
Abstract
Hepatitis C virus (HCV) eradication by antivirals promote fibrosis modification. Whether host genetics determined fibrosis regression in chronic hepatitis C (CHC) patients with sustained virological response (SVR) is to be determined. One hundred and fifty-six SVR patients with paired liver biopsy before and after antivirals were enrolled. Host genetic factors including single nucleotide polymorphism rs17047200 of tolloid-like 1(TLL-1) were analyzed for their association with fibrosis modification. The proportions of improved, unchanged and worsening fibrotic stags were 39.1% (n = 61), 39.1% (n = 61), and 21.8% (n = 34), respectively. The rate of annual fibrotic improvement was 0.16 ± 0.79. There was a significant trend of increased fibrotic improvement rate in patients from F01 to F4 (P < 0.001). However, the rate of improvement seemed more limited in cirrhotic patients among those with advanced liver disease. Patients with fibrotic improvement had a significantly higher proportion of TLL-1 rs17047200 AA genotype compared to those without (92.5% vs. 79.3%, p = 0.039). Logistic regression analysis revealed that the TLL-1 rs17047200 AA genotype was the only independent factor associated with fibrosis improvement (odds ratio/95% confidence intervals: 3.2/1.01–10.12, p = 0.047). Compared with TLL-1 rs17047200 non-AA carriers, a significantly higher proportion of fibrosis improvement in AA genotype carriers was observed among patients with F0-2 (33.3% vs. 0%, p = 0.005) but not with F34 (70% vs. 80%, p = 1). We concluded that TLL-1 genetic variants determined fibrotic improvement in CHC with curative antivirals, particularly in patients with mild liver disease.
Similar content being viewed by others

Introduction
The chronic inflammatory state during hepatitis C virus (HCV) infection drives liver fibrogenesis, which in turns leads to end-stage liver disease. It takes 20–30 years for the development of liver cirrhosis in patients with chronic hepatitis C (CHC). The acceleration of the fibrosis varies among patients. The determining factors largely include environmental factors (e.g., alcohol consumption and diabetics1), virological factors (e.g., HCV genotype 3, hepatitis B virus [HBV] and human immunodeficiency virus co-infection) and host factors (e.g., age at infection and sex2,3). Host genetic predispositions, such as genetic variants of interleukin 28B (IL-28B), patatin-like phospholipase domain-containing 3 (PNPLA3) and other immunogenetic profiles, have also been recognized as potential determinants of liver fibrosis progression1,2,4. The term fibrosis progression rate FPR has been adopted as a measure of the change of fibrotic stage in a given time period2,4. It has been estimated that the median rate of fibrosis progression to be 0.13 fibrosis units per year albeit the progression is more exponential rather than linear5.
On the other hand, successful viral eradication by antivirals would halt liver fibrosis progression and prevent the development of cirrhosis6. In addition, the achievement of sustained virological response (SVR) may also ameliorate portal hypertension7 and prompt fibrosis regression8. The magnitude and extent of fibrosis regression following SVR has been previously studied9. It is estimated that up to 57–94% of patients have improved histology in terms of necroinflammation and fibrosis scores after viral eradication8. However, some patients may have passed “the point of no return” and remain to have fibrosis progression even after viral eradication. Notably, whether host genetic variants would determine fibrotic augmentation after viral eradication has not been clearly addressed. In the current study, we aimed to elucidate the modification of fibrosis after viral eradication in well-characterized CHC patients who received paired liver biopsy before and after achieving SVR. We also aimed to explore the potential contribution of host genetic factors to fibrotic modification in this Taiwanese cohort.
Methods
Patients
CHC patients who received paired liver biopsy before and after interferon-based therapy were consecutively enrolled from 2001 to 2012. Patients who failed to achieve SVR, defined as HCV RNA seronegativity throughout 6 months of post-treatment follow-up period, were excluded. Patients whose paired biopsy interval was less than 1 year; patients with human immunodeficiency virus co-infection and patients with documented alcohol abuse (defined as alcohol consumption >20 gm/day) were also excluded from the current study. This study was conducted according to The Declaration of Helsinki. The institutional review board of the Kaohsiung Medical University Hospital approved the protocols, which conformed to the guidelines of the International Conference on Harmonization for Good Clinical Practice. All patients provided written informed consent. All procedures were followed in accordance with the ethical standards of the responsible conduct of human experimentation and with the Helsinki Declaration of 1975, as revised in 2008.
Laboratory and histological analyses
Biochemistry was measured on a multichannel autoanalyzer (Hitachi Inc., Tokyo, Japan). HCV antibodies (Anti-HCV) were measured by a third-generation enzyme immunoassay (Abbott Laboratories, North Chicago, IL). Hepatitis B surface antigen (HBsAg) was determined using a standard quantitative chemiluminescent microparticle immunoassay (ARCHITECT HBsAg, Abbott Diagnostics). Serum HCV RNA was detected using qualitative real-time polymerase chain reaction (PCR) (COBAS AMPLICOR Hepatitis C Virus Test, ver. 2.0; Roche, Branchburg, NJ, USA, detection limit: 50 IU/ml) and quantification branched DNA assay (Versant HCV RNA 3.0, Bayer, Tarrytown, New Jersey, USA; quantification limit: 615 IU/ml) before 2011. The HCV genotypes were determined using the Okamoto method before 201110. Both HCV RNA and genotype were detected using real-time PCR assay (RealTime HCV; Abbott Molecular, Des Plaines IL, USA; detection limit: 12 IU/ml) since 201211. Serum HBV DNA was detected using a standardized automated quantitative PCR assay (COBAS TaqMan HBV test, Roche Diagnostics, Branchburg, NJ; detection limit 12 IU/ml). A liver biopsy specimen of at least 2 cm in length was obtained and fixed in 10% formalin buffer. Biopsy samples were stained with hematoxylin-eosin, and the results were then reported by one pathologist who was blinded to the treatment of each patient. The liver histology was graded and staged according to the scoring system described by Scheuer12.
Single nucleotide polymorphism (SNP) genetic testing
SNPs rs8099917 of IL-28B, rs738409 of PNPLA3 and rs2596542 of MHC class I polypeptide-related chain A (MICA) were selected as candidate genes as previously described1,13,14. SNP rs17047200 of tolloid-like 1(TLL-1) was selected and determined by ABI TaqMan® SNP genotyping assays (Applied Biosystems, Foster City, CA, USA) by using the pre-designed commercial genotyping assays (ABI Assay ID: C__33773674_10). Briefly, PCR primers and two allelic-specific probes were designed to detect specific SNP target. The PCR reactions were performed in 96-well microplates with ABI 7500 real-time PCR. Allele discrimination was achieved by detecting fluorescence using System SDS software version 1.2.3. All the allele and genotype frequencies were consistent with the Hardy-Weinberg equilibrium.
Statistical analyses
Frequencies were compared between groups using either the χ2 test with the Yates correction or Fisher’s exact test. Group means, presented as the mean values and standard deviations, were compared using analysis of variance and either the Student’s t test or Mann-Whitney U test. The serum HCV RNA levels were expressed after logarithmic transformation of the original values. A significant fibrotic change was defined as ≥1-point modification in Metavir score between biopsies. A stepwise logistic regression analysis was performed to evaluate the independent factors associated with fibrotic improvement or deterioration by analyzing covariates with P values <0.05 in the univariate analysis. The statistical analyses were performed using the SPSS 12.0 statistical package (SPSS, Chicago, IL, USA). All statistical analyses were based on two-tailed hypothesis tests with a significance level of p < 0.05.
Results
Patient profile
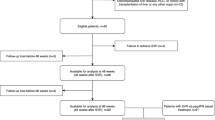
A total of 294 patients who received paired biopsy before and after antiviral therapy were initially enrolled. After excluding those patients without SVR (n = 57) and those patients whose paired biopsy interval was <1 year (n = 81), one hundred and fifty-six patients were included in the current analysis. The mean age was 50.4 years, and males accounted for 50.4% of the population. The median (25, 75 percentile) interval between biopsy was 1.53 (1.34, 1.75) year. The proportion of pretreatment fibrotic stage F0-1, F2, F3 and F4 was 44.9% (n = 70), 28.2% (n = 44), 16.7% (n = 26) and 10.3% (n = 16), respectively (Table 1).
Fibrotic change after achieving SVR
The proportion of improved, unchanged and worsening fibrotic stages was 39.1% (n = 61), 39.1% (n = 61) and 21.8% (n = 34), respectively. The rate of annual fibrotic improvement was 0.16 ± 0.79. The annual fibrotic improvement rate was significantly higher in patients with advanced liver fibrosis (F34) than those with mild liver disease (F0-2) (0.62 ± 0.82 vs. −0.01 ± 0.71, P < 0.001). The rate of fibrotic improvement was substantially higher in patients whose biopsy interval was >1.5 years than in the patients with shorter biopsy intervals. However, this difference did not reach statistical significance. There was a significantly increased trend of fibrotic improvement rate in patients from F01 to F4 (P < 0.001). However, the rate of improvement seemed more limited to patients with cirrhosis compared to those with F3 (Table 2 and Fig. 1).

Rate of annual fibrotic change after achieving sustained virological response, stratified by pretreatment fibrotic status. Each bar represents mean value ± standard error.
Factors associated with fibrotic change
We further analyzed the factors associated with fibrotic improvement or deterioration in this cohort. As shown in Table 3, patients with fibrotic improvement had a significantly higher proportion of TLL-1 rs17047200 AA genotype carriage compared to those without (92.5% vs. 79.3%, p = 0.039). A substantially higher annual fibrosis regression rate was observed in TLL-1 rs17047200 AA genotype carriers compared to those with non-AA genotype (0.18 + 0.77 vs. 0.10 + 0.97), albeit it did not reach statistical difference (P = 0.36). Logistic regression analysis revealed that the carriage of TLL-1 rs17047200 AA genotype was the only independent factor associated with fibrosis improvement (odds ratio [OR]/95% confidence intervals [CI]: 3.2/1.01-10.12, p = 0.047). We further stratified patients by their pretreatment fibrotic stages. Among patients with advanced liver fibrosis (F3-4), the proportion of fibrotic improvement did not differ between patients with TLL-1 rs17047200 AA versus non-AA genotype (70% vs. 80%, P = 1). However, among patients with mild liver disease (F0-2), the proportion of fibrosis improvement was significantly higher in those with the TLL-1 rs17047200 AA genotype compared to those with a non-AA genotype at (33.3% vs. 0%, p = 0.005) (Fig. 2). On the other hand, patients with fibrosis deterioration had a higher r-GT level (94.9 ± 87.7 U/L vs. 60.2 ± 54.6 U/L, p = 0.027). Logistic regression analysis revealed that r-GT was the only independent factor associated with worsening fibrosis (OR/CI: 1.007/1.002-1.013, p = 0.01) (Table 4).

Proportion of fibrotic improvement in patients with different tolloid-like 1 genetic variants, stratified by liver disease severity (fibrotic stage 0–2 and 3–4).
Discussion
Successful HCV eradication may halt and eventually reverse the natural course of liver deterioration in terms of fibrogenesis. In the current study, we demonstrated that the majority of patients have stable or improved liver fibrotic stages after HCV eradication. The extent of fibrotic recovering varied by underlying fibrotic stage. Notably, we identified that TLL-1 genetic variation was the only factor associated with fibrotic improvement. Patients with the TLL-1 rs17047200 AA genotype were more likely to benefit from rapid fibrotic regression after HCV eradication. In addition, this impact of host genetic predisposition on fibrosis improvement was particularly enhanced in patients with mild liver disease.
Although there is ample evidence that supports the beneficial effect of HCV eradication on fibrotic regression, the relentless recover may not be taken for granted across all pupations and studies. The regression slop has been suggested to be very slow by non-invasive modalities15,16. It has also been reported that cirrhosis patients experience only 5% of net fibrosis improvement after 10 years of follow-up15. More convincing evidence may come from studies documenting sequential histological changes by liver biopsy. Pooled data have suggested that the proportion of fibrotic improvement ranged widely from 21–82%, whereas one- to two-thirds of the subjects maintained stable disease with a mean or median observation period of 0.5 to 5.2 years17. The current study is consistent with previous reports that HCV eradication results in suppression of ongoing liver damage and fibrogenesis in the majority patients.
Liver fibrosis progression in viremic patients may be more exponential rather than linear5. The shape of the recovery curve after viral clearance remains unclear since repeated liver biopsy may not be feasible after viral eradication. A more rapid fibrosis regression rate of up to 0.28 to 0.59/year has been proposed9,18. It has also been suggested that regression rate was more evident in patients whose biopsy intervals were more than 3 years compared to those with shorter observation periods4. In the current study, we observed the annual fibrosis regression rate of 0.16, which was similar to the fibrosis progression rate in viremic patients reported by Poynard et al.5 Interestingly, we observed that the recovery rate was more rapid with the advancement of underlying liver fibrosis. This finding echoed the observation that platelet counts, an indicator of liver disease severity, may recover in SVR patients with all stages of liver diseases and were more pronounced in patients with progressed liver fibrosis19. Notably, we observed that the regression rate seemed to slow in cirrhotic patients. Indeed, cirrhosis represents more than merely severe fibrosis17. Semi-quantitative scoring by Metavir or Ishak may not represent the whole spectrum of cirrhotic architecture17,20, which may limit the prognostic prediction on an individual basis21,22. Complex features of different degrees of extracellular matrix deposition vary among cirrhotic patients. This may account for variable reversibility and degradation of collagen and elastin within the cirrhotic tissues17. As a result, some cirrhotic patients may encounter refractory fibrotic down-staging that leads to slower fibrosis regression rate, as was observed in the current study.
The fact that genetic predispositions determine fibrosis progression and liver disease severity has been documented1,23,24. Whether these genetic factors are involved in fibrotic resolution has not yet been explored. Balart et al. have reported different proportions of fibrotic regression between Latinos and non-Latinos who achieved SVR25, giving a hint that genetic variants may also determine fibrosis regression. However, no robust evidence has identified any genetic factors associated with fibrosis regression until recently. In the current study, we identified that SNP rs17047200 of TLL-1 was independently associated with fibrosis resolution. A genome-wide association study has demonstrated the association of this genetic locus with hepatocellular carcinoma occurrence in SVR patients26. In addition, it seems that the impact of SNP rs17047200 of TLL-1 on HCC development is particularly enhanced in patients with mild fibrosis. The elderly who carried a non-AA genotype had a more than 4-fold HCC risk compared to those with the protective AA genotype26. Coincidently, we noted that association of this SNP with fibrosis improvement was also restricted to patients with mild liver disease. None of 16 non-AA genotype carriers with mild liver disease could benefit from fibrotic regression after achieving SVR during the observation period. TLL1 is one of the bone morphogenetic protein 1/tolloid (BMP1/TLD)-like proteinase family. TLL1 and BMP1 has been reported to have a biologically impact on transforming growth factor-β signaling transduction and extracellular matrix assembly regulation27,28. Increased TLL1 mRNA expression has been associated with fibrogenesis both in vivo and in vitro, and patients with unfavorable TLL-1 genetic variants were noted to have a higher expression of TLL-1 short isoform26. Further study is warranted to clarify the potential pathophysiological role of TLL-1 in fibrotic resolution such as extracellular matrix degradations or satellite cell senescence17.
Notably, a minority of patients continued to have fibrosis progression following HCV eradication8,17,18,29. Additional coexisting profibrogenic stimuli, such as alcohol consumption or metabolic disarrangement, may in part account for this result30,31. Progress to decompensated status may even occur in certain cirrhotic patients. Van der Meer et al. have reported an annual risk of approximately 2% for clinical disease progression in cirrhotic patients after a median follow-up period of 5.7 years32. In the current study, one-fifth of the patients had deteriorated liver fibrosis, and a high r-GT level was the only factor associated with this deterioration. r-GT is responsible for the catabolism of extracellular glutathione (GSH) and other γ-glutamyl compounds, and it may serve as a surrogate for oxidative stress. We have previously demonstrated that high pretreatment r-GT level was independently predictive of HCC occurrence in SVR patients, particularly in those without cirrhosis33. As r-GT level has been recognized as a predictor of liver fibrosis progression in CHC34, the current study reinforced its role in fibrotic deterioration in the curative status. One-tenth of the patients were dually infected with inactive HBV in the current population. HBV reactivation after HCV eradication by interferon-based therapy is very rare35,36, and we have reported a cumulative HBsAg seroclearance rate of up to 30.0% after 5 years of post-HCV curative follow-up period37. The impact of HBV dual infection on fibrosis augmentation in the current cohort should be limited.
The current study was limited by the relatively short interval between paired biopsies. To overcome the pitfall, we excluded the patients whose biopsy interval was less than 1 year. There may exist sampling variability and inter/intra-observer bias of biopsy tissues. More quantitative analyses, including immunostaining for cell-specific markers or morphometric analysis, may provide a future solution20,36. As noninvasive tests are widely applied in the directly acting antivirals (DAAs) era, accuracy is challenging and doubtful in the post-curative status16,38. Liver biopsy remains the gold standard to assess liver fibrosis. In conclusion, the majority of Taiwanese CHC patients maintained or had fibrosis regression after HCV eradication. The magnitude of fibrotic improvement varied among patients with different disease severities. Host genetic variation of TLL-1 determined fibrosis improvement in Asian patients, and validation of this finding in different ethnicities is warranted. Since repeated liver biopsy in larger patient cohorts may not be feasible in the DAAs era, the current study may shed light for future fundamental studies in terms of host genetics and fibrotic resolution.
References
Huang, C. F. et al. Association of diabetes and PNPLA3 genetic variants with disease severity of patients with chronic hepatitis C virus infection. Journal of hepatology. 62(3), 512–518 (2015).
Rueger, S. et al. Impact of common risk factors of fibrosis progression in chronic hepatitis C. Gut. 64(10), 1605–1615 (2015).
Ghany, M. G. et al. Predicting clinical and histologic outcomes based on standard laboratory tests in advanced chronic hepatitis C. Gastroenterology. 138(1), 136–146 (2010).
Tamaki, N. et al. Genetic Polymorphisms of IL28B and PNPLA3 Are Predictive for HCV Related Rapid Fibrosis Progression and Identify Patients Who Require Urgent Antiviral Treatment with New Regimens. PloS one. 10(9), e0137351 (2015).
Poynard, T., Bedossa, P. & Opolon, P. Natural history of liver fibrosis progression in patients with chronic hepatitis C. The OBSVIRC, METAVIR, CLINIVIR, and DOSVIRC groups. Lancet. 349(9055), 825–832 (1997).
Huang, J. F. et al. Sustained virological response to interferon reduces cirrhosis in chronic hepatitis C: a 1,386-patient study from Taiwan. Alimentary pharmacology & therapeutics. 25(9), 1029–1037 (2007).
Knop, V. et al. Regression of fibrosis and portal hypertension in HCV-associated cirrhosis and sustained virologic response after interferon-free antiviral therapy. Journal of viral hepatitis. 32(12), 994–1002 (2016).
Omata, M. et al. APASL consensus statements and recommendations for hepatitis C prevention, epidemiology, and laboratory testing. Hepatology international. 10(5), 681–701 (2016).
Shiratori, Y. et al. Histologic improvement of fibrosis in patients with hepatitis C who have sustained response to interferon therapy. Annals of internal medicine. 32(7), 517–524 (2000).
Okamoto, H. et al. Characterization of the genomic sequence of type V (or 3a) hepatitis C virus isolates and PCR primers for specific detection. J Gen Virol. 74(Pt 11), 2385–2390 (1999).
Vermehren, J. et al. Multi-center evaluation of the Abbott RealTime HCV assay for monitoring patients undergoing antiviral therapy for chronic hepatitis C. Journal of clinical virology 52(2), 133–137 (2011).
Scheuer, P. J. Classification of chronic viral hepatitis: a need for reassessment. Journal of hepatology. 13(3), 372–374 (1991).
Huang, C. F. et al. Interleukin-28B genetic variants in identification of hepatitis C virus genotype 1 patients responding to 24 weeks peginterferon/ribavirin. Journal of hepatology. 56(1), 34–40 (2012).
Huang, C. F. et al. Diversity of the association of serum levels and genetic variants of MHC class I polypeptide-related chain A with liver fibrosis in chronic hepatitis C. Oncotarget. 8(20), 32618–32625 (2017).
Poynard, T. et al. Slow regression of liver fibrosis presumed by repeated biomarkers after virological cure in patients with chronic hepatitis C. Journal of hepatology. 59(4), 675–683 (2013).
Facciorusso, A. et al. Long-term liver stiffness assessment in hepatitis C virus patients undergoing antiviral therapy: Results from a 5-year cohort study. Journal of gastroenterology and hepatology. 33(4), 942–949 (2018).
Lee, Y. A. & Friedman, S. L. Reversal, maintenance or progression: what happens to the liver after a virologic cure of hepatitis C? Antiviral research. 107, 23–30 (2014).
Poynard, T. et al. Impact of pegylated interferon alfa-2b and ribavirin on liver fibrosis in patients with chronic hepatitis C. Gastroenterology. 122(5), 1303–1313 (2002).
Huang, C. F. et al. Hepatitis B virus dual infection disrupts platelet count recovery in chronic hepatitis C patients with curative antiviral therapy. Journal of gastroenterology and hepatology. 33(5), 1108–1114 (2018).
D’Ambrosio, R. et al. A morphometric and immunohistochemical study to assess the benefit of a sustained virological response in hepatitis C virus patients with cirrhosis. Hepatology 56(2), 532–543 (2012).
Gomez-Moreno, A. Z. et al. Liver stiffness measurement predicts liver-related events in patients with chronic hepatitis C: A retrospective study. PLoS One. 12(9), e0184404 (2017).
Kim, G. et al. The need for histological subclassification of cirrhosis: a systematic review and meta-analysis. Liver international. 36(6), 847–855 (2016).
Missiha, S. B., Ostrowski, M. & Heathcote, E. J. Disease progression in chronic hepatitis C: modifiable and nonmodifiable factors. Gastroenterology. 134(6), 1699–1714 (2008).
Patin, E. & Kutalik, Z. et al. Genome-wide association study identifies variants associated with progression of liver fibrosis from HCV infection. Gastroenterology. 143(5), 1244–1252 e1241-1212 (2012).
Balart, L. A. et al. Peginterferon alpha-2a plus ribavirin in Latino and Non-Latino Whites with HCV genotype 1: Histologic outcomes and tolerability from the LATINO Study. The American journal of gastroenterology. 105(10), 2177–2185 (2010).
Matsuura, K. et al. Genome-Wide Association Study Identifies TLL1 Variant Associated With Development of Hepatocellular Carcinoma After Eradication of Hepatitis C Virus Infection. Gastroenterology. 152(6), 1383–1394 (2017).
Ge, G. & Greenspan, D. S. BMP1 controls TGFbeta1 activation via cleavage of latent TGFbeta-binding protein. J Cell Biol. 175, 111–120 (2006).
Ge, G. et al. Bone morphogenetic protein-1/tolloid-related metalloproteinases process osteoglycin and enhance its ability to regulate collagen fibrillogenesis. J Biol Chem. 279, 41626–41633 (2004).
van der Meer, A. J. & Berenguer, M. Reversion of disease manifestations after HCV eradication. Journal of hepatology. 65(1 Suppl), S95–108 (2016).
Schuppan, D., Krebs, A., Bauer, M. & Hahn, E. G. Hepatitis C and liver fibrosis. Cell death and differentiation. 10(Suppl 1), S59–67 (2003).
Wiese, M. et al. Evaluation of liver disease progression in the German hepatitis C virus (1b)-contaminated anti-D cohort at 35 years after infection. Hepatology. 59(1), 49–57 (2014).
van der Meer, A. J. et al. Risk of cirrhosis-related complications in patients with advanced fibrosis following hepatitis C virus eradication. Journal of hepatology. 66(3), 485–493 (2017).
Huang, C. F. et al. Baseline gamma-glutamyl transferase levels strongly correlate with hepatocellular carcinoma development in non-cirrhotic patients with successful hepatitis C virus eradication. Journal of hepatology. 61(1), 67–74 (2014).
Everhart, J. E. & Wright, E. C. Association of gamma-glutamyl transferase (GGT) activity with treatment and clinical outcomes in chronic hepatitis C (HCV). Hepatology. 57(5), 1725–1733 (2013).
Yeh, M. L. et al. Reactivation of hepatitis B in patients of chronic hepatitis C with hepatitis B virus infection treated with direct acting antivirals. J Gastroenterol Hepatol. 32(10), 1754–1762 (2017).
Holmes, J. A., Yu, M. L. & Chung, R. T. Hepatitis B reactivation during or after direct acting antiviral therapy - implication for susceptible individuals. Expert Opin Drug Saf. 16(6), 651–672 (2017).
Yu, M. L. et al. Sustained hepatitis C virus clearance and increased hepatitis B surface antigen seroclearance in patients with dual chronic hepatitis C and B during posttreatment follow-up. Hepatology. 57, 2135–2142 (2013).
D’Ambrosio, R. et al. The diagnostic accuracy of Fibroscan for cirrhosis is influenced by liver morphometry in HCV patients with a sustained virological response. Journal of hepatology. 59(2), 251–256 (2013).
Acknowledgements
This work were supported in part by grants from Kaohsiung Medical University (107CM-KMU-10) and Kaohsiung Medical University Hospital (KMUH106-6M02, KMUH105-5R05).
Author information
Authors and Affiliations
Contributions
Conception and design: Ming-Lung Yu and Jyh-Jou Chen Acquisition of data: Chung-Feng Huang, Ming-LunYeh, Ching-I Huang, Jee-Fu Huang, Zu-Yau Lin, Shinn-Cherng Chen Chia-Yen Dai and Wan-Long Chuang. Data analysis and interpretation: Chung-Feng Huang, Ming-LunYeh, and Ming-Lung Yu. Manuscript drafting and critical revising: Chung-Feng Huang, and Ming-Lung Yu. Approval of the final version of the manuscript: Ming-Lung Yu.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, CF., Yeh, ML., Huang, CI. et al. Tolloid-like 1 genetic variants determine fibrosis regression in chronic hepatitis C patients with curative antivirals. Sci Rep 8, 15058 (2018). https://doi.org/10.1038/s41598-018-33448-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-018-33448-1
Keywords
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
