
Abstract
Acquired drug resistance remains a major problem across oncogene-addicted cancers. Elucidation of mechanisms of resistance can inform rational treatment strategies for patients relapsing on targeted therapies while offering insights into tumor evolution. Here, we report acquired MET amplification as a resistance driver in a ROS1-rearranged lung adenocarcinoma after sequential treatment with ROS1 inhibitors. Subsequent combination therapy with lorlatinib plus capmatinib, a MET-selective inhibitor, induced intracranial and extracranial tumor response. At relapse, sequencing of the resistant tumor revealed a MET D1246N mutation and loss of MET amplification. We performed integrated molecular analyses of serial tumor and plasma samples, unveiling dynamic alterations in the ROS1 fusion driver and MET bypass axis at genomic and protein levels and the emergence of polyclonal resistance. This case illustrates the complexity of longitudinal tumor evolution with sequential targeted therapies, highlighting challenges embedded in the current precision oncology paradigm and the importance of developing approaches that prevent resistance.
Similar content being viewed by others

Introduction
Chromosomal rearrangements involving the ROS proto-oncogene 1 (ROS1) gene are potent oncogenic drivers identified across multiple tumor types including in 1–2% of non-small cell lung cancer (NSCLC)1,2. ROS1 rearrangements result in the expression of the ROS1 fusion oncoprotein, constitutive ROS1 kinase activity, and aberrant downstream signaling activation, ultimately leading to dysregulated tumor survival and proliferation3. Patients with metastatic ROS1-rearranged lung cancers typically derive substantial benefit from ROS1-targeted tyrosine kinase inhibitors (TKIs) such as crizotinib and entrectinib, often with marked initial tumor responses4,5. However, the durability of response is inevitably limited in most patients due to acquired drug resistance. Prior studies have elucidated mechanisms of on-target resistance mediated by a spectrum of ROS1 kinase domain mutations (e.g., ROS1 G2032R)6,7,8,9, motivating the clinical development of next-generation ROS1 inhibitors10,11,12,13. In contrast, insights into mechanisms of off-target resistance have remained limited.
Amplification of the mesenchymal-epithelial transition (MET) gene—which encodes a receptor tyrosine kinase involved in promoting mitosis, motility, and survival14—represents one potentially actionable mechanism of off-target resistance to ROS1 inhibitors that has been identified using ROS1-rearranged cell lines and biopsies from patients relapsing on therapy (including after entrectinib and lorlatinib)9,15,16. In EGFR-mutated lung cancers harboring MET amplification-mediated resistance to EGFR TKIs (which occurs in 10–15% of patients relapsing on first-line third-generation EGFR inhibitors)17,18, various combinations of EGFR and MET TKIs have demonstrated clinical activity and tolerability in phase I/II clinical trials19,20, attesting to the actionability on this resistance driver. Similarly, combinations with MET inhibitors have induced tumor responses in ALK- and RET-rearranged lung cancers with MET-driven resistance to TKIs targeting the original oncogene drivers21,22. In ROS1-rearranged disease with MET-mediated resistance, the optimal clinical strategy for co-targeting ROS1 and MET is unknown. One case report described the efficacy of crizotinib monotherapy (a multitargeted ROS1/MET/ALK TKI) in a patient relapsing on entrectinib with MET polysomy23. Crizotinib is a first-generation multikinase inhibitor with limited selectivity, suboptimal coverage of on-target resistance mutations that can emerge on therapy, and poor blood-brain barrier penetrance24, all of which can limit clinical benefit. Therefore, a rationale exists for exploring combination strategies that exploit potent, CNS-penetrant next-generation ROS1 TKIs and selective MET TKIs such as capmatinib or tepotinib.
Of note, across treatment-naïve and -resistant cancers, upregulation of MET signaling occurs through an array of molecular events ranging from protein overexpression to genomic alterations, including amplification, mutations (e.g., exon 14 skipping), or chromosomal rearrangements25. These molecular alterations have distinct functional consequences and clinical implications26. The MET-selective TKIs capmatinib and tepotinib are FDA-approved for the treatment of advanced lung cancers harboring MET exon 14 skipping27,28. However, both TKIs have additionally demonstrated efficacy in MET-amplified NSCLC28,29. By comparison, MET overexpression is not currently considered an actionable biomarker from the perspective of TKI therapy, although this is anticipated to evolve with the advent of non-TKI therapeutic agents targeting MET, such as antibody–drug conjugates (ADCs)30,31.
Here, we report a case of ROS1-rearranged lung cancer overexpressing MET protein at baseline, which then acquired genomic amplification of MET as an off-target mechanism of resistance to ROS1 inhibitors. To our knowledge, this is the first documented case of extracranial and intracranial disease response to a ROS1/MET co-inhibition strategy using lorlatinib plus capmatinib in MET-amplified, ROS1-rearranged NSCLC. The subsequent emergence of an on-target MET resistance mutation and loss of MET amplification highlights the temporal evolution of multi-faceted MET signaling axis activation and complex overall molecular landscape longitudinally as tumor cells acquire resistance to sequential targeted therapies.
Results
Case
A 34-year-old woman with no smoking history presented with persistent dry cough and was found to have a 2.2 cm FDG-avid left lingular lung nodule on chest computed tomography (CT) and positron emission tomography (PET) imaging without evidence of metastatic disease. A biopsy of the lung nodule demonstrated primary lung adenocarcinoma. She underwent left upper lobe segmentectomy but was noted intraoperatively to have pleural and pericardial nodules. Cytology evaluation demonstrated malignant pleural effusion, and pathologic staging was pT2 pNx pM1a. Genotyping via next-generation sequencing (NGS) and ROS1 fluorescence in situ hybridization (FISH) on the resected lung tumor sample revealed a SLC34A2-ROS1 gene rearrangement and CDKN2A H83Y and TP53 V173fs mutations detected on NGS (Supplementary Table 1). ROS1 protein expression was confirmed by immunohistochemistry (IHC). She was initiated on first-line crizotinib, 250 mg twice daily, with the initial response (Fig. 1A). After 14 months, imaging showed evidence of progression with new hepatic metastases. IR-guided liver biopsy confirmed adenocarcinoma with ROS1 rearrangement by NGS testing and FISH (Supplementary Table 1). The patient was started on second-line lorlatinib, dose-reduced to 75 mg for neurocognitive adverse events (AEs). After 30 months, scans showed intrathoracic and abdominal progression with peritoneal carcinomatosis. Thoracentesis of the left pleural effusion revealed recurrent adenocarcinoma. NGS testing again demonstrated the SLC34A2-ROS1 rearrangement, TP53 V173fs, ATM H2075R (VUS), PMS2 E518K (VUS), ERCC2 N92S (VUS) (Supplementary Table 1). Platinum-doublet chemotherapy (carboplatin and pemetrexed) was initiated with continued lorlatinib 50 mg daily.

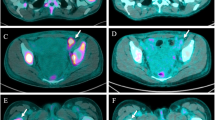
A Timeline summarizing the treatment course and tumor and plasma sampling. Chemo: chemotherapy. B Representative computed tomography (CT) and brain magnetic resonance imaging (MRI) images demonstrating response to, and progression on, lorlatinib plus capmatinib therapy at the indicated time points, including the response of left axillary lymph node (yellow arrow), chest wall disease (yellow arrow), pleural thickening (yellow arrowhead), and liver metastasis (yellow arrow) to lorlatinib plus capmatinib dual therapy at 10 weeks; increasing liver metastasis (red arrow), new abdominal lymphadenopathy (red arrowhead), and new left temporal brain metastasis (red arrowhead). C Measurements on indicated metastatic lesions over the treatment course. Time 0 = baseline scan prior to initiation of lorlatinib plus capmatinib.
After an initial response to chemotherapy plus lorlatinib, the patient presented with intracranial and extracranial progressive disease after 5.5 months (Fig. 1), including new multifocal brain parenchymal lesions, worsening bilateral pleural effusions, enlarging left axillary lymphadenopathy, and a left-sided chest wall soft tissue mass biopsy-proven to be metastatic lung adenocarcinoma without evidence of histologic transformation. DNA-based NGS testing on the chest wall mass revealed the known SLC34A2-ROS1 rearrangement and CDKN2A H83Y and TP53 V173fs mutations (Supplementary Table 1). ROS1 FISH assay confirmed retention of the genomic ROS1 rearrangement (Fig. 3A). However, IHC analysis showed a new loss of ROS1 protein expression (Fig. 2A, B). Additionally, NGS testing revealed a de novo MET amplification (7q31 gain) which was confirmed by MET FISH testing with a MET:CEP7 ratio of 6.1 (Fig. 3A, B). Of note, MET IHC confirmed expression of MET at this time point (H-score 130) (Fig. 2A). However, upon retrospective testing, MET overexpression was detected in all prior biopsies, including in the treatment-naïve lung tumor at initial diagnosis and in the crizotinib- and lorlatinib-resistant tumor specimens (Fig. 2A; H-scores 300 and 300, respectively). In contrast, genomic MET amplification (as tested using MET FISH) was absent in all previously biopsied tumors.

A Representative immunohistochemistry (IHC) images demonstrating the H&E, ROS1, and c-MET immunostaining at indicated time points. Scale bar: 100 µm. B Percentage of tumor cells immunostaining positive for ROS1 at same time points as shown in (A).

A Representative fluorescence in situ hybridization (FISH) images for ROS1 rearrangement (top), EGFR copy number relative to CEP7, and MET copy number relative to CEP7 (bottom). Split signals (white lines) denote ROS1 rearrangement. B The EGFR-to-CEP7 ratio (i.e., the ratio of the green signals to the white signals) and MET-to-CEP7 ratio (i.e., the ratio of the red signals to the white signals) are shown at the indicated time points throughout the disease course. C Guardant360 cell-free DNA (cfDNA) response map showing dynamic changes in variant allele fractions of gene alterations. The evolution of variant alleles over time, represented as % cfDNA, is depicted (bottom). Timepoint 0 = plasma liquid biopsy prior to starting lorlatinib treatment.
Given the evidence of acquired MET amplification, and in view of active disease in the central nervous system (CNS), combination therapy of lorlatinib (dose reduced to 50 mg daily) and capmatinib (400 mg twice daily), an ATP-competitive, potent, and CNS-penetrant type Ib MET TKI28, was pursued. She experienced rapid clinical improvement with palpable shrinkage of left axillary fullness and improvement of fatigue and shortness of breath within 2 weeks. Repeat scans performed 10 weeks after initiation of combination therapy demonstrated partial response per RECIST v1.1 criteria with a 69% reduction in the target tumor lesions (left axillary lymph node, hepatic metastasis) as well as shrinkage of non-target lesions, including the left pleural thickening and chest wall soft tissue disease (Fig. 1B, C, Supplemental Table 1). Furthermore, a brain MRI demonstrated shrinkage of numerous parenchymal CNS metastases, including a 43% reduction in the dominant left temporal lobe lesion, consistent with intracranial partial response per modified RECIST v1.1. She continued the combination for a total of 32 weeks until disease progression, with treatment-emergent AE of grade 1 lower extremity edema and nausea attributed to capmatinib (predominantly) and lorlatinib and grade 2 hyperlipidemia attributed to lorlatinib. No dose interruptions or dose reductions due to treatment-emergent AEs were required.
Subsequently, due to progression, including increased hepatic metastases, portocaval lymphadenopathy, malignant ascites, and recurrence of brain metastases (Fig. 1B, C), the combination regimen was discontinued. Metastatic disease in the left axillary node, left pleura, and left chest wall were continuing to respond, indicating heterogeneity across tumor sites. Repeat liver biopsy showed retained adenocarcinoma histology and persistent loss of ROS1 expression by IHC. NGS testing revealed the original ROS1 rearrangement, and a de novo MET D1246N mutation (Supplementary Table 1), known to confer resistance to type I MET TKIs, including crizotinib, capmatinib, and tepotinib32,33,34. Of note, the previously detected MET amplification was no longer identified on MET FISH testing (MET:CEP7 copy number ratio, 1.1). Liquid biopsy with Guardant360 cell-free DNA (cfDNA) assay demonstrated a SLC34A2-ROS1 rearrangement [variant allele fraction (VAF) 55.6%] and heterogeneous genomic alterations, including MET D1246N (VAF 7.8%) and TP53 V173fd (VAF 45.7%), as well as BRCA2 loss, EGFR amplification (medium-level, plasma copy number 3), and FGFR1 amplification (low-level, plasma copy number 2.4) which had not been identified through tissue NGS or EGFR FISH testing (Fig. 3C, Supplementary Table 1). The patient was offered cabozantinib, a multikinase type II inhibitor with ROS1/MET-targeted activity35, given its potency against the MET D1246N resistance mutation36. The patient received three days of cabozantinib before being hospitalized for worsening ascites and ultimately decided against further treatment and was transitioned to comfort-focused care.
Discussion
This report represents, to the best of our knowledge, the first description of a dramatic—albeit brief—clinical and radiographic tumor response to combined selective inhibition of ROS1 and MET using lorlatinib plus capmatinib in the setting of acquired resistance to sequential ROS1 inhibitor therapy (crizotinib followed by lorlatinib) due to MET amplification. In this case, the combination of lorlatinib plus capmatinib was chosen and preferred over alternative options (crizotinib monotherapy or lorlatinib plus crizotinib) in order to maximize CNS penetration and activity in the context of actively progressing brain metastases. Indeed, the patient experienced both intracranial and extracranial responses to therapy.
The activity of lorlatinib plus capmatinib in this patient with ROS1-rearranged cancer confirms MET amplification as the driver of resistance that is, importantly, actionable. Collective evidence continues to buttress MET amplification as a shared genomic mechanism of off-target resistance and biomarker for subsequent rational combination strategy across oncogene-addicted lung cancers19,21,22, and underscores the clinical value of re-biopsy and comprehensive molecular profiling in therapy-resistant cancers. In addition, here, the combination of half-dose lorlatinib and full-dose capmatinib was tolerated without grade 2 or higher treatment-related AEs or the requirement for further dose modifications. It is impossible to generalize regarding the efficacy or tolerability of lorlatinib plus capmatinib in patients with MET-amplified, ROS1-rearranged lung cancers on the basis of this single case. However, it will not be pragmatically feasible to perform phase Ib/II clinical trials of discrete combination regimens for each individual therapy-resistant, rare molecular subset of lung cancer (e.g., ROS1-, RET-, and NTRK1-3-rearranged NSCLC) and for each distinct genomic mechanism of resistance. Therefore, creative basket trial designs in parallel with added knowledge from smaller case series and extrapolation from the available data in relatively larger molecular subsets of NSCLC (such as EGFR-mutant disease) will be pivotal to effectively inform clinical practice and advance viable treatment strategies. For instance, the field may benefit from shifting the way basket trials are structured to be centered around mechanisms of resistance rather than each original oncogenic driver.
The brief duration of benefit from the combination regimen herein is noteworthy and deemed to reflect the emergent tumor heterogeneity across disease sites under the selective pressure of sequential targeted therapies—a prominent recurring clinical problem within the evolving treatment paradigm37. Indeed, at the time of disease progression on lorlatinib plus capmatinib therapy, a subset of the known metastatic foci of disease were continuing to respond, whereas others had developed resistance—demonstrating clearly discordant inter-metastatic tumor behavior. Furthermore, the serial cfDNA findings were supportive of genotypic polyclonal resistance, with identification in the lorlatinib/capmatinib-resistant sample of new BRCA2 loss, EGFR amplification, and FGFR1 amplification (all likely subclonal events) on top of the known ROS1 rearrangement, TP53 variant, and MET D1246N resistance mutation that had been detected in the contemporaneous liver tumor biopsy. These findings highlight the complementary advantage of liquid biopsy in capturing heterogeneous resistance mechanisms that may exist across distinct metastatic sites within a patient and are missed in single-lession tumor biopsies. In addition, the complex evolution of resistance—as exemplified by (1) the stepwise acquisition of lorlatinib-refractory MET amplification followed then by capmatinib-refractory MET D1246N mutation with loss of MET amplification and (2) the emergence of polyclonal resistance-underscores that therapeutic strategies aimed at delaying or preventing resistance are ultimately more likely to be truly transformative than those aimed at overcoming resistance once it has already developed.
We have additionally uncovered multiple layers of MET axis activation in this case, differentially engaged throughout distinct therapy exposures. MET was overexpressed in the treatment-naïve tumor, and this persisted throughout the patient’s disease course. Despite the protein overexpression of MET, in the absence of additional genomic alterations in MET, tumor cells were ROS1-dependent, as evidenced by the initial durable responses to ROS1 inhibitors crizotinib (14 months) and then lorlatinib (30 months). The subsequent MET amplification event likely served to engage MET signaling as a true bypass pathway, driving the resistance to ROS1-targeted therapy and conferring MET dependency. Thus, this case supports that high-level expression of MET alone without concomitant genomic lesions such as amplification is an insufficient oncogene or resistance driver. Moreover, the case illustrates that distinct mechanisms of MET activation are not functionally equivalent, emphasizing the importance of understanding the differences in various diagnostic methods used (such as differences between FISH, IHC, and NGS testing for MET), to enable accurate interpretation and clinical application of the results.
Intriguingly, we found that ROS1 protein expression was ‘lost’ despite retained genomic ROS1 rearrangement at the time of MET amplification-driven ROS1 TKI resistance. The loss of ROS1 expression in the resistant tumor—while concordant with phenotypic lack of sensitivity to ROS1 TKI—raises important questions about the continued molecular changes involving the original oncogenic driver during targeted therapy. Case reports have detailed the loss of fusion or mutant oncoprotein expression in lineage-transformed TKI-resistant lung cancers, including EGFR-mutant and ROS1-rearranged NSCLC38,39. Furthermore, studies in cell culture models have demonstrated that reduction of ROS1 protein expression or phosphorylation can also occur in the context of ROS1 TKI resistance and activation of off-target bypass pathways15,40. The genetic and epigenetic mechanisms mediating the downregulation or loss of the original oncoprotein expression remain to be elucidated.
In summary, this report confirms that MET amplification is a bona fide driver of ROS1 TKI resistance in ROS1-rearranged lung cancer and that a combination of CNS-penetrant and selective MET and ROS1 inhibitors can induce intra- and extracranial responses. Our findings from the integrated clinical and molecular analyses leveraging multiple serial tumor and plasma samples highlight both the successes and salient challenges embedded in the current era of precision oncology, including the stepwise acquisition of distinct resistance mechanisms with sequential targeted therapies, dynamic and multi-faceted changes involving the original oncogenic driver and the bypass signaling axis, and the emergence of polyclonal resistance resulting in therapy-refractory tumor. Efforts for comprehensive molecular testing at the time of acquired drug resistance will continue to be clinically relevant and potentially guide subsequent therapy selection. Yet these efforts will need to be complemented by discovery efforts focused on developing avenues for fundamentally altering the natural course of cancer and circumventing the evolution of resistance upfront.
Methods
Participant
The patient was treated with commercial supplies of lorlatinib plus capmatinib (off-label use) after a detailed discussion of the potential benefits and risks of the therapeutic regimen. The patient provided informed consent for the treatment. The patient signed written informed consent under an institutional review board (IRB)-approved protocol for electronic medical record review to extract relevant clinicopathologic data, including treatment history and outcomes, and for all indicated sample collections and analyses. The radiographic responses to therapy were assessed by an independent radiologist (S.R.D.) according to the RECIST version 1.1 criteria for extracranial disease and modified RECIST version 1.1 criteria for intracranial disease. AEs were graded according to the CTCAE version 5.0.
Materials and molecular testing
Tumor samples were obtained either by surgical resection, core needle biopsy, or thoracentesis. Formalin-fixed paraffin-embedded specimens derived from tumor samples were reviewed by an MGH pathologist (M.M.K.) to confirm the histopathological diagnosis and to ensure adequate tumor cellularity for analysis. All NGS testing was performed on a clinical basis using extracted DNA and amplicon sequencing with the UPMC Oncomine platform in Pittsburgh, PA. ROS1 rearrangement and EGFR and MET copy number gains were assessed using FISH. Briefly, 5-micron sections of formalin-fixed paraffin-embedded tumor material were prepared, and an H&E section was reviewed to select regions for hybridization that contain a majority of tumor cells. A break-apart probe set targeting the 5′ upstream region of ROS1 (RP11-359J11) and the 3′ downstream region of ROS1 (RP11-1036C2) was hybridized, as previously described41. The number of cells containing break-apart signals or rearrangement was scored out of a total 50-cell count. A ROS1 rearrangement was reported if more than 15% of cells show split signals. For EGFR, we used a dual-color FISH assay using Bacteria Artificial Chromosome probe CTD-211A18 (chromosome 7p EGFR locus) and a copy number control (centromere 7; Abbott-Vysis), and for MET, we used a dual-color FISH assay using Bacterial Artificial Chromosome probe CTB-13N12 (chromosome 7q MET locus) and a copy number control (centromere 7 or CEP7; Abbott-Vysis). Signal quantitation was used to generate an EGFR or MET to centromere 7 ratio. For example, a ratio of MET:CEP7 of 1.8 to 2.2 is considered borderline for amplification. A ratio between 2.2 to 5.0 is considered low-level amplification. A ratio greater than 5.0 or clustered MET signals that are too numerous to count would be considered highly amplified42,43. ROS1 and MET IHC were performed with Automated Stainer Leica Bond III (Leica Biosystems, Danvers, MA) using a ROS1 antibody (clone Rabb4, catalog number 32875, dilution 1:200, Cell Signaling Technology, Danvers, MA) and a c-MET antibody (clone SP44, catalog number 790-4430, Roche Diagnostics, Tucson, AZ), respectively. All procedures were performed in a CLIA-compliant environment.
Ethics
The Dana-Farber/Harvard Cancer Center Institutional Review Board (DFCI #13-416) approved this study.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Data availability
All the data generated for this study are available in this article or from the corresponding author upon request.
References
Gendarme, S., Bylicki, O., Chouaid, C. & Guisier, F. ROS-1 fusions in non-small-cell lung cancer: evidence to date. Curr. Oncol. 29, 641–658 (2022).
Lin, J. J. & Shaw, A. T. Recent advances in targeting ROS1 in lung cancer. J. Thorac. Oncol. 12, 1611–1625 (2017).
Davies, K. D. & Doebele, R. C. Molecular pathways: ROS1 fusion proteins in cancer. Clin. Cancer Res. 19, 4040–4045 (2013).
Shaw, A. T. et al. Crizotinib in ROS1-rearranged advanced non-small-cell lung cancer (NSCLC): updated results, including overall survival, from PROFILE 1001. Ann. Oncol. 30, 1121–1126 (2019).
Drilon, A. et al. Long-term efficacy and safety of entrectinib in ROS1 fusion-positive NSCLC. JTO Clin. Res. Rep. 3, 100332 (2022).
Awad, M. M. et al. Acquired resistance to crizotinib from a mutation in CD74-ROS1. N. Engl. J. Med. 368, 2395–2401 (2013).
McCoach, C. E. et al. Resistance mechanisms to targeted therapies in ROS1(+) and ALK(+) non-small cell lung cancer. Clin. Cancer Res. 24, 3334–3347 (2018).
Zhang, Y. et al. Disease progression patterns and molecular resistance mechanisms to crizotinib of lung adenocarcinoma harboring ROS1 rearrangements. NPJ Precis. Oncol. 6, 20 (2022).
Lin, J. J. et al. Spectrum of mechanisms of resistance to Crizotinib and Lorlatinib in ROS1 fusion-positive lung cancer. Clin. Cancer Res. 27, 2899–2909 (2021).
Drilon, A. et al. Repotrectinib (TPX-0005) is a next-generation ROS1/TRK/ALK inhibitor that potently inhibits ROS1/TRK/ALK solvent-front mutations. Cancer Discov. 8, 1227–1236 (2018).
Drilon, A. et al. NVL-520 is a selective, TRK-sparing, and brain-penetrant inhibitor of ROS1 fusions and secondary resistance mutations. Cancer Discov. 13, 598–615 (2023).
Lin, J. J. et al. Camidge. Intracranial and systemic efficacy of repotrectinib in advanced ROS1 fusion-positive (ROS1+) non-small cell lung cancer (NSCLC) and central nervous system metastases (CNS mets) in the phase 1/2 TRIDENT-1. 2023 ASCO Annual Meeting. J. Clin. Oncol. 41 (suppl 16), abstr 9017 (2023).
Li, W. et al. 14MO Updated efficacy and safety of taletrectinib in patients (pts) with ROS1+ non-small cell lung cancer (NSCLC). Adv. NSCLC https://doi.org/10.1016/S1556-0864(23)00268-X (2023).
Recondo, G., Che, J., Janne, P. A. & Awad, M. M. Targeting MET dysregulation in cancer. Cancer Discov. 10, 922–934 (2020).
Tyler, L. C. et al. MET gene amplification is a mechanism of resistance to entrectinib in ROS1+ NSCLC. Thorac. Cancer 13, 3032–3041 (2022).
Yang, J., Zhou, P., Yu, M. & Zhang, Y. Case report: high-level MET amplification as a resistance mechanism of ROS1-tyrosine kinase inhibitors in ROS1-rearranged non-small cell lung cancer. Front. Oncol. 11, 645224 (2021).
Schoenfeld, A. J. et al. Tumor analyses reveal squamous transformation and off-target alterations as early resistance mechanisms to first-line osimertinib in EGFR-mutant lung cancer. Clin. Cancer Res. 26, 2654–2663 (2020).
Ramalingam, S. S. et al. Mechanisms of acquired resistance to first-line osimertinib: preliminary data from the phase III FLAURA study. Late-Breaking And Deferred Publication Abstracts Nsclc, Metastatic. Ann. Oncol. 29 (Supplement 8), VIII740 (2018).
Sequist, L. V. et al. Osimertinib plus savolitinib in patients with EGFR mutation-positive, MET-amplified, non-small-cell lung cancer after progression on EGFR tyrosine kinase inhibitors: interim results from a multicentre, open-label, phase 1b study. Lancet Oncol. 21, 373–386 (2020).
Hartmaier, R. J. et al. Osimertinib + Savolitinib to overcome acquired MET-mediated resistance in epidermal growth factor receptor-mutated, MET-amplified non-small cell lung cancer: TATTON. Cancer Discov. 13, 98–113 (2023).
Dagogo-Jack, I. et al. MET alterations are a recurring and actionable resistance mechanism in ALK-positive lung cancer. Clin. Cancer Res. 26, 2535–2545 (2020).
Rosen, E. Y. et al. Overcoming MET-dependent resistance to selective RET inhibition in patients with RET fusion-positive lung cancer by combining Selpercatinib with Crizotinib. Clin. Cancer Res. 27, 34–42 (2021).
Takakura, T. et al. Efficacy of Crizotinib after Entrectinib resistance due to MET polysomy in ROS1-rearranged NSCLC: a case report. JTO Clin. Res. Rep. 4, 100523 (2023).
Remon, J. & Soria, J. C. Improving brain penetration of kinase inhibitors in lung cancer patients with oncogene dependency. Ann. Oncol. 28, 196–198 (2017).
Remon, J. et al. MET alterations in NSCLC—current perspectives and future challenges. J. Thorac. Oncol. 18, 419–435 (2023).
Brazel, D., Zhang, S. & Nagasaka, M. Spotlight on Tepotinib and Capmatinib for non-small cell lung cancer with MET exon 14 skipping mutation. Lung Cancer 13, 33–45 (2022).
Paik, P. K. et al. Tepotinib in non-small-cell lung cancer with MET exon 14 skipping mutations. N. Engl. J. Med. 383, 931–943 (2020).
Wolf, J. et al. Capmatinib in MET exon 14-mutated or MET-amplified non-small-cell lung cancer. N. Engl. J. Med. 383, 944–957 (2020).
Le, X. et al. Tepotinib in patients (pts) with advanced non-small cell lung cancer (NSCLC) with MET amplification (METamp). J. Clin. Oncol. 39, 9021–9021 (2021).
Camidge, D. R. et al. Telisotuzumab vedotin (Teliso-V) monotherapy in patients (pts) with previously treated c-Met–overexpressing (OE) advanced non-small cell lung cancer (NSCLC). 2022 ASCO Annual Meeting. J. Clin. Oncol. 40, (suppl 16), abstr 9016 (2022).
Camidge, D. R. et al. Abstract CT214: Preliminary efficacy of telisotuzumab vedotin (Teliso-V) treatment in the 2L/3L setting in MET gene amplified (MET Amp), c-Met protein overexpressing (c-Met OE), non-squamous, non-small cell lung cancer (NSQ NSCLC): retrospective analysis of LUMINOSITY. Cancer Res. https://doi.org/10.1158/1538-7445.AM2023-CT214 (2023).
Li, A. et al. Acquired MET Y1248H and D1246N mutations mediate resistance to MET inhibitors in non-small cell lung cancer. Clin. Cancer Res. 23, 4929–4937 (2017).
Riedel, R. et al. Resistance to MET inhibition in MET-dependent NSCLC and therapeutic activity after switching from type I to type II MET inhibitors. Eur. J. Cancer 179, 124–135 (2023).
Heist, R. S. et al. Acquired resistance to Crizotinib in NSCLC with MET exon 14 skipping. J. Thorac. Oncol. 11, 1242–1245 (2016).
Katayama, R. et al. Cabozantinib overcomes crizotinib resistance in ROS1 fusion-positive cancer. Clin. Cancer Res. 21, 166–174 (2015).
Yakes, F. M. et al. Cabozantinib (XL184), a novel MET and VEGFR2 inhibitor, simultaneously suppresses metastasis, angiogenesis, and tumor growth. Mol. Cancer Ther. 10, 2298–2308 (2011).
Schneider, J. L., Lin, J. J. & Shaw, A. T. ALK-positive lung cancer: a moving target. Nat. Cancer 4, 330–343 (2023).
Lin, J. J. et al. Small cell transformation of ROS1 fusion-positive lung cancer resistant to ROS1 inhibition. NPJ Precis. Oncol. 4, 21 (2020).
Niederst, M. J. et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 6, 6377 (2015).
Davies, K. D. et al. Resistance to ROS1 inhibition mediated by EGFR pathway activation in non-small cell lung cancer. PLoS ONE 8, e82236 (2013).
Dagogo-Jack, I. et al. Molecular analysis of plasma from patients with ROS1-positive NSCLC. J. Thorac. Oncol. 14, 816–824 (2019).
Kwak, E. L. et al. Molecular heterogeneity and receptor coamplification drive resistance to targeted therapy in MET-amplified esophagogastric cancer. Cancer Discov. 5, 1271–1281 (2015).
Lennerz, J. K. et al. MET amplification identifies a small and aggressive subgroup of esophagogastric adenocarcinoma with evidence of responsiveness to crizotinib. J. Clin. Oncol. 29, 4803–4810 (2011).
Acknowledgements
We are grateful to the patient and family who generously contributed to this research. We would like to acknowledge the FISH team from the Center of Integrated Diagnostics at MGH. This work was supported in part by A Breath of Hope Lung Foundation and American Cancer Society Institutional Research Grant (to J.L.S.), by NIH/NCI R01CA164273 (to J.J.L.), and by the Massachusetts General Hospital Targeting a Cure for Lung Cancer Foundation.
Author information
Authors and Affiliations
Contributions
All authors meet all four criteria, including (1) Substantial contributions to the conception or design of the work or the acquisition, analysis, or interpretation of the data, (2) drafting the work or revising it critically for important intellectual content, (3) final approval of the completed version, (4) accountability for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Corresponding author
Ethics declarations
Competing interests
J.L.S. has received honoraria from the Academy of Continued Healthcare Learning, Springer Healthcare, Targeted Oncology, and Dava Oncology. S.R.D. has received honorarium from Elsevier, Siemens; received research funding from Lunit Inc., GE, QURE AI, Vuno; is a member of the Speakers’ Bureau for Siemens; and provides independent image analysis for hospital-contracted clinical trials programs for Merck, Pfizer, Bristol Myers Squibb, Novartis, Roche, Polaris, Cascadian, Abbvie, Gradalis, Bayer, Zai Laboratories, Biengen, Resonance, Analise. J.F.G. has served as a compensated consultant for Amgen, Arcus Biosciences, AI Proteins, AstraZeneca, Beigene, Blueprint Medicines, Bristol Myers Squibb, Genentech/Roche, EMD Serono, InterVenn Biosciences, Gilead Sciences, iTeos Therapeutics, Jounce Therapeutics, Karyopharm Therapeutics, Lilly, Loxo, Merus, Mirati Therapeutics, Pfizer, Sanofi, Silverback Therapeutics, Merck, Moderna Therapeutics, Mariana Oncology, Takeda; has received honorarium from Merck, Pfizer, Novartis, Pfizer, Takeda; has received research funding from Adaptimmune, Alexo Therapeutics, Array BioPharma, AstraZeneca, Blueprint Medicines, Bristol Myers Squibb, Genentech, Jounce Therapeutics, Merck, Moderna Therapeutics, Novartis, Tesaro; has an immediate family member who is an employee with stock and other ownership interests in Ironwood Pharmaceuticals; and has equity in AI Proteins. M.M.K. has acted as a paid consultant for AstraZeneca, Janssen Oncology, Repapre, Innate, Sanofi, Daiichi-Sankyo, and AbbVie and received royalties from Elsevier but declares no non-financial competing interests. T.F.B. has served as compensated consultant for DSMB: Advarra, Inc. (Lantern Pharma), Scientific Advisory Board: (no stock) Janssen Scientific Affairs, LLC, Jazz Pharmaceuticals Inc., Blueprint Medicines Corporation (BPMC), Emd Serono Inc., Mirati Therapeutics, Inc., Amgen, AstraZeneca, and Takeda Pharmaceuticals USA, Inc., and received institutional research funds from Novartis. JJL has served as a compensated consultant for Genentech, Regeneron, C4 Therapeutics, Blueprint Medicines, Nuvalent, Bayer, Elevation Oncology, Novartis, Mirati Therapeutics, Ellipses, CLaiM Therapeutics, Pfizer, Turning Point Therapeutics, and received institutional research funds from Hengrui Therapeutics, Turning Point Therapeutics, Neon Therapeutics, Relay Therapeutics, Bayer, Elevation Oncology, Roche, Linnaeus Therapeutics, Nuvalent, and Novartis. The remaining authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Schneider, J.L., Shaverdashvili, K., Mino-Kenudson, M. et al. Lorlatinib and capmatinib in a ROS1-rearranged NSCLC with MET-driven resistance: tumor response and evolution. npj Precis. Onc. 7, 116 (2023). https://doi.org/10.1038/s41698-023-00464-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41698-023-00464-y
This article is cited by
-
Multiple drugs
Reactions Weekly (2024)
