
Abstract
Liver cancer is the fourth leading cause of cancer-related death. Hepatocellular carcinoma (HCC) is a primary liver cancer that results from chronic hepatitis caused by multiple predisposing factors such as viral infection, alcohol consumption, and non-alcoholic fatty liver disease. Accumulating studies have indicated that dysfunction of the gut epithelial barrier and hepatic translocation of gut microbes may be implicated in the pathogenesis of HCC. However, the translocated bacteria in HCC patients remains unclear. Here, we characterised tumour-associated microbiota in patients with liver cancer and focused on HCC. We observed that the number of amplicon sequence variants in tumour-associated microbiota was significantly higher compared with that in non-tumour regions of the liver. The tumour-associated microbiota consisted of Bacteroidetes, Firmicutes, and Proteobacteria as the dominant phyla. We identified an unclassified genus that belonged to the Bacteroides, Romboutsia, uncultured bacterium of Lachnospiraceae as a signature taxon for primary liver cancer. Additionally, we identified Ruminococcus gnavus as a signature taxon for HCC patients infected with hepatitis B and/or hepatitis C viruses. This study suggests that tumour microbiota may contribute to the pathology of HCC.
Similar content being viewed by others


Introduction
Liver cancer is the fourth leading cause of cancer-related death worldwide (approximately 78,700 cases/year, 8.2% of all cancers)1. The prevalence of hepatocellular carcinoma (HCC), which accounts for more than 80% of primary liver cancers2, is associated with multiple environmental factors such as viral infection with hepatitis B virus (HBV) or hepatitis C virus (HCV), excessive alcohol consumption, and non-alcoholic fatty liver disease (NAFLD). These factors synergistically cause chronic inflammation in the liver, which eventually leads to HCC. In previous cohort studies, < 15% of HBV-infected Taiwanese participants had developed HCC within 13 years3, whereas the rate of HCC was 3% among HCV-infected individuals2. Similarly, the incidence rate of HCC was also at most 30% among patients with either alcohol-related liver disease or NAFLD4. These studies indicate that multiple rather than single predisposing factors are required to promote chronic inflammation.
Accumulating studies have suggested that gut epithelial barrier functions and microbiota are associated with the development of HCC5,6. In the intestines, a single layer of epithelial cells forms a physical barrier to prevent leakage of luminal contents including gut microbes into the body. Notably, patients with chronic liver diseases, such as alcoholic hepatitis, cirrhosis, and HCC, exhibit higher serum lipopolysaccharide (LPS) levels compared with healthy subjects, which indicates increased permeability of the gut epithelial barrier7. An animal study also demonstrated that chemical disruption of the epithelial barrier promotes tumorigenesis in the liver8. Thus, increased gut permeability has been implicated in tumorigenesis in patients with chronic liver diseases. This pathological event largely depends on Toll-like receptor 4 (TLR4) activation by LPS in hepatocytes, because genetic ablation of Tlr4 suppresses HCC development in mice9. Moreover, epithelial barrier dysfunction facilitates translocation of certain bacteria into the liver. For example, mice transplanted with stool from alcohol hepatitis patients show translocation of cytolysin-positive Enterococcus faecalis from the gut to the liver upon administration of alcohol10. Detection of the cytolysin-positive E. faecalis strain is positively associated with mortality of alcohol hepatitis patients. These observations suggest that hepatic translocation of gut microbes is involved in the pathogenesis of other liver diseases such as HCC.
Recently, tumour tissues of several cancer types were found to be colonised with bacteria referred to as cancer microbiota, such as colorectal cancer and stomach cancer as well as tissues considered to be sterile, which included breast cancer and pancreatic ductal adenocarcinoma11,12,13. Cancer microbiota affects cancer pathology by regulating tumorigenesis, cancer progression, and resistance against chemotherapy11,12,14. Fusobacterium nucleatum, which is found in tumour tissues of colorectal cancer, increases proinflammatory cytokines in colorectal cancer of both rodent models and patients14. These studies suggest that cancer microbiota enhances inflammation in the cancer microenvironment.
Similar to heathy human gut microbiota, most cancer microbiotas belong to phyla Proteobacteria, Firmicutes, Actinobacteria, and Bacteroides, which indicates that a major source of cancer microbiota is the gut microbiota13. Considering that the liver is anatomically connected to the intestines via the portal vein and that gut bacteria translocate to the liver in chronic liver disease patients, HCC might be also colonised with cancer microbiota. However, the presence of microbiota in liver cancer and its clinical implications remain unclear.
In this study, to investigate whether bacteria are present in liver cancer, especially HCC, we examined microbiota in liver cancer tissues including both primary HCC and metastatic liver cancer. We observed an increase of the diversity in tumour tissues compared with adjacent non-tumour tissues. This trend was evident in patients without HBV or HCV infection. Linear discriminant analysis revealed Ruminococcus gnavus as a putative biomarker for viral HCC. This study implies the presence of bacteria in liver cancer tissues and an association with hepatic virus infection.
Results
Distinct microbial compositions between tumour and non-tumour regions in the liver
To investigate microbial signatures of liver cancers, we analysed tumour and non-tumoural adjacent regions that were surgically resected from patients with primary (i.e., HCC and cholangiocarcinoma) and metastatic liver cancers (Fig. 1). Quantitative PCR (qPCR) analysis revealed no significant differences in bacterial load between tumour regions and adjacent non-tumoural regions both in primary and metastatic liver cancers (Fig. 2a,b). We subsequently performed 16S ribosomal RNA (rRNA) gene sequencing to dissect the bacterial compositions. To eliminate interference by host-derived reads, we filtered human-associated reads from the 16S rRNA sequencing data in advance. For quality control of the analysis, we sequenced PCR amplicons of negative controls (NCs), namely, sterile water or DNA extracts from empty samples. We confirmed that bacterial families in the NCs were only minimally detected in the microbiota of the tumour and non-tumour regions (Supplemental Fig. S1).

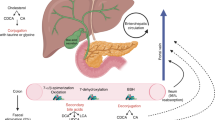
A scheme of the analysis of tumour-associated microbiota in the liver. CC cholangiocarcinoma, HCC hepatocellular carcinoma.

Microbial signature of tumour and non-tumour tissues dissected from primary and metastatic liver cancers. (a,b) Bacterial load in the non-tumour and tumour regions of primary (a) or metastatic liver cancers (b). Bacterial load was assessed by qPCR for 16S rRNA gene. The Y axis indicates the number of bacteria as the logarithm (base 10). (c,d) Species richness (Observed ASVs) of the microbiota in the non-tumour and tumour regions of primary (c) or metastatic liver cancers (d). (e,f) Principal component analysis of unweighted UniFrac distances among the microbiota in the non-tumour and tumour regions of primary (e) or metastatic liver cancers (f). (g) The bacterial composition of the microbiota at the phylum level in the non-tumour (NT) and tumour (T) regions of primary and metastatic liver cancers. Each bar represents an individual sample. (h,i) Discriminating taxa between the non-tumour and tumour regions of primary (h) or metastatic liver cancers (i) determined by LefSe analysis15. The prefix of “uncl_” stands for unclassified bacterium on the SILVA_132 reference database (SSURef_NR99_132_SILVA)36,37,38. Each boxplot represents median, interquartile range (IQR), the lowest and highest values within 1.5 IQRs of the first and third quartiles, and the outliers shown with dots (primary liver cancers: n = 37/group; non-tumour regions of metastatic liver cancers: n = 12; tumour regions of metastatic liver cancers n = 16). Each bar represents mean, and the values of each sample are indicated with dots. Significant differences are indicated at the top of the panels (*p < 0.05; Wilcoxon rank sum test).
We observed that the number of amplicon sequence variants (ASVs) of tumour regions was significantly higher than that of non-tumour regions in primary liver cancers (Fig. 2c). The similar tendency was also observed in metastatic liver cancers, albeit without a significant difference (Fig. 2d). We further assessed beta-diversity by conducting principal component analysis with multiple distance metrics: unweighted- and weighted UniFrac distance, and Bray–Curtis distance. The beta-diversities of tumour regions in primary cancers were also statistically different from those of the non-tumour counterparts in any metric [unweighted UniFrac distance: p = 0.006, weighted UniFrac distance: p = 0.001, Bray–Curtis distance: p = 0.001 (PERMANOVA, number of permutation = 999)] and in metastatic cancer [unweighted UniFrac distance: p = 0.002, weighted UniFrac distance: p = 0.002, Bray–Curtis distance: p = 0.006 (PERMANOVA, number of permutation = 999)] (Fig. 2e,f, and Supplementary Fig. S2a–d).
Taxonomic assignment demonstrated that the hepatic microbiota mainly consisted of bacteria that belonged to Actinobacteria, Bacteroidetes, Firmicutes, and Proteobacteria phyla in patients with both of primary and metastatic liver cancers (Fig. 2g, Supplemental Fig. S1, and Supplemental Data 1). To profile microbes specific for primary or metastatic liver cancers, we analysed the bacterial compositions at the genus level by the linear discriminant analysis effect size (LEfSe) method15. We identified Bacteroides genus, uncultured bacterium that belonged to Lachnospiraceae family, and Romboutsia genus as the feature taxa of the primary liver cancer (Fig. 2h). Moreover, an unclassified genus that belonged to Lachnospiraceae family, Lachnospiraceae NK4A136 group, and uncultured bacterium that belonged to Muribaculaceae family were identified as signature taxa for the metastatic liver cancer (Fig. 2i).
Ruminococcus gnavus as a biomarker for the tumour region in viral HCC patients
Approximately half of the tested HCC patients were infected with HBV and/or HCV (HBV: 13.04%; HCV: 19.57%; Both: 8.70% in HCC samples). We further explored similarities and differences in the tumour-associated microbiota between viral HCC and non-HBV/non-HCV (NBNC) HCC patients. In both viral HCC and NBNC HCC patients, the number of ASVs was significantly greater in the tumour region than in the non-tumour region (Fig. 3a,b). Furthermore, the beta-diversity of microbiota was significantly different between tumour and non-tumour regions in both NBNC HCC [unweighted UniFrac distance: p = 0.044, weighted UniFrac distance: p = 0.013, Bray–Curtis distance: p = 0.028 (PERMANOVA, number of permutations = 999)] and viral HCC [unweighted UniFrac distance: p = 0.014, weighted UniFrac distance: p = 0.085, Bray–Curtis distance: p = 0.005 (PERMANOVA, number of permutations = 999)] patients (Fig. 3c,d, and Supplementary Fig. S3a–d). To identify characteristic taxa of viral or non-viral HCC, we compared the bacterial compositions by the LEfSe method15. We detected the Lachnoclostridium genus as the only characteristic taxon for viral HCC (Fig. 4a). However, phylogenetic analysis by NCBI MOLE-BLAST revealed that the sequences of the Lachnoclostridium genus were homologous to those of Ruminococcus gnavus with more than 97% identity (Fig. 4b,c). Occupation of R. gnavus was 0.5–2% in viral HCC patients, although this genus was not detected in the non-tumour region of viral HCC patients or in any regions of NBNC HCC (Fig. 4d). Thus, R. gnavus is a potential marker taxon to discriminate viral HCC from NBNC HCC.

Microbial signature of the non-tumour and tumour regions of viral and NBNC HCC. (a,b) Species richness (observed ASVs) of the microbiota in the non-tumour and tumour regions of viral (a) or NBNC HCC (b). (c,d) Principal component analysis of unweighted UniFrac distances among the microbiota in the non-tumour and tumour regions of viral (c) or NBNC HCC (d). Each bar represents mean, and the values of each sample are indicated with dots. Significant differences are indicated at the top of the panels (*p < 0.05; Wilcoxon rank sum test).

R. gnavus is a marker taxon for viral HCC. (a) Discriminating taxa between the non-tumour and tumour regions both of viral and NBNC HCC determined by LefSe analysis15. (b,c) Related species of feature sequences assigned as Lachnoclostridium genus. Phylogenetic tree based on 16S rRNA gene sequences shows species homologous to the feature sequences (b), and the table displays sequence identity of the feature sequences to R. gnavus 16S rRNA gene sequence (c). (d) Relative abundance of R. gnavus in the non-tumour (NT) and tumour (T) regions both of viral and NBNC HCC. Each boxplot represents median, interquartile range (IQR), the lowest and highest values within 1.5 IQRs of the first and third quartiles, and the outliers shown with dots. Significant differences are indicated at the top of the panels (*p < 0.05; the Kruskal–Wallis test followed by the pairwise t test with Benjamini–Hochberg method). [NBNC (NT): n = 18; NBNC (T): n = 23; viral (NT): n = 15; viral (T): n = 12].
Discussion
The tumour-associated microbiotas of primary and metastatic liver cancers largely consisted of Bacteroidetes, Firmicutes, and Proteobacteria whose frequencies were highly diverse among the specimens. This observation could be a common feature of tumour-associated microbiota because these three phyla are also major taxa in various tumour tissues such as bone, breast, colon, lung, and ovarian cancers13,16,17,18,19,20. Furthermore, non-gastrointestinal tumour tissues such as lung, ovary, and breast cancers abundantly harbour Actinobacteria, another major phylum of gut microbiota13,16,17,18,19,20. However, our data showed that liver tumour-associated microbiota had little, if any, Actinobacteria, similar to the gastrointestinal tumour-associated microbiota13. Additionally, we found several marker taxa which are associated with primary or metastatic liver cancers. Among them, we detected Bacteroides genera as a marker taxon for the tumour regions of primary liver cancers. The abundance of Bacteroides genera is increased in the gut microbiome of patients with NAFLD, non-alcoholic steatohepatitis, and cirrhosis21,22,23. Based on these studies and our data, the overrepresentation of Bacteroides in primary liver cancers might be implicated in the development of liver cancers, although further investigations are necessary to clarify this notion.
Bacteroidetes, Firmicutes, and Proteobacteria detected in the liver cancer-associated microbiota are major members of the gut commensal microbiota in humans24. Considering the spatial proximity of the liver to the intestines via the portal vein, liver cancer-associated bacteria are most likely attributed to gut microbiota. In support of this view, dysfunction of the gut epithelial barrier is often observed in patients with chronic liver diseases such as alcoholic hepatitis25 and cirrhosis7,23, both of which are predisposing factors for liver cancer. Many studies have shown that gut-derived LPS promotes HCC, whereas our data provide evidence that certain bacteria translocate from the gut to the liver. This event may be implicated in the pathology of carcinogenesis, although further investigations are required to verify this notion.
Accumulating evidence has shown that NBNC HCC patients develop intestinal dysbiosis characterised by overrepresentation of proinflammatory bacteria, such as Escherichia-Shigella and Enterococcus, and underrepresentation of butylate-producing bacteria such as Faecalibacterium, Ruminococcus, and Ruminoclostridium26. Such microbial alterations may contribute to the pathogenesis of NBNC HCC. Similarly, chronic hepatitis patients infected with HBV or HCV also show intestinal dysbiosis. For example, genus Bacteroides, Veillonella were overrepresented in the intestinal microbiota of HBV-positive HCC patients, whereas that of HCV-positive HCC patients was characterised by reduction of microbial diversity in association with overrepresentation of genus Streptococcus and Lactobacillus27,28,29. Additionally, we found that R. gnavus was associated with the tumour region of viral HCC patients. The abundance of R. gnavus increases in patients with inflammatory bowel diseases, especially Crohn’s disease30,31,32. R. gnavus produces glucorhamnan that functions as a ligand of TLR4 and eventually induces production of tumor necrosis factor-alpha (TNF-α) from dendritic cells33. Because TNF-α promotes hepatocellular carcinogenesis34, R. gnavus may contribute to the development of viral HCC. Considering that some patients with metastatic liver cancer carry viable bacteria in tumour tissues35, investigation of bacterial viability in HCC would facilitate understanding the pathological role of tumour-associated microbiota. However, we acknowledge the limitation of our study, which includes the small sample size for a clinical study, unmatched cohort, and insufficient grouping of NBNC HCC based on habitual data such as alcoholic intake. Therefore, further investigations with larger sample size and matched cohort are needed to establish marker taxa for HCC.
In conclusion, we have characterised tumour-associated microbiota in primary and metastatic liver cancers and identified R. gnavus as a marker taxon for viral HCC. Our findings shed light on the microbiome profile of liver cancer, which should facilitate understanding the pathological contribution of tumour-associated microbiota in the liver.
Methods
Sample collection
We enrolled 65 patients who underwent hepatectomy from January 2018 to February 2019 at the National Center for Global Health and Medicine (NCGM). Paired liver tumour and adjacent non-tumour tissues were collected from 19 patients with metastatic liver tumours (16 primary colorectal cancer, 2 primary gastric cancers, and 1 primary endometrial cancer). From the other patients with primary liver cancers, 47 hepatocellular carcinoma samples and 15 cholangiocarcinoma samples were collected. These tissue samples were dissected in operating rooms using a sterile surgical knife and tweezers. We renewed a surgical knife and tweezers before dissecting different regions to avoid cross-contamination. The specimens were collected in sterile cryotubes, immediately frozen with liquid nitrogen, and stored at − 80 °C until the analyses. This study was approved by the NCGM research ethics committees (#2464) and Keio University (#190118-1, 190118-2) and informed consent were obtained before sample collection from these patients. All steps were carried out in accordance with national guidelines and regulations.
DNA extraction and 16S rRNA gene sequencing
Genomic DNA was extracted from liver tissue samples with a DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germany) or QIAamp PowerFecal Pro DNA Kit (Qiagen) in accordance with the manufacturer’s protocols. Before extracting genomic DNA with the DNeasy Blood and Tissue Kit, the tissue samples were homogenised for 10 min with Shake Master Neo (Biomedical Sciences, Tokyo, Japan) and 0.70-mm Garnet PowerBeads (Qiagen). A 16S rRNA genomic library was constructed in accordance with the protocol of the Illumina technical note with some modifications. Briefly, the extracted DNA samples were used as DNA templates in a polymerase chain reaction (PCR) carried out in a reaction mixture of template DNA, KAPA HiFi HotStart Ready Mix, and primers specific for the 16S rRNA V3–V4 region under the following conditions: initial denaturation at 95 °C for 3 min, followed by 35 cycles at 95 °C for 30 s, 55 °C for 30 s, and 72 °C for 30 s. A final elongation step was performed at 72 °C for 5 min. The amplicons were purified with AMPure XP beads (Beckman Coulter, Brea, CA, USA) and attached to dual indices by index PCR with a Nextera XT Index kit (Illumina, San Diego, CA, USA). The libraries were purified with AMPure XP beads (Beckman Coulter). The purified libraries were diluted to 4 nM with Tris–HCl buffer and pooled. The libraries were then sequenced on a Miseq (Illumina) with 300 bp paired-end reads. As negative controls, we sequenced PCR amplicons of negative controls: sterile water or DNA extracts from empty samples.
Real-time quantitative PCR to estimate bacterial load
Real-time quantitative PCR (qPCR) to estimate bacterial load in liver tissues was performed in accordance with a previously published protocol11. Briefly, 2 µl of the extracted DNA was added to a mixture of 5 µl SsoAdvanced Universal SYBR Green Supermix, 2.5 µl sterile water, and 0.5 µl primer mixture containing a forward primer (5ʹ-CCTACGGGNGGCWGCAG-3ʹ) and reverse primer (5ʹ-GACTACHVGGGTATCTAATCC-3ʹ) for the 16S rRNA V3–V4 gene region. The qPCR was performed on a CFX Connect real-time PCR analysis system (Bio-Rad, Tokyo, Japan) at 98 °C for 3 min with 40 cycles at 94 °C for 15 s, 55 °C for 10 s, and 60 °C for 1 min, and completed with melting curve analysis. Each sample was quantified in duplicate. A standard curve was produced with a dilution series (0, 0.05, 0.1, 0.2, 0.8, 3.2, 12.8, and 51.8 pg/µl) of Escherichia coli DNA. The standard curve was used to calculate the bacterial number in each sample with the estimation that a bacterium contains 0.005 pg DNA. The Ct values of each sample were compared with that of the standard curve and the bacterial number was calculated as the common log.
Microbiome analysis
After removing human-associated contaminants using Bowtie2 with the GRch37 index, FASTQ files were analysed using the QIIME2 pipeline (QIIME2 version 2020.2). After conversion to the qza format, the sequence data were demultiplexed and summarised using QIIME2 paired-end-demux. Then, the sequences were trimmed and denoised with the dada2 plugin for QIIME2. Taxonomic assignment was performed using a naïve Bayes fitted classifier trained on the SILVA_132 reference database (SSURef_NR99_132_SILVA) with the feature-classifier plugin for QIIME236,37,38. The phylogenetic tree for diversity analysis was reconstructed using QIIME2 align-to-tree-mafft-fasttree. Diversity analysis ware performed with QIIME2 core-metrics-phylogenetic. Relative abundances of each taxon were calculated using the taxa collapse QIIME2 plugin.
Statistics
To analyse the number of observed ASVs, bacterial load, and the abundance of each bacterium, statistical differences were examined by the Student’s t test for comparisons between two groups or Tukey’s multiple comparison test to compare multiple groups in the case of homogenous variance. In the case of heterogeneous variance, statistical differences were examined by the Wilcoxon rank-sum test for two groups or the Kruskal–Wallis test followed by the pairwise t test with Benjamini–Hochberg method. Identification of feature taxa was performed by the LEfSe method15. Statistical analyses with the exception of LefSe analysis and visualisation were performed with R version 4.0.2.
Data availability
Datasets from DDBJ (accession# DRA 011436) are publicly available. The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Bray, F. et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 68, 394–424 (2018).
El-Serag, H. B. Epidemiology of viral hepatitis and hepatocellular carcinoma. Gastroenterology 142, 1264-1273.e1 (2012).
Chen, C. J. et al. Risk of hepatocellular carcinoma across a biological gradient of serum hepatitis B virus DNA Level. J. Am. Med. Assoc. 295, 65–73 (2006).
Pocha, C. & Xie, C. Hepatocellular carcinoma in alcoholic and non-alcoholic fatty liver disease—One of a kind or two different enemies?. Transl. Gastroenterol. Hepatol. 4, 72–72 (2019).
Tripathi, A. et al. The gut–liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 15, 397–411 (2018).
Yu, L. X. & Schwabe, R. F. The gut microbiome and liver cancer: Mechanisms and clinical translation. Nat. Rev. Gastroenterol. Hepatol. 14, 527–539 (2017).
Lin, R. S. et al. Endotoxemia in patients with chronic liver diseases: Relationship to severity of liver diseases, presence of esophaegeal varices, and hyperdynamic circulation. J. Hepatol. 22, 165–172 (1995).
Achiwa, K. et al. DSS colitis promotes tumorigenesis and fibrogenesis in a choline-deficient high-fat diet-induced NASH mouse model. Biochem. Biophys. Res. Commun. 470, 15–21 (2016).
Dapito, D. H. et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 21, 504–516 (2012).
Duan, Y. et al. Bacteriophage targeting of gut bacterium attenuates alcoholic liver disease. Nature 575, 505–511 (2019).
Geller, L. T. et al. Potential role of intratumor bacteria in mediating tumor resistance to the chemotherapeutic drug gemcitabine. Science 357, 1156–1160 (2017).
Riquelme, E. et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell 178, 795-806.e12 (2019).
Nejman, D. et al. The human tumor microbiome is composed of tumor type-specific intracellular bacteria. Science 368, 973–980 (2020).
Kostic, A. D. et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 14, 207–215 (2013).
Segata, N. et al. Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60 (2011).
Banerjee, S. et al. Distinct microbial signatures associated with different breast cancer types. Front. Microbiol. 9, 951 (2018).
Banerjee, S. et al. The ovarian cancer oncobiome. Oncotarget 8, 36225–36245 (2017).
Greathouse, K. L. et al. Interaction between the microbiome and TP53 in human lung cancer. Genome Biol. 19, 123 (2018).
Urbaniak, C. et al. Microbiota of human breast tissue. Appl. Environ. Microbiol. 80, 3007–3014 (2014).
Urbaniak, C. et al. The microbiota of breast tissue and its association with breast cancer. Appl. Environ. Microbiol. 82, 5039–5048 (2016).
Loomba, R. et al. Gut microbiome-based metagenomic signature for non-invasive detection of advanced fibrosis in human nonalcoholic fatty liver disease. Cell Metab. 25, 1054-1062.e5 (2017).
Boursier, J. et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 63, 764–775 (2016).
Ponziani, F. R. et al. Hepatocellular carcinoma is associated with gut microbiota profile and inflammation in nonalcoholic fatty liver disease. Hepatology 69, 107–120 (2019).
Arumugam, M. et al. Enterotypes of the human gut microbiome. Nature 473, 174–180 (2011).
Bishehsari, F. et al. Alcohol and gut-derived inflammation. Alcohol Res. Curr. Rev. 38, e-1-e-9 (2017).
Liu, Q. et al. Alteration in gut microbiota associated with hepatitis B and non-hepatitis virus related hepatocellular carcinoma. Gut Pathog. 11, 1 (2019).
Zeng, Y. et al. Gut microbiota dysbiosis in patients with hepatitis B virus-induced chronic liver disease covering chronic hepatitis, liver cirrhosis and hepatocellular carcinoma. J. Viral Hepat. 27, 143–155 (2020).
Wang, J. et al. Gut microbial dysbiosis is associated with altered hepatic functions and serum metabolites in chronic hepatitis B patients. Front. Microbiol. 8, 2222 (2017).
Inoue, T. et al. Gut dysbiosis associated with hepatitis C virus infection. Clin. Infect. Dis. 67, 869–877 (2018).
Willing, B. P. et al. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology 139, 1844-1854.e1 (2010).
Hall, A. B. et al. A novel Ruminococcus gnavus clade enriched in inflammatory bowel disease patients. Genome Med. 9, 103 (2017).
Nishino, K. et al. Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. J. Gastroenterol. 53, 95–106 (2018).
Henke, M. T. et al. Ruminococcus gnavus, a member of the human gut microbiome associated with Crohn’s disease, produces an inflammatory polysaccharide. Proc. Natl. Acad. Sci. U.S.A. 116, 12672–12677 (2019).
Jing, Y. et al. Tumor necrosis factor-α promotes hepatocellular carcinogenesis through the activation of hepatic progenitor cells. Cancer Lett. 434, 22–32 (2018).
Bullman, S. et al. Analysis of Fusobacterium persistence and antibiotic response in colorectal cancer. Science 358, 1443–1448 (2017).
Glöckner, F. O. et al. 25 years of serving the community with ribosomal RNA gene reference databases and tools. J. Biotechnol. 261, 169–176 (2017).
Yilmaz, P. et al. The SILVA and ‘all-species Living Tree Project (LTP)’ taxonomic frameworks. Nucleic Acids Res. 42, D643–D648 (2014).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596 (2013).
Acknowledgements
We thank Masato Hirota and Yusuke Kinashi for technical assistance. This study was supported by grants from the National Center for Global Health and Medicine (30-1021 to NT and YIK, 30-1006 and 20A1007 to YIK), the Japan Society for the Promotion of Science (20H00509 and 20H05876 to KH), AMED-Crest (18gm1010004h0103, 19gm1010004s0104, 20gm1010004h0105, and 20gm1310009h000 to KH), JST-Crest (20-191030457 to KH), Yakult Foundation (KH), SECOM Science and Technology Foundation (KH), Takeda Science Foundation (KH), The Asahi Glass Foundation (KH), The Science Research Promotion Fund, and The Promotion and Mutual Aid Corporation for Private Schools of Japan (KH).
Author information
Authors and Affiliations
Contributions
Y.I.K. and K.H. conceived the study. T.Y. designed experiments. N.T, N.K., and Y.I.K collected the samples and provided the sample information. S.K. and T.Y. performed the experiments. S.K analysed the data with support from T.Y. S.K wrote the manuscript. T.Y. and K.H. revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Komiyama, S., Yamada, T., Takemura, N. et al. Profiling of tumour-associated microbiota in human hepatocellular carcinoma. Sci Rep 11, 10589 (2021). https://doi.org/10.1038/s41598-021-89963-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-89963-1
This article is cited by
-
Cryptogenic non-cirrhotic HCC: Clinical, prognostic and immunologic aspects of an emerging HCC etiology
Scientific Reports (2024)
-
Causal relationship between gut microbiota and cancers: a two-sample Mendelian randomisation study
BMC Medicine (2023)
-
Gut dysbiosis in Thai intrahepatic cholangiocarcinoma and hepatocellular carcinoma
Scientific Reports (2023)
-
Western diet contributes to the pathogenesis of non-alcoholic steatohepatitis in male mice via remodeling gut microbiota and increasing production of 2-oleoylglycerol
Nature Communications (2023)
-
Awareness of intratumoral bacteria and their potential application in cancer treatment
Discover Oncology (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
