
Abstract
Socially affected traits are affected by direct genetic effects (DGE) and social genetic effects (SGE). DGE and SGE of an individual directly quantify the genetic influence of its own phenotypes and the phenotypes of other individuals, respectively. In the current study, a total of 3,276 Large White pigs from different pens were used, and each pen contained 10 piglets. DGE and SGE were estimated for six socially affected traits, and then a GWAS was conducted to identify SNPs associated with DGE and SGE. Based on the whole-genome re-sequencing, 40 Large White pigs were genotyped and 10,501,384 high quality SNPs were retained for single-locus and multi-locus GWAS. For single-locus GWAS, a total of 54 SNPs associated with DGE and 33 SNPs with SGE exceeded the threshold (P < 5.00E-07) were detected for six growth traits. Of these, 22 SNPs with pleiotropic effects were shared by DGE and SGE. For multi-locus GWAS, a total of 72 and 110 putative QTNs were detected for DGE and SGE, respectively. Of these, 5 SNPs with pleiotropic effects were shared by DGE and SGE. It is noteworthy that 2 SNPs (SSC8: 16438396 for DGE and SSC17: 9697454 for SGE) were detected in single-locus and multi-locus GWAS. Furthermore, 15 positional candidate genes shared by SGE and DGE were identified because of their roles in behaviour, health and disease. Identification of genetic variants and candidate genes for DGE and SGE for socially affected traits will provide a new insight to understand the genetic architecture of socially affected traits in pigs.
Similar content being viewed by others

Introduction
Social interactions between individuals are plentiful, which could contribute to pig’s growth and welfare1,2,3,4. Social cooperation among pigs (such as group behaviour) would result in positive effects, whereas the competition (such as tail biting) may result in negative effects. Considering the social interaction in the genetic evaluation model, the phenotypic value should be decomposed into a direct effect due to its own characteristic and a social effect originating from others5. Both effects can be further decomposed into a heritable component and a non-heritable component. The heritable component of the social effect its called the social genetic effect (SGE)6,7. Griffing et al. (1967) first reported that the social effects among group individuals distinctly affected the selection processes in a group selection5. Amelie Baud et al. (2017) reported that the social genetic effect explained up to 29% of phenotypic variance for health and disease traits in mice, that showed the contribution of the social genetic effect was more than the direct genetic effect (explained 21% of phenotypic variance) in some cases8. However, the classical quantitative genetic model ignored the SGE among group members in the past several decades. In that case, the genetic variance for the socially affected trait would be incorrectly estimated due to the ignorance of SGE5,8.
Previous studies have been demonstrated that the SGE plays an important role in genetic evaluation4 and that their genetic parameters for important traits were estimated in pigs4,5,9. For average daily gain (ADG), the genetic correlation between direct genetic effect and social effect was low to moderate (0.24) in pigs10. The model ignored the pen effects and showed that the heritability of SGE was approximately were about 0.00311 and 0.000712 for ADG. The reported social heritabilities were 0.007, 0.001, and 0.003 for body weight, carcass backfat depth, and carcass lean meat content in heavy pigs13, respectively. Moreover, Rostellato et al. (2015) reported that the social genetic effect contributed largely to total genetic variance for the carcass traits (ranging from 33.2% to 35%) in pigs13. Canario et al. (2017) showed that the heritability of SGE was 0.001 to 0.004 for pig growth rate and its genetic correlations among direct and SGE were −0.28 to 0.12 in a Swedish pig population3.
In general, the pigs were grouped into different pens, and the social effects among members strongly affected pig productive performance. In pigs, the estimation of the social genetic effect was mainly focused on ADG14, feed intake, backfat and muscle depth7. Several models that considered different fixed factors were built to estimate SGE. The animal model was first extended to SGE in Japanese quail15. This model incorporated the competitive effects in classical BLUP (best linear unbiased prediction), and substantially resulted in an extra selection response. Muir16 and Bijma et al.17,18 presented a full model that simultaneously contained both direct genetic effects and social genetic effects. They estimated SGEs for growth traits and found that SGE contributed the main part of genetic variance in growth rate and feed intake in pigs. In summary, the previous studies on SGE mainly focused on the development of an evaluation model and the estimation of genetic parameters. The genetic architecture and causal pathway for SGE are still unknown.
Pig growth traits are affected by direct genetic effects (DGE) and SGE. To investigate the genetic architecture of DGE and SGE, this study conducted a GWAS for DGE and SGE of six growth traits using the whole-genome re-sequenced data from the Large White pig population. The purpose of this study was to estimate the DGE and SGE for six socially affected traits, and reveal the genomic variation and candidate genes for DGE and SGE of six growth traits in pigs.
Results
Summary of estimated DGE and SGE
Using this full model Y = Xb + ZDaD + ZSaS + Wl + Vg + e (see Materials and Methods), the DGE and SGE were estimated for each socially affected trait in Large White pigs. Table S1 listed the describe statistics about the DGE and SGE for these six traits. The DGE and SGE contained residual effects of 40 genotyped pigs were separately used to conduct GWAS. Variance components were presented in Table 1 for six socially affected traits.
The single-locus GWAS results using GEMMA
For DGE
A single marker test was performed to identify SNPs associated with DGE of 6 growth traits. The Table 2 and Fig. 1 show the GWAS results for DGE of 6 growth traits. And Q-Q plots are shown in Supplementary Fig. 1. A total of 54 SNPs and 20 genes were detected for DGE in this study.

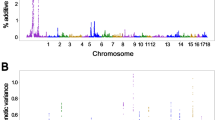
Manhattan plots of genome-wide association analysis results associated to DGE for six growth traits using GEMMA software. The threshold for genome-wide and suggestive significance was set at P = 5.00 × 10−8 and P = 5.00 × 10−7, respectively.
At the genome-wide threshold (P < 5 × 10−8), 1 SNP was associated with dgeADG (Fig. 1A) and 1 with dgeB100 (Fig. 1C). For dgeADG, the genome-wide significant chromosome region was located in SSC14: 98.69–98.73 Mb. The top SNP (SSC14: 98711408 bp, P = 2.05 × 10−8) was located in the PRKG1 gene. For dgeB100, the most significant chromosome region was located in SSC6: 39.75–39.79 Mb, and the top SNP (SSC6: 39767887 bp, P = 1.49 × 10−8) was located in POP4 and the PLEKHF1 gene.
At the suggestive threshold (P < 5 × 10−7), 9 SNPs were associated with dgeADG, 19 with dgeD100, 6 with dgeB100, 2 with dgeADFI, 4 with dgeFCR, and 12 with dgeRFI (Table 2). Of these, 5 top SNPs (including SSC17: 65146021 bp for dgeADG (Fig. 1A); SSC2: 14306048 bp, SSC4: 15405575 bp and SSC7: 124864925 bp for dgeD100 (Fig. 2B); SSC3: 129695008 bp for dgeRFI (Fig. 1F)) were also interesting, since the P-value of these SNPs were closed to genome-wide threshold. In the region of SSC2: 14.29–14.33 Mb, the top SNP (SSC2: 14306048 bp, P = 5.99 × 10−8) was located in LOC110259270 gene for dgeD100. In addition, a clear peak was observed within the region SS16: 23.20–23.23 Mb for dgeD100, however, no candidate gene was identified in this region.

Manhattan plots of genome-wide association analysis results associated to SGE for six growth traits using GEMMA software. The threshold for genome-wide and suggestive significance was set at P = 5.00 × 10−8 and P = 5.00 × 10−7, respectively.
For SGE
To explore the potential genetic variants associated with SGE, a whole-genome re-sequencing association study was conducted for SGE of 6 growth traits. All of the Manhattan plots are shown in Fig. 1 and the Q-Q plots are shown in Supplementary Fig. 2. At the genome-wide threshold (P < 5 × 10−8), 4 identified SNPs were associated with SGE of days to 100 kg (sgeD100), 7 with sgeRFI. At the lenient threshold (P < 5 × 10−7) for suggestive associations, 1 SNP was associated with SGE of average daily gain (sgeADG), 14 with sgeD100, 2 with sgeADFI, 4 with sgeFCR, and 1 with sgeRFI, but no association was found for sgeB100 (Table 3 and Fig. 2).
For sgeADG, only one suggestive SNP (P = 3.80 × 10−7) was distributed on SSC14 (Table 3 and Fig. 2A). One candidate gene (PRKG1) was found for sgeADG within the ±20 kb region of the top SNP (SSC14: 98711408 bp).
The results of sgeD100 are shown in Table 3 and Fig. 2B. The most significant locus with 4 consecutive SNPs was identified on SSC7, and the P-value of the top SNP (SSC7: 124864925 bp) was 1.03 × 10−8. Furthermore, a total of 14 suggestive significant SNPs (P < 5 × 10−7) for sgeD100 was characterized on SSC2, SSC3, SSC4, SSC8, SSC9, SSC16 and SSC17. On SSC2, the SNP (SSC2: 14306048 bp) was located within the region of the uncharacterized gene (LOC110259270). Two genes (GAR1, and CFI) near the SNP (SSC8: 112431517 bp, P = 3.41 × 10−7) were found for sgeD100.
A total of 2 suggestive significant SNPs (P < 5 × 10−7) on SSC8 and SSC14 were detected for sgeADFI (Table 3 and Fig. 2D). In these two significant chromosome regions (SSC8: 88.65–88.69 Mb and SSC14: 116.10–116.14 Mb), two genes (SLC7A11, SORCS3) were found.
For sgeFCR, a total of 4 associations reached the suggestive significant threshold (P < 5 × 10−7) on SSC1 and SSC6 (Table 3 and Fig. 2E). In the significant chromosome region (SSC1: 57.25–57.29 Mb), the top SNP (SSC3: 57267261, P = 2.83 × 10−7) was located in the GABRR2 gene.
A total of 8 SNPs were found on SSC1 and SSC16 for sgeRFI (Table 3 and Fig. 2F). Two SNPs located on SSC1 and five SNPs located on SSC16 were found to reach the genome-wide significance threshold (P < 5 × 10−8). The top SNPs (SSC16: 43805766) with the lowest P-value of 3.72 × 10−9 showed the strongest association effect in the region of 43.79–43.83 Mb. In the significant region of SSC16: 43.79–43.83 Mb, five consecutive SNPs and one promising candidate gene (ADAMTS6) were identified for sgeRFI. The second top SNP (SSC1: 312947811) with a low P-value of 1.18 × 10−8 was found in the region of 312.93–312.97 Mb. In addition, one candidate gene (LPAR1) related to disease and healthy were found for sgeRFI in the chromosome regions of SSC1: 251.97–252.01 Mb.
A total of 15 significant SNPs were shared by DGE and SGE that were distributed on SSC1, SSC2, SSC3, SSC4, SSC7, SSC9, SSC14 and SSC16 (Table 4). In the region of SSC14: 98.69–98.73 Mb, the top SNP (SSC14: 98711408 bp) was located in the PRKG1 gene for dgeADG and sgeADG. In the region of SSC2: 14.29–14.33 Mb, the top SNP (SSC2: 14306048 bp) was located in the LOC110259270 gene, and in the region of SSC8:112.41–112.45 Mb, the top SNP (SSC8: 112431517 bp) was located in GAR1 and the CFI gene for dgeD100 and sgeD100, respectively.
The multi-locus GWAS results using FarmCPU
The summary of GWAS results using the FarmCPU approach is listed in Tables S2 and S3. In total, the current study identified 51 and 101 SNPs for DGE and SGE, respectively. For DGE, 4 SNPs were associated with dgeB100, 45 with dgeD100 and 2 with dgeFCR (Table S2), respectively. For SGE, 3 SNPs were associated with sgeADG, 63 with sgeB100, 18 with sgeD100, 7 with sgeADFI, 5 with sgeFCR and 4 with sgeRFI (Table S3), respectively. Among these SNPs, a total of 7 SNPs were shared by dgeD100 and sgeD100 (Table 4). Additionally, three candidate genes were found at 20 kb around these top SNPs for dgeD100 and sgeD100. Among theme, two genes, NR3C2 and PKD2, were separately located in SSC8: 80.56–80.60 Mb and 131.00–131.04 Mb. One gene, LYPLAL1, was located in the region of SSC10: 8.96–9.00 Mb.
The multi-locus GWAS results using FASTmrEMMA
The final results of the FASTmrEMMA method are shown in Tables S4 and S5. For DGE, 16, 18 and 12 putative QTNs were detected for dgeADFI, dgeB100 and dgeD100 (Table S4), respectively. Of these, a QTN SSC8: 16438396 bp located in the region 16.42–16.46 Mb was detected by single-locus GWAS and multi-locus GWAS for dgeB100. This QTN explained 33.19% of the phenotypic variance and included −3.30 QTN effect. For SGE, 19, 15, 14, 5, 15 and 8 putative QTNs were identified for sgeRFI, sgeFCR, sgeD100, sgeB100, sgeADG and sgeADFI (Table S5), respectively. In the region of SSC17: 9.68–9.72 Mb, a QTN located on SSC17: 9697454 bp was identified by single-locus GWAS and multi-locus GWAS for sgeD100. This QTN with 6.04 QTN effect explained 14.30% of the phenotypic variance. Furthermore, a total of 5 putative QTNs were shared by SGE and DGE of each trait (Table 4). For the ADFI, B100 and D100 trait, the QTN SSC10: 11015344 was shared by DGE and SGE. Four QTNs including SSC4: 140729546 bp, SSC8: 33116699 bp, SSC11: 15329359 bp and SSC13: 21796761 bp, were detected in both dgeD100 and sgeD100.
Interestingly common loci in the three methods
This study performed a GWAS for DGE and SGE using the GEMMA, FarmCPU and FASTmrEMMA methods. Interestingly, two identified SNPs were validated in three methods (Table 5). One SNP (SSC8: 16438396 bp) was associated with dgeB100. One SNP (SSC17: 9697454 bp) was associated with sgeD100.
Discussion
The estimation of genetic effect
Many studies have reported that the social genetic effect are widespread in hens19, mice20, and pigs7. However, at present, very few GWAS for SGE are conducted in pigs because the genetic parameters of SGE are difficult to measure. In past several decades, the heritability of social effects were estimated to be very low in pigs11,21,22 using the common REML method. Therefore, social effects were often ignored in past pig breeding. Instead of common heritability of social effects, the ratio between total heritable variance and phenotypic variance was used as a measure of the importance of SGE. On that basis, the social effect was found to contribute the heritable variance in growth rate and feed intake23. This study used the full model (including DGE and SGE) described by Bergsma et al.23 to estimate DGE and SGE of six growth traits. In this model, on one hand, the fixed effects contained sex, tested year and month, the random litter effect and the random group effect were corrected. On the other hand, additional 3236 phenotypic records for ADG, D100, B100, ADFI, FCR and RFI and pedigree information were added into mixed model equations, which improved the prediction accuracy of DGE and SGE for these traits.
The genetic architecture for SGE
Currently, the genomic information has increased the understanding of complex traits, but the majority of studies have focused only on the DGE of studied traits. Published studies demonstrated that ignoring SGE may severely bias estimates of DGE, and the genetic basis of SGE may be different from the genetic basis of DGE8. If SGE contributes to the studied traits, the study of genetic architecture should simultaneously consider DGE and SGE for socially-affected traits. However, few studies have focused on the genetic architecture of SGE. This study simultaneously quantified the SGE and DGE, and performed GWAS to reveal the genetic architecture of DGE and SGE for six growth traits in Large White pigs. Based on the Illumina PorcineSNP60 v2 BeadChip panel, Hong et al.24 identified several genomic regions and candidate genes associated with SGE for ADG in Landrace pigs. In their study, the individual ADG and the average ADG of unrelated pen-mates were directly used as DGE and SGE, respectively. In our study, the SGE and DGE were estimated for six growth traits, using the full animal model. Then, (1) pseudo phenotype values including DGE and residual effects were used to perform GWAS for DGE of six growth traits; and (2) for SGE of six growth traits, pseudo phenotype values including SGE and residual effects were used to perform a GWAS. Using these pseudo phenotype values, this study revealed the genetic architecture of DGE and SGE for six growth traits in Large White pigs. This study detected 151 and 205 SNPs for DGE and SGE, respectively. These results did not confirm the results with Hong et al.24. In the present study, the power of QTL detection could be limited by the low estimated heritability of SGE and the small population size in this study. Furthermore the assessment of SGE was different between our study and the previous study24. In a previous study, the average ADG of unrelated pen mates was used to detect genetic variants of SGE. However, in our study, the full model Y = Xb + ZDaD + ZSaS + Wl + Vg + e (see Materials and Methods) were used to estimate the SGE of each trait, and then the estimated SGE wasused to perform GWAS for SGE of each trait. Our study offered a new insight and effective method for the genetic architecture of socially affected traits in pigs.
Comparing the single-locus and multi-locus GWAS
To minimize the number of false positives and obtain credible results, the SNPs with MAF > 0.1 were used to perform single-locus and multi-locus GWAS using GEMMA25, FarmCPU26 and FASTmrEMMA27,28 software. Due to the multiple tests, the single-locus GWAS required Bonferroni correction29. However, the quantitative traits are controlled by numerous polygenes with large or minor effects, and the Bonferroni correction is overly conservative to identify important loci and may result in false negative results27 for complex traits. Recently, some multi-locus GWAS were proposed to detect significant SNPs for complex traits30,31. The significant threshold of the multi-locus GWAS is less stringent than single-locus GWAS. Because of the multi-locus and shrinkage natures of this method, a less stringent selection criterion is used in the multi-locus GWAS31. Although the multi-locus GWAS used a less stringent criterion, the study used simulated and real data to demonstrated that the multi-locus GWAS using the FASTmrEMMA model is more powerful and has less bias of QTN effect estimation than other methods31,32. The FASTmrEMMA method with high power, low false-positive rate and low computing time in QTN detection were used to perform multi-locus GWAS. Using GEMMA, 54 and 33 SNPs reached the threshold of 5.00 × 10−7 for DGE and SGE, respectively. Using FarmCPU, 51 and 101 SNPs reached the threshold of 5.00 × 10−7 for DGE and SGE, respectively. For FASTmrEMMA, a total of 46 and 71 putative QTNs were detected for DGE and SGE, respectively. Clearly, different methods provided different results. However, all these results show that numerous identified SNPs are associated with pleiotropic effects for multiple socially affected traits. Importantly, two SNPs were detected in three methods. Among them, one SNP (SSC8: 16438396 bp) was located in the region 16.42–16.46 Mb for dgeB100. In this region, four reported QTLs were associated with the B100 trait in pigs33,34. One SNP (SSC17: 9697454 bp) was detected in the region of 9.68–9.72 Mb for sgeD100. A previous study reported that numerous QTLs were associated with body weight35, behaviour36, immune capacity37 and disease38 in pigs. This SNP may contribute to weight, behaviour, immune and disease traits, and further affect socially affected traits. Thus this study suggested that the SNP SSC17: 9697454 bp was an informative causal locus for SGE in socially affected traits.
The promising candidate genes identified by GWAS
To investigate the genomic features that contribute to SGE and DGE in pigs, 40 Large White pigs were re-sequenced and 6 socially affected traits were studied. Based on the whole-genome re-sequencing data, this GWAS revealed the genetic architecture for 6 socially affected traits. The previous study only reported a total of 5 QTLs related to socially affected traits in pigs39. To date, no genes with their biological functions were reported to associate with SGE in animals. Thus, it is essential to reveal the candidate genes and understand the genetic architecture of DGE and SGE for future pig breeding programmes. In the current study, using a GWAS study, many candidate genes associated with DGE and SGE were found. A total of 20 genes for DGE and 9 genes for SGE were found using single-locus GWAS. A total of 42 and 54 genes were found for DGE and SGE using multi-locus GWAS, respectively. All of these genes were first reported to be associated with SGE in pigs. Additionally, a total of 15 candidate genes associated with pleiotropism for socially affected traits. Among them, 4 genes for single-locus GWAS (including PRKG1, uncharacterized LOC110259270, GABRR2 and ADAMTS6 genes) and 11 genes for multi-locus GWAS (including SLC26A2, HMGXB3, SPTB, PLEKHG3, SLC30A9, NRSN2, FOXO1, LYPLAL1, NR3C2, PKD2 and TRIB3 genes) were shared by DGE and SGE, which may imply the pleiotropism of these shared genes for socially affected traits in pigs. These candidate genes were play a role in behaviour, disease and health.
The PRKG1 gene is a protein coding gene that acts as a key mediator of the cyclic guanosine monophosphate (cGMP) signalling pathway and plays an important role in the cellular signal transduction process. In platelets, hippocampal neurons, smooth muscle and cerebellar Purkinje cells, the PRKG1 gene was found to be strongly expressed40,41. This gene is known to be associated with behaviour41, circadian rhythms42, sleep42, learning42 and memory41. In humans, the PRKG1 plays an important role in stress response related traits43.
SORCS3 is an orphan receptor of the VPS10P domain receptor family, a group of sorting and signalling receptors central to many pathways in the control of neuronal viability and function. SORCS3 plays an important role in the nervous system. This gene was associated with behavioural activities in mice44.
On the region of SSC1: 57.25–57.29 Mb, GABRR2 was found to be associated with dgeFCR and sgeFCR in pigs. GABRR2 is a protein coding gene that encodes a receptor for gamma-aminobutyric acid (GABA). The GABA receptor regulates the neurotransmitter in the brain45, and plays an important role in the behavioral stress response and physiology in animals and humans45,46,47,48. In mice, the GABA was associated with the aggressiveness and sociability towards conspecifics47. The performance and physical condition were affected by GABA in heat-stressed Roman hens49.
The strongest association (SSC16: 43805766, P = 4.85 × 10−9) was located in a disintegrin and metalloproteinase with thrombospondin motifs 6 (ADAMTS6). The ADAMTS6 gene is a member of the ADAMTS protein family and is regulated by the cytokine TNF-alpha50. The functions of this gene were still not clearly elucidated and mainly reported about aetiology and played a role in the turnover of the extracellular matrix51. ADAMTS6 was expressed differently in each tissue, but the accurate function and the substrates of ADAMTS6 protein were not clearly demonstrated by a previous study. In past decades, numerous association studies have found the ADAMTS6 gene to be associated with complex traits in humans. Inguinal hernias52 and central corneal thickness and keratoconus53 were both identified ADAMTS6 in a GWAS, which suggested that the ADAMTS6 gene would affected the collagen homoeostasis in tissues and disorders and lung function. A previous study also suggested that this gene was related to osteosarcoma54. Some studies also demonstrated that the ADAMTS6 gene was significantly upregulated in cancer cells and stromal cells55. Importantly, this gene affected the intelligence and growth development, and the balanced translocation disruption of the ADAMTS6 gene was resulted in short stature and intellectual disability56. Given previous studies, ADAMTS6 gene was associated with growth and disease in biological organisms. The ADAMTS6 gene was first identified to associate with the sgeRFI. The sgeRFI was not only affected the feed efficiency, but also affected the growth and health of livestock. Considering the pleiotropic effect of this gene, the ADAMTS6 gene with a potential effect on socially affected traits would be identified as a prominent candidate gene.
In summary, numerous SNPs and genes were identified for DGE and SGE of six traits using single-locus and multi-locus GWAS in Large White pigs. These findings were particularly interesting to better understand the genetic and physiologic mechanisms of both DGE and SGE. Although, a limited number of individuals were used in this study, this study provided a new insight to investigate socially affected traits. Further study using a large population size should contribute to validating the genetic mechanisms for socially affected traits in pigs.
Materials and Methods
Ethics statement
All experimental procedures were performed in accordance with the Institutional Review Board (IRB14044) and the Institutional Animal Care and Use Committee of the Sichuan Agricultural University under permit number DKY-B20140302.
Animals and Trait Recorded
The phenotypic data was collected in 2017, including ADG, days to 100 kg (D100), backfat thickness to 100 kg (B100), average daily feed intake (ADFI), residual feed intake (RFI) and feed conversion ratio (FCR). Initially, the tested pigs were selected between 120 and 130 day of age, with the body weight (BW) about 60 kg. Then a total of 40 Large White pigs from 4 pens were used for this research, containing 27 female and 13 male pigs, and each pen contained 10 piglets homogenous in body weight and age in this study. In this test, these pigs were grouped in the standard commercial pens, which were fed ad libitum by an automatic feeder (The feed intake recording equipment of OSBOREN). The test began at about 65 kg (BW1) and the ended at about 110 kg (BW2) in the feeding trial.
During the test period, we measured the phenotype data every ten days, and calculated the average value in finally. The ADG, ADFI and FCR were directly calculated from the collected data. The D100 and B100 were calculated as below (Kennedy and Chesnais, unpublished data):
where CF is the correction coefficient in pig breeding program. Furthermore, the RFI was computed according to the formula (neglecting the lean meat content and dressing percentage)57:
The average metabolic body weight (AMW) was calculated for each individual using the following classical formula58:
Calculate the DGE and SGE
For a socially affected traits, the phenotypic value of an individual can be denoted using following model5,16:
where Pi is the phenotypic value of individual i, AD,i is the direct breeding value of individual i, ED,i is the direct non-genetic effect of individual i, AS,j is the social breeding value of the group member j, ES,j is the social non-genetic effect of the group mate j, and the n is the group size. The ED,i and \(\,\sum _{j\ne i}^{n-1}\,{E}_{S,j}\) are combined into a residual component. So,
To investigate the direct and social genetic effects, the following full model was built for each trait in this study.
where Y is the vector of phenotypic observations; b is the vector of fixed effects, including sex, tested year and month; aD is a vector of DGE; aS is a vector of SGE; l is vector of random litter effects; g is vector of random group effects; e is random residual vector; X, ZD, ZS, W and V are incidence matrix of b, aD, aS, l and g, respectively. The variance-covariance matrix of aD and aS is denoted as:
where A is additive genetic correlation matrix; \({{\boldsymbol{\sigma }}}_{{{\boldsymbol{A}}}_{{\boldsymbol{d}}}}^{2}\), \({{\boldsymbol{\sigma }}}_{{{\boldsymbol{A}}}_{{\boldsymbol{ds}}}}^{2}\) and \({{\boldsymbol{\sigma }}}_{{{\boldsymbol{A}}}_{{\boldsymbol{s}}}}^{2}\) are genetic variance and covariance between direct effects and social effects. At the ith row of ZS, the group members of individual i were set to 1 and the others were set to 0. For improving the estimation accuracy of DGE and SGE, other 3,236 phenotypic records about ADG, D100, B100, ADFI, FCR and RFI that were obtained from previous performance test also were added into this analysis. These traits were analyzed using the average information restricted maximum likelihood (AI-REML) algorithm by DMU software59. Supplementary Table 1 lists the describe statistics about the DGE and SGE for these six traits.
Genotyping
The pig’s ear tissue was collected from Large White pigs and stored in 75% alcohol. The genomic DNA was extracted by the standard phenol/chloroform method. The Nanodrop-2000 spectrophotometer was used to control the quality of genomic DNA. To obtain nucleotide polymorphism information, a total of 40 Large White pigs were re-sequenced using the Illumina HiSeq PE150 platform, then about 2,000 Gb sequence data was obtained in total. The average sequencing depth of these samples were close to 20×. All the sequence reads were filtered for data quality and mapped to the Sscrofa11.1 reference sequence using BWA software60. The mapped reads were realigned by GATK software61. A total of 21,104,245 variants were identified using GATK software61. In order to minimize the number of false positives in this limited population size, the SNP with MAF > 0.1 were retained for further analysis. Thus, the nucleotide variants were filter based on the quality requirement with minor allele frequency (MAF > 0.1), Missing rate (Miss < 0.1), Hardy-Weinberg equilibrium (HWE < 1.0 × 10−6), and read depth (dp > 6) using VCFtools 4.262. Then, the SNPs on the sex chromosome and scaffolds were removed. After the quality control, a total of 10,501,384 SNPs across 18 autosomes were analyzed for association study.
Single-locus association analysis
The genome-wide efficient mixed-model analysis (GEMMA)25 with a univariate liner mixed model were used to perform the single-locus association for DGE and SGE. The analysis models was written as follows:
where y is a vector of DGE or SGE contained residual effects from previous model (1), X is the incidence matrix of SNP effects, m is the vector of SNP effects, W is the incidence matrix of residual polygene effects, a is the vector of residual polygene effects, e is the random residual vector. Notably, where y is a vector that phenotype value was corrected by fixed effect, DGE (for SGE), SGE (for DGE), random litter and random group effects.
Recently, several statistical methods have been contributed to determine the significance threshold, such as Bonferroni correction63, False discovery rate64, and Sidak correction method65. In published literatures, the traditional Bonferroni correction method was routinely adopted, but this threshold was not absolute. When identified SNPs exceeded the accepted genome-wide statistical significance threshold P < 1.0 × 10−8, the GWAS results are most reliable66,67. The identified associations (5.00 × 10−8 < P ≤ 5.00 × 10−7) also could be replicated from subsequent studies28. The proper significant threshold is various due to different populations, different traits, and different genotypic data68,69,70. Furthermore, the Bonferroni correction is too conservative due to the ignorance of linkages between SNPs, especially for the sequencing data where many adjacent SNPs are highly linked. In the sequencing data, the number of markers used in the Bonferroni correction should be more relevant to the number of segments of chromosome. Thus, the whole-genome significance threshold 5.00 × 10−8 was established71,72 and the suggestive significant threshold was set at 5.00 × 10−7 in this study73.
Multi-locus association analysis
The multi-locus association analysis were performed by FarmCPU26 and the fast multi-locus random-SNP-effect EMMA (FASTmrEMMA)31,74. FarmCPU iteratively used fixed effect model and random effect model. Using the FASTmrEMMA, this study performed a GWAS for DGE and SGE for six traits in pigs. In this multi-locus GWAS, the random-SNP-effect and multi-locus model methods were used to improve the power and decrease the false-positive rate. At the first stage, the putative quantitative trait nucleotides (QTNs) with P ≤ 0.005 were selected for further analysis. Then the selected putative QTNs were analyzed using a multi-locus model for true QTN detection.
Selecting candidate genes
The bioinformatics databases BioMart (http://www.ensembl.org/) and NCBI (https://www.ncbi.nlm.nih.gov/) were implemented to screen the candidate genes located the significant loci. Only the genes located at the region of ±20 kb around the significant SNPs were considered in this study.
Data Availability
The authors affirm that all of the data necessary for confirming the conclusions made within this article are contained within the article. The dataset used in this study is available from the corresponding author upon reasonable request.
References
Schrøder-Petersen, D. L. & Simonsen, H. B. Tail Biting in Pigs. Veterinary Journal 162, 196–210 (2001).
Mm, V. K. The prevention and control of feather pecking in laying hens: identifying the underlying principles. Worlds Poultry Science Journal 69, 361–374 (2013).
Canario, L., Lundeheim, N. & Bijma, P. The early-life environment of a pig shapes the phenotypes of its social partners in adulthood. Heredity 118 (2017).
Ellen, E. D. et al. The prospects of selection for social genetic effects to improve welfare and productivity in livestock. Front Genet 5, 377 (2014).
Griffing, B. Selection in reference to biological groups. I. Individual and group selection applied to populations of unordered groups. Australian Journal of Biological Sciences 20, 127–139 (1967).
Moore, A. J. & Wolf, J. B. Interacting Phenotypes and the Evolutionary Process: I. Direct and Indirect Genetic Effects of Social Interactions. Evolution 51, 1352 (1997).
Bergsma, R., Kanis, E., Knol, E. F. & Bijma, P. The contribution of social effects to heritable variation in finishing traits of domestic pigs (Sus scrofa). Genetics 178, 1559 (2008).
Baud, A. et al. Genetic Variation in the Social Environment Contributes to Health and Disease. Plos Genetics 13 (2017).
Bijma, P. & Wade, M. J. The joint effects of kin, multilevel selection and indirect genetic effects on response to genetic selection. Journal of Evolutionary Biology 21, 1175–1188 (2008).
Chen, C. Y., Kachman, S. D., Johnson, R. K., Newman, S. & Van Vleck, L. D. Estimation of genetic parameters for average daily gain using models with competition effects. Journal of Animal Science 86, 2525–2530 (2008).
Chen, C. Y., Johnson, R. K., Newman, S., Kachman, S. D. & Van Vleck, L. D. Effects of social interactions on empirical responses to selection for average daily gain of boars. Journal of Animal Science 87, 844–849 (2009).
Bouwman, A. C., Bergsma, R., Duijvesteijn, N. & Bijma, P. Maternal and social genetic effects on average daily gain of piglets from birth until weaning. Journal of Animal Science 88, 2883–2892 (2010).
Rostellato, R., Sartori, C., Bonfatti, V., Chiarot, G. & Carnier, P. Direct and social genetic effects on body weight at 270 days and carcass and ham quality traits in heavy pigs. Journal of Animal Science 93, 1 (2015).
Bouwman, A. C., Bergsma, R., Duijvesteijn, N. & Bijma, P. Maternal and social genetic effects on average daily gain of piglets from birth till weaning. Journal of Animal Science 88, 2883 (2010).
Muir, W. M. & Schinckel, A. Incorporation of competitive effects in breeding programs to improve productivity and animal well being (ed. Muir and Schinckel). [Macmillan] (2002).
Muir, W. M. Incorporation of Competitive Effects in Forest Tree or Animal Breeding Programs. Genetics 170, 1247–1259 (2005).
Bijma, P., Muir, W. M. & Van Arendonk, J. A. Multilevel Selection 1: Quantitative Genetics of Inheritance and Response to Selection. Genetics 175, 277–288 (2007).
Bijma, P., Muir, W. M., Ellen, E. D., Wolf, J. B. & Van Arendonk, J. A. Multilevel Selection 2: Estimating the Genetic Parameters Determining Inheritance and Response to Selection. Genetics 175, 289 (2007).
Ellen, E. D., Visscher, J., Van Arendonk, J. A. M. & Bijma, P. Survival of Laying Hens: Genetic Parameters for Direct and Associative Effects in Three Purebred Layer Lines. Poult Sci 87, 233–239 (2008).
Wilson, A. J., Gelin, U., Perron, M. C. & Réale, D. Indirect genetic effects and the evolution of aggression in a vertebrate system. Proc Biol Sci 276, 533–541 (2009).
Cassady, J. P. & Vleck, L. D. V. Estimation of (co) variance components due to genetic competition effects in pigs. Journal of Animal Science 83, 6–7 (2005).
Arango, J., Misztal, I., Tsuruta, S., Culbertson, M. & Herring, W. Estimation of variance components including competitive effects of Large White growing gilts. Journal of Animal Science 83, 1241–1246 (2005).
Bergsma, R., Kanis, E., Knol, E. F. & Bijma, P. The Contribution of Social Effects to Heritable Variation in Finishing Traits of Domestic Pigs (Sus scrofa). Molecular & Cellular Biology 12, 4375–4383 (2008).
Hong, J. K. et al. A genome-wide association study of social genetic effects in Landrace pigs. Asian-Australasian Journal of Animal Sciences 31, 784–790 (2018).
Zhou, X. & Stephens, M. Genome-wide Efficient Mixed Model Analysis for Association Studies. Nature Genetics 44, 821 (2012).
Liu, X., Huang, M., Fan, B., Buckler, E. S. & Zhang, Z. Iterative Usage of Fixed and Random Effect Models for Powerful and Efficient Genome-Wide Association Studies. Plos Genetics 12, e1005767 (2016).
Johnson, R. C. et al. Accounting for multiple comparisons in a genome-wide association study (GWAS). Bmc Genomics 11, 724 (2010).
Panagiotou, O. A. & Ioannidis, J. P. A. What should the genome-wide significance threshold be? Empirical replication of borderline genetic associations. International Journal of Epidemiology 41, 273 (2012).
Wang, S. B. et al. Mapping small-effect and linked quantitative trait loci for complex traits in backcross or DH populations via a multi-locus GWAS methodology. Sci Rep 6, 29951 (2016).
Wang, S. B. et al. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology. Sci Rep 6, 19444 (2016).
Wen, Y. J. et al. Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Briefings in Bioinformatics 19, 700–712 (2018).
Sant’Ana, G. C. et al. Genome-wide association study reveals candidate genes influencing lipids and diterpenes contents in Coffea arabica L. Scientific Reports 8 (2018).
Vidal, O. et al. Identification of carcass and meat quality quantitative trait loci in a Landrace pig population selected for growth and leanness. Journal of Animal Science 83, 293–300 (2005).
De Koning, D. J. et al. Full pedigree quantitative trait locus analysis in commercial pigs using variance components1. Journal of Animal Science 81, 2155–2163 (2003).
Guo, Y. M., Lee, G. J., Archibald, A. L. & Haley, C. S. Quantitative trait loci for production traits in pigs: a combined analysis of two Meishan × Large White populations. Animal Genetics 39, 486–495 (2008).
Chen, C. et al. Association and haplotype analysis of candidate genes in five genomic regions linked to sow maternal infanticide in a white Duroc × Erhualian resource population. BMC Genetics, 12, 1 (2011-02-09) 12, 24 (2011).
Reiner, G. et al. Quantitative trait loci for white blood cell numbers in swine. Animal Genetics 39, 163–168 (2010).
Reiner, G. et al. Identification of QTL affecting resistance/susceptibility to acute Actinobacillus pleuropneumoniae infection in swine. Mammalian Genome 25, 180–191 (2014).
Reiner, G. et al. Mapping of quantitative trait loci affecting behaviour in swine. Animal Genetics 40, 366–376 (2009).
Lohmann, S. M. & Walter, U. Tracking functions of cGMP-dependent protein kinases (cGK). Frontiers in Bioscience A Journal & Virtual Library 10, 1313 (2005).
Kleppisch, T. & Feil, R. cGMP Signalling in the Mammalian Brain: Role in Synaptic Plasticity and Behaviour. Handbook of Experimental Pharmacology 191, 549–579 (2009).
Corradini, E. et al. Alterations in the cerebellar (Phospho)proteome of a cyclic guanosine monophosphate (cGMP)-dependent protein kinase knockout mouse (2014).
Ashley-Koch, A. E. et al. Genome-wide association study of posttraumatic stress disorder in a cohort of Iraq–Afghanistan era veterans. Journal of Affective Disorders 184, 225–234 (2015).
Breiderhoff, T. et al. Sortilin-related receptor SORCS3 is a postsynaptic modulator of synaptic depression and fear extinction. Plos One 8, e75006 (2013).
Aki, T., Akiko, S., Boyson, C. O., Debold, J. F. & Miczek, K. A. GABA(B) receptor modulation of serotonin neurons in the dorsal raphé nucleus and escalation of aggression in mice. Journal of Neuroscience the Official Journal of the Society for Neuroscience 30, 11771–11780 (2010).
Almeida, R. M. M. D., Ferrari, P. F., Parmigiani, S. & Miczek, K. A. Escalated aggressive behavior: Dopamine, serotonin and GABA. European Journal of Pharmacology 526, 51–64 (2005).
Poshivalov, V. P. GABA-ergic correlates between aggressiveness and sociability in isolated mice. Bulletin of Experimental Biology & Medicine 91, 648–650 (1981).
Takahashi, A. et al. Behavioral characterization of escalated aggression induced by GABA(B) receptor activation in the dorsal raphe nucleus. Psychopharmacology 224, 155–166 (2012).
Min, Z., Xiao-Ting, Z., Hui, L., Xin-Yang, D. & Wenjing, Z. Effect of dietary γ-aminobutyric acid on laying performance, egg quality, immune activity and endocrine hormone in heat-stressed Roman hens. Animal Science Journal 83, 141–147 (2012).
Kelwick, R., Desanlis, I., Wheeler, G. N. & Edwards, D. R. The ADAMTS (A Disintegrin and Metalloproteinase with Thrombospondin motifs) family. Genome Biology 16, 113 (2015).
Bevitt, D. J. et al. Analysis of full length ADAMTS6 transcript reveals alternative splicing and a role for the 5′ untranslated region in translational control ☆. Gene 359, 99 (2005).
Jorgenson, E. et al. A genome-wide association study identifies four novel susceptibility loci underlying inguinal hernia. Nature Communications 6, 10130 (2015).
Lu, Y. et al. Genome-wide association analyses identify multiple loci associated with central corneal thickness and keratoconus. Nature Genetics 45, 155 (2013).
Savage, S. A. et al. Genome-wide association study identifies two susceptibility loci for osteosarcoma. Nature Genetics 45, 799–803 (2013).
Liu, L., Yang, Z., Ni, W. & Xuan, Y. ADAMTS-6 is a predictor of poor prognosis in patients with esophageal squamous cell carcinoma. Experimental & Molecular Pathology (2018).
Malli, T. et al. Disruption of the ARID1B and ADAMTS6 loci due to a t(5; 6) (q12.3; q25.3) in a patient with developmental delay. American Journal of Medical Genetics Part A 164A, 3126 (2014).
Saintilan, R. et al. Genetics of residual feed intake in growing pigs: Relationships with production traits, and nitrogen and phosphorus excretion traits. Journal of Animal Science 91, 2542–2554 (2013).
Noblet, J., Karege, C., Dubois, S. & Van, M. J. Metabolic utilization of energy and maintenance requirements in growing pigs: effects of sex and genotype. Journal of Animal Science 77, 1208–1216 (1999).
Madsen, P., Jensen, J., Labouriau, R., Christensen, O. F. & Sahana, G. DMU - A Package for Analyzing Multivariate Mixed Models in quantitative Genetics and Genomics. World Congress on Genetics Applied to Livestock (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows–Wheeler transform. (Oxford University Press, 2009).
Depristo, M. A. et al. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nature Genetics 43, 491–498 (2011).
Danecek, P. et al. The variant call format and VCFtools. Bioinformatics 27, 2156 (2011).
Bland, J. M. & Altman, D. G. Multiple significance tests: the Bonferroni method (1995).
Storey, J. D. & Tibshirani, R. Statistical significance for genomewide studies (2003).
Sidak, Z. Rectangular Confidence Regions for the Means of Multivariate Normal Distributions. Publications of the American Statistical Association 62, 626–633 (1967).
Manolio, T. A. Genomewide association studies and assessment of the risk of disease. New England Journal of Medicine 363, 166 (2010).
Itsik, P. E., Roman, Y., David, A. & Daly, M. J. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genetic Epidemiology 32, 381–385 (2010).
Hoggart, C. J., Clark, T. G., Iorio, M. D., Whittaker, J. C. & Balding, D. J. Genome-wide significance for dense SNP and resequencing data. Genetic Epidemiology 32, 179 (2008).
Daetwyler, H. D. et al. Whole-genome sequencing of 234 bulls facilitates mapping of monogenic and complex traits in cattle. Nature Genetics 46, 858–865 (2014).
Fontanesi, L. et al. A genome wide association study for backfat thickness in Italian Large White pigs highlights new regions affecting fat deposition including neuronal genes. Bmc Genomics 13, 583 (2012).
Michael, O. A haplotype map of the human genome. Nature 437, 1299–1320 (2005).
Pe’Er, I., Yelensky, R., Altshuler, D. & Daly, M. J. Estimation of the multiple testing burden for genomewide association studies of nearly all common variants. Genetic Epidemiology 32, 381–385 (2008).
Burton, P. R. et al. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature 447, 661–678 (2007).
Zhang,Y. M. et al. Mapping quantitative trait loci using naturally occurring genetic variance among commercial inbred lines of maize (Zea mays L.). Genetics 169, 2267–2275 (2005).
Acknowledgements
The study was supported by grants from the National Key R&D Program of China #2018YFD0501204, National Natural Science Foundation of China (31530073), National Natural Science Foundation of China #C170102, Chinese National Science and Tech Support 360 Program (Nos 2015BAD03B01, 2015GA810001), earmarked fund for the China Agriculture Research System (No. CARS-35-01A).
Author information
Authors and Affiliations
Contributions
G.T., P.W. and K.W. performed experiments; P.W., K.W., Q.Y., J.Z., D.C., Y.L. and P.L. analyzed data and prepared figures and tables; G.T. and P.W. edited and revised manuscript; G.T., P.W., J.M., Q.T., L.J., W.X., A.J., Y.J., L.Z., M.L. and X.L. conceived, designed research and wrote this paper. P.W., K.W., Q.Y., J.Z., D.C., Y.L., J.M., Q.T., L.J., W.X., P.L., A.J., Y.J., L.Z., M.L., X.L. and G.T. approved final version of this manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wu, P., Wang, K., Yang, Q. et al. Whole-genome re-sequencing association study for direct genetic effects and social genetic effects of six growth traits in Large White pigs. Sci Rep 9, 9667 (2019). https://doi.org/10.1038/s41598-019-45919-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-45919-0
This article is cited by
-
Shared alleles and genetic structures in different Thai domestic cat breeds: the possible influence of common racial origins
Genomics & Informatics (2024)
-
A combined GWAS approach reveals key loci for socially-affected traits in Yorkshire pigs
Communications Biology (2021)
-
Single-step genome-wide association study for social genetic effects and direct genetic effects on growth in Landrace pigs
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
