy diagnosis and treatment. Curr. Opin. Neurol. 30, 193–199 (2017).
Demarest, S. T. & Brooks-Kayal, A. From molecules to medicines: the dawn of targeted therapies for genetic epilepsies. Nat. Rev. Neurol. 14, 735–745 (2018).
Duncan, J. S., Sander, J. W., Sisodiya, S. M. & Walker, M. C. Adult epilepsy. Lancet 367, 1087–1100 (2006).
Scharfman, H. E. The neurobiology of epilepsy. Curr. Neurol. Neurosci. Rep. 7, 348–354 (2007).
Lytton, W. W. Computer modelling of epilepsy. Nat. Rev. Neurosci. 9, 626–637 (2008).
Zilberter, Y., Popova, I. & Zilberter, M. Unifying mechanism behind the onset of acquired epilepsy. Trends Pharmacol. Sci. 43, 87–96 (2022).
Marder, E. & Goaillard, J.-M. Variability, compensation and homeostasis in neuron and network function. Nat. Rev. Neurosci. 7, 563–574 (2006).
Calabrese, R. L. Inconvenient truth to principle of neuroscience. Trends Neurosci. 41, 488–491 (2018).
Drion, G., O’Leary, T. & Marder, E. Ion channel degeneracy enables robust and tunable neuronal firing rates. Proc. Natl Acad. Sci. 112, E5361–E5370 (2015).
Mishra, P. & Narayanan, R. Ion-channel degeneracy: multiple ion channels heterogeneously regulate intrinsic physiology of rat hippocampal granule cells. Physiol. Rep. 9, e14963 (2021).
Lerche, H. et al. Ion channels in genetic and acquired forms of epilepsy. J. Physiol. 591, 753–764 (2013).
Wolfart, J. & Laker, D. Homeostasis or channelopathy? Acquired cell type-specific ion channel changes in temporal lobe epilepsy and their antiepileptic potential. Front. Physiol. 6, 168 (2015).
Oyrer, J. et al. Ion channels in genetic epilepsy: from genes and mechanisms to disease-targeted therapies. Pharmacol. Rev. 70, 142–173 (2018).
Klassen, T. et al. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell 145, 1036–1048 (2011).
Kaplan, D. I., Isom, L. L. & Petrou, S. Role of sodium channels in epilepsy. Cold Spring Harb. Perspect. Med. 6, a022814 (2016).
Thomas, E. A., Reid, C. A., Berkovic, S. F. & Petrou, S. Prediction by modeling that epilepsy may be caused by very small functional changes in ion channels. Arch. Neurol. 66, 1225–1232 (2009).
Onasch, S. & Gjorgjieva, J. Circuit stability to perturbations reveals hidden variability in the balance of intrinsic and synaptic conductances. J. Neurosci. 40, 3186–3202 (2020).
Schneider, M., Gidon, A., Triesch, J., Jedlicka, P. & Cuntz, H. Biological complexity facilitates tuning of the neuronal parameter space. bioRxiv (2021).
Larkum, M. E., Wu, J., Duverdin, S. A. & Gidon, A. The guide to dendritic spikes of the mammalian cortex in vitro and in vivo. Neuroscience 489, 15–33 (2022).
Manita, S., Miyakawa, H., Kitamura, K. & Murayama, M. Dendritic spikes in sensory perception. Front. Cell. Neurosci. 11, 29 (2017).
Johnston, D. & Narayanan, R. Active dendrites: colorful wings of the mysterious butterflies. Trends Neurosci. 31, 309–316 (2008).
Sjostrom, P. J., Rancz, E. A., Roth, A. & Hausser, M. Dendritic excitability and synaptic plasticity. Physiol. Rev. 88, 769–840 (2008).
Harnett, M. T., Magee, J. C. & Williams, S. R. Distribution and function of hcn channels in the apical dendritic tuft of neocortical pyramidal neurons. J. Neurosci. 35, 1024–1037 (2015).
Basak, R. & Narayanan, R. Spatially dispersed synapses yield sharply-tuned place cell responses through dendritic spike initiation. J. Physiol. 596, 4173–4205 (2018).
Johnston, D., Hoffman, D. A. & Poolos, N. P. Potassium channels and dendritic function in hippocampal pyramidal neurons. Epilepsia 41, 1072–1073 (2000).
Su, H. et al. Upregulation of a t-type ca2+ channel causes a long-lasting modification of neuronal firing mode after status epilepticus. J. Neurosci. 22, 3645–3655 (2002).
Bernard, C. et al. Acquired dendritic channelopathy in temporal lobe epilepsy. Science 305, 532–535 (2004).
Shah, M. M., Anderson, A. E., Leung, V., Lin, X. & Johnston, D. Seizure-induced plasticity of h channels in entorhinal cortical layer iii pyramidal neurons. Neuron 44, 495–508 (2004).
Jung, S. et al. Progressive dendritic hcn channelopathy during epileptogenesis in the rat pilocarpine model of epilepsy. J. Neurosci. 27, 13012–13021 (2007).
Shin, M., Brager, D., Jaramillo, T. C., Johnston, D. & Chetkovich, D. M. Mislocalization of h channel subunits underlies h channelopathy in temporal lobe epilepsy. Neurobiol. Dis. 32, 26–36 (2008).
Jung, S. et al. Downregulation of dendritic hcn channel gating in epilepsy is mediated by altered phosphorylation signaling. J. Neurosci. 30, 6678–6688 (2010).
Jung, S., Warner, L. N., Pitsch, J., Becker, A. J. & Poolos, N. P. Rapid loss of dendritic hcn channel expression in hippocampal pyramidal neurons following status epilepticus. J. Neurosci. 31, 14291–14295 (2011).
Poolos, N. P. & Johnston, D. Dendritic ion channelopathy in acquired epilepsy. Epilepsia 53, 32–40 (2012).
Arnold, E. C., McMurray, C., Gray, R. & Johnston, D. Epilepsy-induced reduction in hcn channel expression contributes to an increased excitability in dorsal, but not ventral, hippocampal ca1 neurons. eNeuro 6, ENEURO.0036-19 (2019).
Koch, N. A., Sonnenberg, L., Hedrich, U. B., Lauxmann, S. & Benda, J. Loss or gain of function? neuronal firing effects of ion channel mutations depend on cell type. bioRxiv 2023–01 (2023).
Masala, N. et al. Targeting aberrant dendritic integration to treat cognitive comorbidities of epilepsy. Brain https://doi.org/10.1093/brain/awac455. https://academic.oup.com/brain/advance-article-pdf/doi/10.1093/brain/awac455/47446961/awac455.pdf (2022).
Narayanan, R. & Johnston, D. Functional maps within a single neuron. J. Neurophysiol. 108, 2343–2351 (2012).
Rathour, R. K. & Narayanan, R. Homeostasis of functional maps in active dendrites emerges in the absence of individual channelostasis. Proc. Natl Acad. Sci. USA 111, E1787–E1796 (2014).
Rathour, R. K., Malik, R. & Narayanan, R. Transient potassium channels augment degeneracy in hippocampal active dendritic spectral tuning. Sci. Rep. 6, 24678 (2016).
Migliore, R. et al. The physiological variability of channel density in hippocampal ca1 pyramidal cells and interneurons explored using a unified data-driven modeling workflow. PLoS Comput Biol. 14, e1006423 (2018).
Basak, R. & Narayanan, R. Robust emergence of sharply tuned place-cell responses in hippocampal neurons with structural and biophysical heterogeneities. Brain Struct. Funct. 225, 567–590 (2020).
Roy, A. & Narayanan, R. Spatial information transfer in hippocampal place cells depends on trial-to-trial variability, symmetry of place-field firing, and biophysical heterogeneities. Neural Netw. 142, 636–660 (2021).
Roy, R. & Narayanan, R. Ion-channel degeneracy and heterogeneities in the emergence of complex spike bursts in ca3 pyramidal neurons. J. Physiol. https://www.ncbi.nlm.nih.gov/pubmed/36201674 (2022).
Rich, S., Chameh, H. M., Lefebvre, J. & Valiante, T. A. Loss of neuronal heterogeneity in epileptogenic human tissue impairs network resilience to sudden changes in synchrony. Cell Rep. 39, 110863 (2022).
Cunha, A. O. S. et al. Intrinsic and synaptic properties of hippocampal ca1 pyramidal neurons of the wistar audiogenic rat (war) strain, a genetic model of epilepsy. Sci. Rep. 8, 1–11 (2018).
Whitebirch, A. C. et al. Enhanced excitability of the hippocampal ca2 region and its contribution to seizure activity in a mouse model of temporal lobe epilepsy. Neuron 110, 3121–3138 (2022).
Padmanabhan, K. & Urban, N. N. Intrinsic biophysical diversity decorrelates neuronal firing while increasing information content. Nat. Neurosci. 13, 1276–1282 (2010).
Sanabria, E. R., Su, H. & Yaari, Y. Initiation of network bursts by ca2+-dependent intrinsic bursting in the rat pilocarpine model of temporal lobe epilepsy. J. Physiol. 532, 205–216 (2001).
Yaari, Y., Yue, C. & Su, H. Recruitment of apical dendritic t-type ca2+ channels by backpropagating spikes underlies de novo intrinsic bursting in hippocampal epileptogenesis. J. Physiol. 580, 435–450 (2007).
Beck, H. & Yaari, Y. Plasticity of intrinsic neuronal properties in cns disorders. Nat. Rev. Neurosci. 9, 357–369 (2008).
Lopez-Santiago, L. F. et al. Neuronal hyperexcitability in a mouse model of scn8a epileptic encephalopathy. Proc. Natl Acad. Sci. 114, 2383–2388 (2017).
Niday, Z., Hawkins, V. E., Soh, H., Mulkey, D. K. & Tzingounis, A. V. Epilepsy-associated kcnq2 channels regulate multiple intrinsic properties of layer 2/3 pyramidal neurons. J. Neurosci. 37, 576–586 (2017).
Soh, H., Pant, R., LoTurco, J. J. & Tzingounis, A. V. Conditional deletions of epilepsy-associated kcnq2 and kcnq3 channels from cerebral cortex cause differential effects on neuronal excitability. J. Neurosci. 34, 5311–5321 (2014).
Shruti, S., Clem, R. L. & Barth, A. L. A seizure-induced gain-of-function in bk channels is associated with elevated firing activity in neocortical pyramidal neurons. Neurobiol. Dis. 30, 323–330 (2008).
Kole, M. H., Bräuer, A. U. & Stuart, G. J. Inherited cortical hcn1 channel loss amplifies dendritic calcium electrogenesis and burst firing in a rat absence epilepsy model. J. Physiol. 578, 507–525 (2007).
Santoro, B. et al. Increased seizure severity and seizure-related death in mice lacking hcn1 channels. Epilepsia 51, 1624–1627 (2010).
Albertson, A. J., Yang, J. & Hablitz, J. J. Decreased hyperpolarization-activated currents in layer 5 pyramidal neurons enhances excitability in focal cortical dysplasia. J. Neurophysiol. 106, 2189–2200 (2011).
Ottolini, M., Barker, B. S., Gaykema, R. P., Meisler, M. H. & Patel, M. K. Aberrant sodium channel currents and hyperexcitability of medial entorhinal cortex neurons in a mouse model of scn8a encephalopathy. J. Neurosci. 37, 7643–7655 (2017).
Szulczyk, B. & Nurowska, E. Valproic acid inhibits ttx-resistant sodium currents in prefrontal cortex pyramidal neurons. Biochem. Biophys. Res. Commun. 491, 291–295 (2017).
Kispersky, T. J., Caplan, J. S. & Marder, E. Increase in sodium conductance decreases firing rate and gain in model neurons. J. Neurosci. 32, 10995–11004 (2012).
Mishra, P. & Narayanan, R. Ion-channel regulation of response decorrelation in a heterogeneous multi-scale model of the dentate gyrus. Curr. Res. Neurobiol. 2, 100007 (2021).
Bock, T., Honnuraiah, S. & Stuart, G. J. Paradoxical excitatory impact of sk channels on dendritic excitability. J. Neurosci. 39, 7826–7839 (2019).
Dyhrfjeld-Johnsen, J., Morgan, R. J. & Soltesz, I. Double trouble? potential for hyperexcitability following both channelopathic up-and downregulation of ih in epilepsy. Front. Neurosci. 3, 5 (2009).
Noam, Y., Bernard, C. & Baram, T. Z. Towards an integrated view of hcn channel role in epilepsy. Curr. Opin. Neurobiol. 21, 873–879 (2011).
Mäki-Marttunen, T. & Mäki-Marttunen, V. Excitatory and inhibitory effects of hcn channel modulation on excitability of layer v pyramidal cells. PLoS Comput. Biol. 18, e1010506 (2022).
Kessi, M. et al. The contribution of hcn channelopathies in different epileptic syndromes, mechanisms, modulators, and potential treatment targets: a systematic review. Front. Mol. Neurosci. 15, 807202 (2022).
Rho, Y.-A. & Prescott, S. A. Identification of molecular pathologies sufficient to cause neuropathic excitability in primary somatosensory afferents using dynamical systems theory. PLoS Comput. Biol. 8, e1002524 (2012).
Ratté, S., Zhu, Y., Lee, K. Y. & Prescott, S. A. Criticality and degeneracy in injury-induced changes in primary afferent excitability and the implications for neuropathic pain. Elife 3, e02370 (2014).
Multani, P., Myers, R., Blume, H., Schomer, D. & Sotrel, A. Neocortical dendritic pathology in human partial epilepsy: a quantitative golgi study. Epilepsia 35, 728–736 (1994).
Arisi, G. M. & Garcia-Cairasco, N. Doublecortin-positive newly born granule cells of hippocampus have abnormal apical dendritic morphology in the pilocarpine model of temporal lobe epilepsy. Brain Res. 1165, 126–134 (2007).
Vannini, E. et al. Altered sensory processing and dendritic remodeling in hyperexcitable visual cortical networks. Brain Struct. Funct. 221, 2919–2936 (2016).
Narayanan, R. & Chattarji, S. Computational analysis of the impact of chronic stress on intrinsic and synaptic excitability in the hippocampus. J. Neurophysiol. 103, 3070–3083 (2010).
Dhupia, N., Rathour, R. K. & Narayanan, R. Dendritic atrophy constricts functional maps in resonance and impedance properties of hippocampal model neurons. Front. Cell. Neurosci. 8, 456 (2015).
Mainen, Z. F. & Sejnowski, T. J. Influence of dendritic structure on firing pattern in model neocortical neurons. Nature 382, 363–366 (1996).
Bekkers, J. M. & Häusser, M. Targeted dendrotomy reveals active and passive contributions of the dendritic tree to synaptic integration and neuronal output. Proc. Natl Acad. Sci. 104, 11447–11452 (2007).
Vetter, P., Roth, A. & Häusser, M. Propagation of action potentials in dendrites depends on dendritic morphology. J. Neurophysiol. 85, 926–937 (2001).
Van Ooyen, A., Duijnhouwer, J., Remme, M. W. & van Pelt, J. The effect of dendritic topology on firing patterns in model neurons. Netw.: Comput. neural Syst. 13, 311 (2002).
van Elburg, R. A. & van Ooyen, A. Impact of dendritic size and dendritic topology on burst firing in pyramidal cells. PLoS Comput. Biol. 6, e1000781 (2010).
Cuntz, H. et al. A general principle of dendritic constancy: a neuron’s size-and shape-invariant excitability. Neuron 109, 3647–3662 (2021).
Gutenkunst, R. N. et al. Universally sloppy parameter sensitivities in systems biology models. PLoS Comput. Biol. 3, e189 (2007).
Häusser, M. Synaptic function: dendritic democracy. Curr. Biol. 11, R10–R12 (2001).
Tejada, J., Arisi, G. M., Garcia-Cairasco, N. & Roque, A. C. Morphological alterations in newly born dentate gyrus granule cells that emerge after status epilepticus contribute to make them less excitable. PloS one 7, e40726 (2012).
Tejada, J., Garcia-Cairasco, N. & Roque, A. C. Combined role of seizure-induced dendritic morphology alterations and spine loss in newborn granule cells with mossy fiber sprouting on the hyperexcitability of a computer model of the dentate gyrus. PLoS Comput. Biol. 10, e1003601 (2014).
Taylor, A. L., Goaillard, J.-M. & Marder, E. How multiple conductances determine electrophysiological properties in a multicompartment model. J. Neurosci. 29, 5573–5586 (2009).
Medlock, L. et al. Multiscale computer model of the spinal dorsal horn reveals changes in network processing associated with chronic pain. J. Neurosci. 42, 3133–3149 (2022).
Mishra, P. & Narayanan, R. Disparate forms of heterogeneities and interactions among them drive channel decorrelation in the dentate gyrus: Degeneracy and dominance. Hippocampus 29, 378–403 (2019).
Grashow, R., Brookings, T. & Marder, E. Compensation for variable intrinsic neuronal excitability by circuit-synaptic interactions. J. Neurosci. 30, 9145–9156 (2010).
Goaillard, J.-M., Taylor, A. L., Schulz, D. J. & Marder, E. Functional consequences of animal-to-animal variation in circuit parameters. Nat. Neurosci. 12, 1424–1430 (2009).
Seenivasan, P. & Narayanan, R. Efficient information coding and degeneracy in the nervous system. Curr. Opin. Neurobiol. 76, 102620 (2022).
Lothman, E. W., Stringer, J. L. & Bertram, E. H. The dentate gyrus as a control point for seizures in the hippocampus and beyond. Epilepsy Res. Suppl. 7, 301–313 (1992).
Heinemann, U. et al. The dentate gyrus as a regulated gate for the propagation of epileptiform activity. Epilepsy Res. Suppl. 7, 273–280 (1992).
Krook-Magnuson, E. et al. In vivo evaluation of the dentate gate theory in epilepsy. J. Physiol. 593, 2379–2388 (2015).
Dengler, C. G. & Coulter, D. A. Chapter 6 - Normal and epilepsy-associated pathologic function of the dentate gyrus. In Rossignol, E., Carmant, L. & Lacaille, J.-C. (eds.) Neurobiology of Epilepsy, vol. 226, 155–178 (Elsevier, 2016). https://www.sciencedirect.com/science/article/pii/S0079612316300097.
Krueppel, R., Remy, S. & Beck, H. Dendritic integration in hippocampal dentate granule cells. Neuron 71, 512–28 (2011).
Stegen, M., Young, C. C., Haas, C. A., Zentner, J. & Wolfart, J. Increased leak conductance in dentate gyrus granule cells of temporal lobe epilepsy patients with Ammon’s horn sclerosis. Epilepsia 50, 646–653 (2009).
Stegen, M. et al. Adaptive intrinsic plasticity in human dentate gyrus granule cells during temporal lobe epilepsy. Cereb. Cortex 22, 2087–2101 (2012).
Young, C. C. et al. Upregulation of inward rectifier K+ (Kir2) channels in dentate gyrus granule cells in temporal lobe epilepsy. J. Physiol. 587, 4213–4233 (2009).
Surges, R. et al. Hyperpolarization-activated cation current ih of dentate gyrus granule cells is upregulated in human and rat temporal lobe epilepsy. Biochem. Biophys. Res. Commun. 420, 156–160 (2012).
Kirchheim, F., Tinnes, S., Haas, C. A., Stegen, M. & Wolfart, J. Regulation of action potential delays via voltage-gated potassium kv1. 1 channels in dentate granule cells during hippocampal epilepsy. Front. Cell. Neurosci. 7, 248 (2013).
Janz, P. et al. Synaptic remodeling of entorhinal input contributes to an aberrant hippocampal network in temporal lobe epilepsy. Cereb. Cortex 27, 2348–2364 (2017).
Dey, D. et al. A potassium leak channel silences hyperactive neurons and ameliorates status epilepticus. Epilepsia 55, 203–213 (2014).
Coulter, D. A. & Carlson, G. C. Functional regulation of the dentate gyrus by gaba-mediated inhibition. Prog. Brain Res. 163, 235–812 (2007).
Freund, T. F. & Buzsáki, G. Interneurons of the hippocampus. Hippocampus 6, 347–470 (1996).
Houser, C. R. Interneurons of the dentate gyrus: an overview of cell types, terminal fields and neurochemical identity. Prog. Brain Res. 163, 217–811 (2007).
Hainmueller, T. & Bartos, M. Dentate gyrus circuits for encoding, retrieval and discrimination of episodic memories. Nat. Rev. Neurosci. 21, 153–168 (2020).
Degro, C. E., Bolduan, F., Vida, I. & Booker, S. A. Interneuron diversity in the rat dentate gyrus: An unbiased in vitro classification. Hippocampus 32, 310–331 (2022).
Dudok, B., Klein, P. M. & Soltesz, I. Toward understanding the diverse roles of perisomatic interneurons in epilepsy. Epilepsy Curr. https://doi.org/10.1177/15357597211053687 (2021).
Lee, C.-T. et al. Causal evidence for the role of specific gabaergic interneuron types in entorhinal recruitment of dentate granule cells. Sci. Rep. 6, 1–13 (2016).
Walker, M. C. & Kullmann, D. M. Tonic gabaa receptor-mediated signaling in epilepsy. Jasper’s Basic Mechanisms of the Epilepsies [Internet]. 4th edition, (National Center for Biotechnology Information (US), 2012).
Li, Z.-X., Yu, H.-M. & Jiang, K.-W. Tonic gaba inhibition in hippocampal dentate granule cells: its regulation and function in temporal lobe epilepsies. Acta Physiol. 209, 199–211 (2013).
Sloviter, R. S., Bumanglag, A. V., Schwarcz, R. & Frotscher, M. Abnormal dentate gyrus network circuitry in temporal lobe epilepsy. Jasper’s Basic Mechanisms of the Epilepsies [Internet]. 4th edition, (National Center for Biotechnology Information (US), 2012).
Neuberger, E. J., Gupta, A., Subramanian, D., Korgaonkar, A. A. & Santhakumar, V. Converging early responses to brain injury pave the road to epileptogenesis. J. Neurosci. Res. 97, 1335–1344 (2019).
Scharfman, H. E. The dentate gyrus and temporal lobe epilepsy: an “exciting” era. Epilepsy Curr. 19, 249–255 (2019).
Dudek, F. E. & Sutula, T. P. Epileptogenesis in the dentate gyrus: a critical perspective. In The dentate gyrus: A comprehensive guide to structure, function, and clinical implications (Scharfman, H. E. B. T. P. i. B. R. (ed.) vol. 163, 755–773 (Elsevier, 2007). https://www.sciencedirect.com/science/article/pii/S0079612307630416.
Scharfman, H. E. & Brooks-Kayal, A. R. Is plasticity of gabaergic mechanisms relevant to epileptogenesis? Adv Exp Med Biol. 813, 133–150 (2014).
Alexander, A., Maroso, M. & Soltesz, I. Organization and control of epileptic circuits in temporal lobe epilepsy. Prog. Brain Res. 226, 127–154 (2016).
Moore, Y. E., Kelley, M. R., Brandon, N. J., Deeb, T. Z. & Moss, S. J. Seizing control of KCC2: A new therapeutic target for epilepsy. Trends Neurosci. 40, 555–571 (2017).
Wittner, L. et al. Preservation of perisomatic inhibitory input of granule cells in the epileptic human dentate gyrus. Neuroscience 108, 587–600 (2001).
Kobayashi, M. & Buckmaster, P. S. Reduced inhibition of dentate granule cells in a model of temporal lobe epilepsy. J. Neurosci. 23, 2440–2452 (2003).
Hofmann, G., Balgooyen, L., Mattis, J., Deisseroth, K. & Buckmaster, P. S. Hilar somatostatin interneuron loss reduces dentate gyrus inhibition in a mouse model of temporal lobe epilepsy. Epilepsia 57, 977–983 (2016).
Wittner, L. & Maglóczky, Z. Synaptic reorganization of the perisomatic inhibitory network in hippocampi of temporal lobe epileptic patients. BioMed. Res. Int. 2017, 7154295 (2017).
Sloviter, R. S., Zappone, C. A., Harvey, B. D. & Frotscher, M. Kainic acid-induced recurrent mossy fiber innervation of dentate gyrus inhibitory interneurons: possible anatomical substrate of granule cell hyperinhibition in chronically epileptic rats. J. Comp. Neurol. 494, 944–960 (2006).
Mukherjee, S. et al. Neuroinflammatory mechanisms of post-traumatic epilepsy. J. Neuroinflamm. 17, 193 (2020).
Morgan, R. J., Santhakumar, V. & Soltesz, I. Modeling the dentate gyrus. Prog. Brain Res. 163, 639–658 (2007).
Yu, J., Proddutur, A., Elgammal, F. S., Ito, T. & Santhakumar, V. Status epilepticus enhances tonic gaba currents and depolarizes gaba reversal potential in dentate fast-spiking basket cells. J. Neurophysiol. 109, 1746–1763 (2013).
Proddutur, A., Yu, J., Elgammal, F. S. & Santhakumar, V. Seizure-induced alterations in fast-spiking basket cell gaba currents modulate frequency and coherence of gamma oscillation in network simulations. Chaos: Interdiscip. J. Nonlinear Sci. 23, 046109 (2013).
Jessberger, S. & Parent, J. M. Epilepsy and adult neurogenesis. Cold Spring Harb. Perspect. Biol. 7, a020677 (2015).
Danzer, S. C. Adult neurogenesis in the development of epilepsy. Epilepsy Curr. 19, 316–320 (2019).
Sutula, T. P. & Dudek, F. E. Unmasking recurrent excitation generated by mossy fiber sprouting in the epileptic dentate gyrus: an emergent property of a complex system. Prog. Brain Res. 163, 541–563 (2007).
Buckmaster, P. S. Does mossy fiber sprouting give rise to the epileptic state? Adv Exp Med Biol. 813, 161–168 (2014).
Cavarsan, C. F., Malheiros, J., Hamani, C., Najm, I. & Covolan, L. Is mossy fiber sprouting a potential therapeutic target for epilepsy? Front. Neurol. 9, 1023 (2018).
Ratzliff, A. H., Santhakumar, V., Howard, A. & Soltesz, I. Mossy cells in epilepsy: rigor mortis or vigor mortis? Trends Neurosci. 25, 140–144 (2002).
Sloviter, R. S. et al. "dormant basket cell” hypothesis revisited: relative vulnerabilities of dentate gyrus mossy cells and inhibitory interneurons after hippocampal status epilepticus in the rat. J. Comp. Neurol. 459, 44–76 (2003).
Scharfman, H. E. The enigmatic mossy cell of the dentate gyrus. Nat. Rev. Neurosci. 17, 562–575 (2016).
Oakley, J. C., Kalume, F. & Catterall, W. A. Insights into pathophysiology and therapy from a mouse model of dravet syndrome. Epilepsia 52, 59–61 (2011).
Cheah, C. S. et al. Specific deletion of nav1. 1 sodium channels in inhibitory interneurons causes seizures and premature death in a mouse model of dravet syndrome. Proc. Natl Acad. Sci. 109, 14646–14651 (2012).
Dutton, S. B. et al. Preferential inactivation of scn1a in parvalbumin interneurons increases seizure susceptibility. Neurobiol. Dis. 49, 211–220 (2013).
Rubinstein, M. et al. Dissecting the phenotypes of dravet syndrome by gene deletion. Brain 138, 2219–2233 (2015).
Chancey, J. H. & Howard, M. A. Synaptic integration in ca1 pyramidal neurons is intact despite deficits in gabaergic transmission in the scn1a haploinsufficiency mouse model of dravet syndrome. Eneuro 9, ENEURO.0080-22.2022 (2022).
Jiao, J. et al. Modeling dravet syndrome using induced pluripotent stem cells (ipscs) and directly converted neurons. Hum. Mol. Genet. 22, 4241–4252 (2013).
Liu, Y. et al. Dravet syndrome patient-derived neurons suggest a novel epilepsy mechanism. Ann. Neurol. 74, 128–139 (2013).
Mattis, J. et al. Corticohippocampal circuit dysfunction in a mouse model of Dravet syndrome. eLife 11, e69293 (2022).
Hawkins, N. A. et al. Screening of conventional anticonvulsants in a genetic mouse model of epilepsy. Ann. Clin. Transl. Neurol. 4, 326–339 (2017).
Brunklaus, A. et al. Scn1a variants from bench to bedside-improved clinical prediction from functional characterization. Hum. Mutat. 41, 363–374 (2020).
Sanders, S. J. et al. Progress in understanding and treating scn2a-mediated disorders. Trends Neurosci. 41, 442–456 (2018).
Burman, R. J. et al. Excitatory gabaergic signalling is associated with benzodiazepine resistance in status epilepticus. Brain 142, 3482–3501 (2019).
Codadu, N. K. et al. Divergent paths to seizure-like events. Physiol. Rep. 7, e14226 (2019).
Rich, S. et al. Inhibitory network bistability explains increased interneuronal activity prior to seizure onset. Front. neural circuits 13, 81 (2020).
Lillis, K. P., Kramer, M. A., Mertz, J., Staley, K. J. & White, J. A. Pyramidal cells accumulate chloride at seizure onset. Neurobiol. Dis. 47, 358–366 (2012).
Muldoon, S. F. et al. Gabaergic inhibition shapes interictal dynamics in awake epileptic mice. Brain 138, 2875–2890 (2015).
Elahian, B. et al. Low-voltage fast seizures in humans begin with increased interneuron firing. Ann. Neurol. 84, 588–600 (2018).
Jirsa, V. K., Stacey, W. C., Quilichini, P. P., Ivanov, A. I. & Bernard, C. On the nature of seizure dynamics. Brain 137, 2210–2230 (2014).
Kramer, M. A. et al. Human seizures self-terminate across spatial scales via a critical transition. Proc. Natl Acad. Sci. 109, 21116–21121 (2012).
Saggio, M. L. et al. A taxonomy of seizure dynamotypes. Elife 9, e55632 (2020).
Raikov, I. & Soltesz, I. A Master Plan for the Epilepsies? Toward a General Theory of Seizure Dynamics. Epilepsy Curr. 15, 133–135 (2015).
Chizhov, A. V., Zefirov, A. V., Amakhin, D. V., Smirnova, E. Y. & Zaitsev, A. V. Minimal model of interictal and ictal discharges “epileptor-2”. PLoS Comput. Biol. 14, e1006186 (2018).
Proix, T., Bartolomei, F., Chauvel, P., Bernard, C. & Jirsa, V. K. Permittivity coupling across brain regions determines seizure recruitment in partial epilepsy. J. Neurosci. 34, 15009–15021 (2014).
Lux, H. D., Heinemann, U. & Dietzel, I. Ionic changes and alterations in the size of the extracellular space during epileptic activity. Adv. Neurol. 44, 619–639 (1986).
Fröhlich, F., Bazhenov, M., Iragui-Madoz, V. & Sejnowski, T. J. Potassium dynamics in the epileptic cortex: new insights on an old topic. Neuroscientist. 14, 422–433 (2008).
Raimondo, J. V., Burman, R. J., Katz, A. A. & Akerman, C. J. Ion dynamics during seizures. Front. Cell. Neurosci. 9, 419 (2015).
Chizhov, A. V., Zefirov, A. V., Amakhin, D. V., Smirnova, E. Y. & Zaitsev, A. V. Minimal model of interictal and ictal discharges “Epileptor-2”. PLOS Comput. Biol. 14, e1006186 (2018).
Kajikawa, Y. & Schroeder, C. E. How local is the local field potential?. Neuron 72, 847–858 (2011).
Linden, H. et al. Modeling the spatial reach of the lfp. Neuron 72, 859–872 (2011).
Buzsaki, G., Anastassiou, C. A. & Koch, C. The origin of extracellular fields and currents–eeg, ecog, lfp and spikes. Nat. Rev. Neurosci. 13, 407–420 (2012).
Einevoll, G. T., Kayser, C., Logothetis, N. K. & Panzeri, S. Modelling and analysis of local field potentials for studying the function of cortical circuits. Nat. Rev. Neurosci. 14, 770–785 (2013).
Leski, S., Linden, H., Tetzlaff, T., Pettersen, K. H. & Einevoll, G. T. Frequency dependence of signal power and spatial reach of the local field potential. PLoS Comput. Biol. 9, e1003137 (2013).
Hagen, E., Naess, S., Ness, T. V. & Einevoll, G. T. Multimodal modeling of neural network activity: Computing lfp, ecog, eeg, and meg signals with lfpy 2.0. Front. Neuroinform. 12, 92 (2018).
Pesaran, B. et al. Investigating large-scale brain dynamics using field potential recordings: analysis and interpretation. Nat. Neurosci. 21, 903–919 (2018).
Martinez-Canada, P., Ness, T. V., Einevoll, G. T., Fellin, T. & Panzeri, S. Computation of the electroencephalogram (eeg) from network models of point neurons. PLoS Comput. Biol. 17, e1008893 (2021).
Sinha, M. & Narayanan, R. Active dendrites and local field potentials: biophysical mechanisms and computational explorations. Neuroscience 489, 111–142 (2022).
Reimann, M. W. et al. A biophysically detailed model of neocortical local field potentials predicts the critical role of active membrane currents. Neuron 79, 375–390 (2013).
Sinha, M. & Narayanan, R. Hcn channels enhance spike phase coherence and regulate the phase of spikes and lfps in the theta-frequency range. Proc. Natl Acad. Sci. USA 112, E2207–E2216 (2015).
Ness, T. V., Remme, M. W. H. & Einevoll, G. T. Active subthreshold dendritic conductances shape the local field potential. J. Physiol.-Lond. 594, 3809–3825 (2016).
Ness, T. V., Remme, M. W. H. & Einevoll, G. T. h-type membrane current shapes the local field potential from populations of pyramidal neurons. J. Neurosci. 38, 6011–6024 (2018).
Gibbs, F. A., Davis, H. & Lennox, W. G. The electro-encephalogram in epilepsy and in conditions of impaired consciousness. Arch. Neurol. Psychiatry 34, 1133–1148 (1935).
Dichter, M. A. Basic mechanisms of epilepsy: targets for therapeutic intervention. Epilepsia 38, S2–6 (1997).
Bragin, A., Engel, J. J., Wilson, C. L., Fried, I. & Buzsaki, G. High-frequency oscillations in human brain. Hippocampus 9, 137–142 (1999).
Bragin, A., Engel, J. J., Wilson, C. L., Fried, I. & Mathern, G. W. Hippocampal and entorhinal cortex high-frequency oscillations (100–500 hz) in human epileptic brain and in kainic acid–treated rats with chronic seizures. Epilepsia 40, 127–137 (1999).
McCormick, D. A. & Contreras, D. On the cellular and network bases of epileptic seizures. Annu Rev. Physiol. 63, 815–846 (2001).
Uhlhaas, P. J. & Singer, W. Neural synchrony in brain disorders: relevance for cognitive dysfunctions and pathophysiology. Neuron 52, 155–168 (2006).
Urrestarazu, E., Chander, R., Dubeau, F. & Gotman, J. Interictal high-frequency oscillations (100–500 hz) in the intracerebral eeg of epileptic patients. Brain 130, 2354–2366 (2007).
Liu, S. & Parvizi, J. Cognitive refractory state caused by spontaneous epileptic high-frequency oscillations in the human brain. Sci. Transl. Med. 11, eaax7830 (2019).
Sparks, F. T. et al. Hippocampal adult-born granule cells drive network activity in a mouse model of chronic temporal lobe epilepsy. Nat. Commun. 11, 6138 (2020).
Farrell, J. S., Nguyen, Q. A. & Soltesz, I. Resolving the micro-macro disconnect to address core features of seizure networks. Neuron 101, 1016–1028 (2019).
El Houssaini, K., Bernard, C. & Jirsa, V. K. The epileptor model: a systematic mathematical analysis linked to the dynamics of seizures, refractory status epilepticus, and depolarization block. eNeuro 7, ENEURO.0485-18.2019 (2020).
Weng, Y. et al. Macroscale and microcircuit dissociation of focal and generalized human epilepsies. Commun. Biol. 3, 244 (2020).
Buzsaki, G. Theta oscillations in the hippocampus. Neuron 33, 325–340 (2002).
Buzsaki, G.Rhythms of the brain (Oxford University Press, New York, 2006).
Goutagny, R., Jackson, J. & Williams, S. Self-generated theta oscillations in the hippocampus. Nat. Neurosci. 12, 1491–1493 (2009).
Colgin, L. L. Mechanisms and functions of theta rhythms. Annu. Rev. Neurosci. 36, 295–312 (2013).
Bezaire, M. J., Raikov, I., Burk, K., Vyas, D. & Soltesz, I. Interneuronal mechanisms of hippocampal theta oscillations in a full-scale model of the rodent ca1 circuit. Elife 5, e18566 (2016).
Colgin, L. L. Rhythms of the hippocampal network. Nat. Rev. Neurosci. 17, 239–249 (2016).
Pernia-Andrade, A. J. & Jonas, P. Theta-gamma-modulated synaptic currents in hippocampal granule cells in vivo define a mechanism for network oscillations. Neuron 81, 140–152 (2014).
Colgin, L. L. & Moser, E. I. Gamma oscillations in the hippocampus. Physiology 25, 319–329 (2010).
Buzsaki, G. & Wang, X. J. Mechanisms of gamma oscillations. Annu Rev. Neurosci. 35, 203–225 (2012).
Krook-Magnuson, E. & Soltesz, I. Beyond the hammer and the scalpel: selective circuit control for the epilepsies. Nat. Neurosci. 18, 331–338 (2015).
Zhang, L. et al. Fdg-pet and neun-gfap immunohistochemistry of hippocampus at different phases of the pilocarpine model of temporal lobe epilepsy. Int. J. Med. Sci. 12, 288 (2015).
Weissberg, I. et al. Albumin induces excitatory synaptogenesis through astrocytic tgf-β/alk5 signaling in a model of acquired epilepsy following blood–brain barrier dysfunction. Neurobiol. Dis. 78, 115–125 (2015).
Dingledine, R., Varvel, N. H. & Dudek, F. E. When and how do seizures kill neurons, and is cell death relevant to epileptogenesis? Adv Exp Med Biol. 813, 109–122 (2014).
Kenny, E. M. et al. Ferroptosis contributes to neuronal death and functional outcome after traumatic brain injury. Crit. Care Med. 47, 410 (2019).
Pitkänen, A. & Immonen, R. Epilepsy related to traumatic brain injury. Neurotherapeutics 11, 286–296 (2014).
Ostergard, T., Sweet, J., Kusyk, D., Herring, E. & Miller, J. Animal models of post-traumatic epilepsy. J. Neurosci. Methods 272, 50–55 (2016).
Xanthos, D. N. & Sandkühler, J. Neurogenic neuroinflammation: inflammatory cns reactions in response to neuronal activity. Nat. Rev. Neurosci. 15, 43–53 (2014).
Vezzani, A., Balosso, S. & Ravizza, T. Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat. Rev. Neurol. 15, 459–472 (2019).
Li, G. et al. Cytokines and epilepsy. Seizure 20, 249–256 (2011).
Klein, P. et al. Commonalities in epileptogenic processes from different acute brain insults: Do they translate? Epilepsia 59, 37–66 (2018).
Zengeler, K. E. & Lukens, J. R. Innate immunity at the crossroads of healthy brain maturation and neurodevelopmental disorders. Nat. Rev. Immunol. 21, 1–15 (2021).
González, H., Elgueta, D., Montoya, A. & Pacheco, R. Neuroimmune regulation of microglial activity involved in neuroinflammation and neurodegenerative diseases. J. Neuroimmunol. 274, 1–13 (2014).
Deczkowska, A., Amit, I. & Schwartz, M. Microglial immune checkpoint mechanisms. Nat. Neurosci. 21, 779–786 (2018).
Badimon, A. et al. Negative feedback control of neuronal activity by microglia. Nature 586, 417–423 (2020).
Stellwagen, D. & Malenka, R. C. Synaptic scaling mediated by glial tnf-α. Nature 440, 1054–1059 (2006).
Nikolic, L. et al. Blocking tnfα-driven astrocyte purinergic signaling restores normal synaptic activity during epileptogenesis. Glia 66, 2673–2683 (2018).
Shlosberg, D., Benifla, M., Kaufer, D. & Friedman, A. Blood–brain barrier breakdown as a therapeutic target in traumatic brain injury. Nat. Rev. Neurol. 6, 393–403 (2010).
Yong, H. Y., Rawji, K. S., Ghorbani, S., Xue, M. & Yong, V. W. The benefits of neuroinflammation for the repair of the injured central nervous system. Cell. Mol. Immunol. 16, 540–546 (2019).
Van Vliet, E. et al. Blood–brain barrier leakage may lead to progression of temporal lobe epilepsy. Brain 130, 521–534 (2007).
Rüber, T. et al. Evidence for peri-ictal blood–brain barrier dysfunction in patients with epilepsy. Brain 141, 2952–2965 (2018).
Kim, S. Y., Buckwalter, M., Soreq, H., Vezzani, A. & Kaufer, D. Blood–brain barrier dysfunction–induced inflammatory signaling in brain pathology and epileptogenesis. Epilepsia 53, 37–44 (2012).
Batulin, D., Lagzi, F., Vezzani, A., Jedlicka, P. & Triesch, J. A mathematical model of neuroimmune interactions in epileptogenesis for discovering treatment strategies. Iscience 25, 104343 (2022).
Kirkman, N. J., Libbey, J. E., Wilcox, K. S., White, H. S. & Fujinami, R. S. Innate but not adaptive immune responses contribute to behavioral seizures following viral infection. Epilepsia 51, 454–464 (2010).
Brackhan, M. et al. Serial quantitative tspo-targeted pet reveals peak microglial activation up to 2 weeks after an epileptogenic brain insult. J. Nucl. Med. 57, 1302–1308 (2016).
Patel, D. C. et al. Hippocampal tnfα signaling contributes to seizure generation in an infection-induced mouse model of limbic epilepsy. Eneuro 4, ENEURO.0105-17.2017 (2017).
Cobos, I. et al. Mice lacking dlx1 show subtype-specific loss of interneurons, reduced inhibition and epilepsy. Nat. Neurosci. 8, 1059–1068 (2005).
Cossart, R. et al. Dendritic but not somatic gabaergic inhibition is decreased in experimental epilepsy. Nat. Neurosci. 4, 52–62 (2001).
Lytton, W. W. et al. Multiscale modeling in the clinic: diseases of the brain and nervous system. Brain Inf. 4, 219–230 (2017).
Knox, A. T., Glauser, T., Tenney, J., Lytton, W. W. & Holland, K. Modeling pathogenesis and treatment response in childhood absence epilepsy. Epilepsia 59, 135–145 (2018).
Sakurai, A., Tamvacakis, A. N. & Katz, P. S. Hidden synaptic differences in a neural circuit underlie differential behavioral susceptibility to a neural injury. eLife 3, e02598 (2014).
Haddad, S. A. & Marder, E. Circuit robustness to temperature perturbation is altered by neuromodulators. Neuron 100, 609–623.e3 (2018).
Günay, C., Edgerton, J. R. & Jaeger, D. Channel density distributions explain spiking variability in the globus pallidus: a combined physiology and computer simulation database approach. J. Neurosci. 28, 7476–7491 (2008).
Sekulic, V., Lawrence, J. J. & Skinner, F. K. Using multi-compartment ensemble modeling as an investigative tool of spatially distributed biophysical balances: application to hippocampal oriens-lacunosum/moleculare (O-LM) cells. PloS One 9, e106567 (2014).
Shoval, O. et al. Evolutionary trade-offs, pareto optimality, and the geometry of phenotype space. Science 336, 1157–1160 (2012).
Szekely, P., Sheftel, H., Mayo, A. & Alon, U. Evolutionary tradeoffs between economy and effectiveness in biological homeostasis systems. PLoS Comput. Biol. 9, e1003163 (2013).
Remme, M. W., Rinzel, J. & Schreiber, S. Function and energy consumption constrain neuronal biophysics in a canonical computation: Coincidence detection. PLoS Comput. Biol. 14, e1006612 (2018).
Alon, U. Multi-objective optimality in biology. In An Introduction to Systems Biology: Design Principles of Biological Circuits, chap. (Alon, U. ed.) 14, 249–272 (Chapman and Hall/CRC, Boca Raton, London, New York, 2020), 2 edn. https://doi.org/10.1201/9780429283321.
Pallasdies, F., Norton, P., Schleimer, J.-H. & Schreiber, S. Neural optimization: Understanding trade-offs with pareto theory. Curr. Opin. Neurobiol. 71, 84–91 (2021).
Jedlicka, P., Bird, A. D. & Cuntz, H. Pareto optimality, economy-effectiveness trade-offs and ion channel degeneracy: improving population modelling for single neurons. Open Biol. 12, 220073 (2022).
Goaillard, J.-M. & Dufour, M. A. Neuropathic pain: the pros and cons of degeneracy. Elife 3, e02615 (2014).
Allam, S. L., Rumbell, T. H., Hoang-Trong, T., Parikh, J. & Kozloski, J. R. Neuronal population models reveal specific linear conductance controllers sufficient to rescue preclinical disease phenotypes. Iscience 24, 103279 (2021).
Beaulieu-Laroche, L. et al. Enhanced dendritic compartmentalization in human cortical neurons. Cell 175, 643–651 (2018).
Kalmbach, B. E. et al. h-channels contribute to divergent intrinsic membrane properties of supragranular pyramidal neurons in human versus mouse cerebral cortex. Neuron 100, 1194–1208 (2018).
Rich, S., Moradi Chameh, H., Sekulic, V., Valiante, T. A. & Skinner, F. K. Modeling reveals human–rodent differences in h-current kinetics influencing resonance in cortical layer 5 neurons. Cereb. Cortex 31, 845–872 (2021).
Moradi Chameh, H. et al. Diversity amongst human cortical pyramidal neurons revealed via their sag currents and frequency preferences. Nat. Commun. 12, 1–15 (2021).
Gidon, A. et al. Dendritic action potentials and computation in human layer 2/3 cortical neurons. Science 367, 83–87 (2020).
Testa-Silva, G. et al. High synaptic threshold for dendritic nmda spike generation in human layer 2/3 pyramidal neurons. Cell Rep. 41, 111787 (2022).
Olah, G. et al. Accelerated signal propagation speed in human neocortical microcircuits. bioRxiv 2022–09 (2022).
Eyal, G. et al. Human cortical pyramidal neurons: from spines to spikes via models. Front. Cell. Neurosci. 12, 181 (2018).
Fişek, M. & Häusser, M. Are human dendrites different? Trends Cogn. Sci. 24, 411–412 (2020).
Guet-McCreight, A. et al. Age-dependent increased sag amplitude in human pyramidal neurons dampens baseline cortical activity. Cerebral Cortex 33, 4360–4373 (2023).
Mishra, P. & Narayanan, R. High-conductance states and a-type k+ channels are potential regulators of the conductance-current balance triggered by hcn channels. J. Neurophysiol. 113, 23–43 (2015).
Dyhrfjeld-Johnsen, J., Morgan, R. J., Földy, C. & Soltesz, I. Upregulated h-current in hyperexcitable ca1 dendrites after febrile seizures. Front. Cell. Neurosci. 2, 2 (2008).
Poolos, N. P., Migliore, M. & Johnston, D. Pharmacological upregulation of h-channels reduces the excitability of pyramidal neuron dendrites. Nat. Neurosci. 5, 767–774 (2002).
Reid, C. A., Phillips, A. M. & Petrou, S. Hcn channelopathies: pathophysiology in genetic epilepsy and therapeutic implications. Br. J. Pharmacol. 165, 49–56 (2012).
Buchin, A. et al. Multi-modal characterization and simulation of human epileptic circuitry. Cell Rep. 41, 111873 (2022).
Kwan, P., Sills, G. J. & Brodie, M. J. The mechanisms of action of commonly used antiepileptic drugs. Pharmacol. Therap. 90, 21–34 (2001).
Löscher, W. Single-target versus multi-target drugs versus combinations of drugs with multiple targets: Preclinical and clinical evidence for the treatment or prevention of epilepsy. Front. Pharmacol. 12, 730257 (2021).
Brodie, M. J. & Sills, G. J. Combining antiepileptic drugs-rational polytherapy? Seizure 20, 369–375 (2011).
Brigo, F., Ausserer, H., Tezzon, F. & Nardone, R. When one plus one makes three: the quest for rational antiepileptic polytherapy with supraadditive anticonvulsant efficacy. Epilepsy Behav. 27, 439–442 (2013).
Verrotti, A. et al. The role of polytherapy in the management of epilepsy: suggestions for rational antiepileptic drug selection. Expert Rev. Neurotherap. 20, 167–173 (2020).
Li, M. C. & Cook, M. J. Deep brain stimulation for drug-resistant epilepsy. Epilepsia 59, 273–290 (2018).
Regenmortel, M. H. V. Reductionism and complexity in molecular biology: scientists now have the tools to unravel biological complexity and overcome the limitations of reductionism. EMBO Rep. 5, 1016–1020 (2004).
Maffei, A. & Turrigiano, G. G. Multiple modes of network homeostasis in visual cortical layer 2/3. J. Neurosci. 28, 4377–4384 (2008).
Watt, A. J. & Desai, N. S. Homeostatic plasticity and stdp: keeping a neuron’s cool in a fluctuating world. Front. Synaptic Neurosci. 2, 5 (2010).
Turrigiano, G. Too many cooks? intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu. Rev. Neurosci. 34, 89–103 (2011).
Mishra, P. & Narayanan, R. Stable continual learning through structured multiscale plasticity manifolds. Current Opinion in Neurobiology. 70, 51–63 (2021).
Lazar, A., Pipa, G. & Triesch, J. Sorn: a self-organizing recurrent neural network. Front. Comput. Neurosci. 3, 23 (2009).
Cannon, J. & Miller, P. Synaptic and intrinsic homeostasis cooperate to optimize single neuron response properties and tune integrator circuits. J. Neurophysiol. 116, 2004–2022 (2016).
Mason, P. H. Degeneracy at multiple levels of complexity. Biol. Theory 5, 277–288 (2010).
Del Giudice, M. & Crespi, B. J. Basic functional trade-offs in cognition: an integrative framework. Cognition 179, 56–70 (2018).
Laughlin, S. B. & Sejnowski, T. J. Communication in neuronal networks. Science 301, 1870–1874 (2003).
Sterling, P. & Laughlin, S.Principles of neural design (MIT press, 2015).
Deistler, M., Macke, J. H. & Gonçalves, P. J. Energy-efficient network activity from disparate circuit parameters. Proc. Natl Acad. Sci. 119, e2207632119 (2022).
Yang, J., Shakil, H., Ratté, S. & Prescott, S. A. Minimal requirements for a neuron to coregulate many properties and the implications for ion channel correlations and robustness. eLife 11, e72875 (2022).
Sajid, N., Parr, T., Hope, T. M., Price, C. J. & Friston, K. J. Degeneracy and redundancy in active inference. Cereb. Cortex 30, 5750–5766 (2020).
Balcioglu, A. et al. Mapping thalamic innervation to individual l2/3 pyramidal neurons and modeling their ‘readout’of visual input. Nat. Neurosci. 26, 470–480 (2023).
Tripathy, S. J., Padmanabhan, K., Gerkin, R. C. & Urban, N. N. Intermediate intrinsic diversity enhances neural population coding. Proc. Natl Acad. Sci. 110, 8248–8253 (2013).
Stearns, S. C. & Medzhitov, R.Evolutionary medicine (Sinauer Associates, Incorporated, Publishers, 2016).
Beggs, J. M. The criticality hypothesis: how local cortical networks might optimize information processing. Philos. Trans. R. Soc. A: Math., Phys. Eng. Sci. 366, 329–343 (2008).
Destexhe, A. & Touboul, J. D. Is there sufficient evidence for criticality in cortical systems? Eneuro 8, ENEURO.0551-20.2021 (2021).
Casaril, A. M., Katsalifis, A., Schmidt, R. M. & Bas-Orth, C. Activated glia cells cause bioenergetic impairment of neurons that can be rescued by knock-down of the mitochondrial calcium uniporter. Biochem. Biophys. Res. Commun. 608, 45–51 (2022).
Acknowledgements
The work was supported by the grant LOEWE CePTER - Center for Personalized Translational Epilepsy Research (D.B., T.M.S., J.T., P.J.). D.B. is supported by the International Max Planck Research School (IMPRS) for Neural Circuits. J.T. is supported by the Johanna Quandt Foundation. R.N. is supported by a senior fellowship (IA/S/16/2/502727) from the DBT-Wellcome Trust India Alliance. P.J. is supported by the BMBF grant (No. 031L0229) and funds from the von Behring Röntgen Foundation.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
All authors, T.M.S., D.B., J.T., R.N., P.J., contributed to conceptualizing and writing the manuscript. R.N., D.B. and T.M.S. created the figures.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Ethics statement
This work follows the ethical guidelines of Nature Portfolio journals.
Peer review
Peer review information
Communications Biology thanks Caren Armstrong and the other, anonymous reviewers for their contribution to the peer review of this work. Primary Handling Editor: George Inglis. Peer reviewer reports are available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stöber, T.M., Batulin, D., Triesch, J. et al. Degeneracy in epilepsy: multiple routes to hyperexcitable brain circuits and their repair. Commun Biol 6, 479 (2023). https://doi.org/10.1038/s42003-023-04823-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-023-04823-0
This article is cited by
-
A high-efficiency model indicating the role of inhibition in the resilience of neuronal networks to damage resulting from traumatic injury
Journal of Computational Neuroscience (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
