
Abstract
Photosynthetic antenna proteins can be thought of as “programmed solvents”, which bind pigments at specific mutual orientations, thus tuning the overall energetic landscape and ensuring highly efficient light-harvesting. While positioning of chlorophyll cofactors is well understood and rationalized by the principle of an “energy funnel”, the carotenoids still pose many open questions. Particularly, their short excited state lifetime (<25 ps) renders them potential energy sinks able to compete with the reaction centers and drastically undermine light-harvesting efficiency. Exploration of the orientational phase-space revealed that the placement of central carotenoids minimizes their interaction with the nearest chlorophylls in the plant antenna complexes LHCII, CP26, CP29 and LHCI. At the same time we show that this interaction is highly sensitive to structural perturbations, which has a profound effect on the overall lifetime of the complex. This links the protein dynamics to the light-harvesting regulation in plants by the carotenoids.
Similar content being viewed by others
Introduction
Photosystem I (PSI) and Photosystem II (PSII) are large, integral membrane protein super-complexes in plants and green algae1,2. They are the key components of the light reactions of photosynthesis. While PSII performs water oxidation to build a transmembrane proton gradient and induce electron transfer3, PSI primarily produces the universal redox carrier NADPH4. From the functional perspective, the photosystem super-complexes are divided into the core sites of actual photochemical reactions, called reaction centers (RCs), and accessory light-harvesting complexes (LHCs). Even though the RC sub-units capture light themselves, the peripheral LHC antenna proteins of the Lhcb (in PSII) and Lhca (in PSI) families are necessary to increase the absorption cross-section and ensure optimal performance5. The antenna complexes transfer excitation energy with remarkable efficiency, enabling near unity quantum yield of PSI/II (one photochemical reaction per one photon captured)6. This is due to the fine tuning of the relative positions, orientations and excitation energies of chlorophyll (Chl) cofactors coordinated by the residues, which is the reason why LHC proteins are sometimes referred to as “programmed solvent”2. The current precision of pigment placement resolved from the crystal structures7,8,9,10 allows for highly detailed models, describing both the initial steps of exciton transfer11,12 and the subsequent charge separation in the RCs13. However, such models largely account only for the Chls, while the second major building block of the pigment arrays, the carotenoids (Cars), are usually disregarded.
Cars are typically included in photodynamic models of bacterial systems only, where they are significant light-harvesters14, while their light-harvesting role in plants is minor compared to the photoprotective function15,16. The latter is performed primarily by quenching the Chl triplet states17, which would otherwise sensitize molecular oxygen to form harmful singlet oxygen species18. Alternatively, Cars may directly act as singlet oxygen scavengers19. However, an accumulating body of knowledge points to even deeper involvement of Cars in photoprotection, already suppressing the formation of Chl triplet states: a part of a process termed non-photochemical quenching (NPQ)20. Even though several molecular NPQ mechanisms are proposed21, Cars are especially appealing agent-candidates because of their extremely short-lived (10–25 ps) lowest singlet excited state S1 22, which was demonstrated to yield significant quenching in artificial caroteno-phthalocyanine dyads23. However, the optically dark nature of S1 24 makes it nearly impossible to be observed directly and poses considerable challenge in describing its properties from first principles25. These two features largely prevented the full inclusion of Cars in the photodynamic models in plants, an issue which is only currently being addressed26,27.
The fact that the lifetime of the S1 state is comparable to the typical time-scales of the energy transfer between the protein sub-units (or even shorter)11 raises a conceptual question: How can such pigments be incorporated into a light-harvesting system without hindering its function by wasteful dissipation of the captured energy? Or alternatively, how can such a dissipative channel play a specific integral role in regulating light-harvesting in a fluctuating light environment? In this study we analyze the nearest Chl–Car pairs in the plant antenna complexes LHCII/LHCI, CP26 and CP29 with particular focus on the mutual orientation. We emphasize the observation of repetitive conformation pattern within these systems. The study of the coupling strength between the lowest singlet excited states of the two pigments revealed that, within the phase-space of possible mutual orientations, the configuration of minimal coupling is assumed. Furthermore, we show that within such a configuration, Chl-to-Car excitation transfer rate is highly sensitive to the mutual orientation, which can be driven from excitation-preserving to quenching configurations within physiologically reasonable boundaries. This not only supports the idea of Cars acting as one of the agents regulating energy density in the photosystems under high-light conditions, but also presents the most feasible molecular switching pathway.
Results
Clearly-expressed preferred mutual orientation of closest co-facial Chl–Car pairs
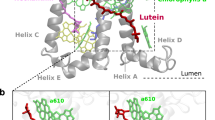
We inspected Car placement within the major and minor antenna proteins of the PSII supercomplex (PDB: 3JCU)9 and the antenna proteins of the PSI supercomplex (PDB: 4XK8)8. We focused on the Lhcb and Lhca sub-units that comprise LHCII (Lhcb1 (shown in Fig. 1a–c), Lhcb2 and Lhcb3 forming a trimer, Fig. 1d), CP29 (Lhcb4), CP26 (Lhcb5) and LHCI (Lhca1–4). Each of these homologous sub-units host several Car binding sites. We identify the Chls that are closest to the middle section of the Cars (C16=C37 bond), because that is the center of their transition density (which governs their coupling capability, see Supplementary Information). Additionally, we look for the Chl that has its chlorin ring maximally parallel to the conjugation plane of the Car, because that is a condition for maximal interaction due to the overlap of transition densities28. These conditions are best met by two Cars, each bound in an elongated groove on the two sides of the central transmembrane helices A and B of the Lhca/Lhcb apoproteins (Fig. 1a–c). The Car within the groove ending at the lumenal side helix D, called the L1 site (nomenclature according to Kühlbrandt et al.29), is assigned as lutein (Lut). The second site, L2, ending at the second short helix E, is occupied either by Lut (in Lhcb1–3 and 5) or by violaxanthin (Vio; in Lhcb4, Lhca1–4). The distance from Chl (central Mg atom) to Car (central bond) is between 5.7–6.7 Å in all Lhcas/Lhcbs.

Mutual orientation of Cars and their closest co-facial Chl partners in the L1/L2 sites. (a–c) Positioning of Lhcb1 within the membrane (PDB: 3JCU)9. The views are along the membrane plane (a,b) and from the stromal side (c). A close-up view of the L1/L2 sites in b shows the pigment pairs and the naming nomenclature according to Liu et al.7. Transmembrane helices A, B, C and amphipathic helices D and E are shown in light-blue. The remaining pigment composition of Lhcb1 is shown in c: 8 Chl a’s are shown in green, 6 Chl b’s are in red, Vio and neoxanthin are shown in pink and yellow, accordingly (molecules and proteins plotted with VMD31). (d) The full trimer of the LHCII antenna (lumenal view). Monomers are emphasized by colors. (e,f) Superposition of all the L1 and L2 pairs from Lhcb1-5 and Lhca1-4 protein units. The front view (e) and the side view (f) also show the coordinate system associated with the Chl. Cars are shown as ghost atoms to emphasize the distribution rather than the actual positions, except for Lut 620 from L1 site of Lhcb1 which is highlighted for comparison. Only Chl a612 from the L1 site of Lhcb1 represents all the Chls for clarity because the other Chls from the pairs largely differ only by the conformation of their phytol tails, which do not contribute to the transition density (Supplementary Fig. 1a).
Additionally, there are sites N1 (binds neoxanthin in Lhcbs1-5 or all-trans β-carotene in all Lhcas and, as recently suggested, Lhcb610) and V1 (only in Lhcb1-3, possess either Vio, astaxanthin or zeaxanthin). However, since we are particularly interested in the possible role of Cars as singlet energy acceptors, we disregard these two sites. The N1 Cars are not taken into account since they interact almost exclusively with Chl b’s30 (which rapidly transfer energy to the Chl a pool), while the V1 site is disregarded because the surrounding Chls are only close to the Car end-groups, which barely contribute to the transition density of the S1 state (Supplementary Fig. 1b,c). Cars within V1 site were also shown to have their S1 state negligibly coupled to the nearest Chls because of unfavorable edge-on orientations and larger distances (>9 Å)26.
We have superimposed all L1 and L2 Chl–Car dimers onto one another, so as to achieve maximal coincidence of the four nitrogen atoms within the Chls (Fig. 1e,f). Interestingly, the orientations are strictly preserved among both the Lhcb/Lhca apoproteins and the two sites. Specifically, the Cars seem to be located at one preferred side of their Chl partner. In order to rationalize such preferred Chl and Car binding orientation, we investigated the dependence of the energy-transfer inducing electronic coupling on the mutual orientation of the two pigments. The coupling, also termed resonance interaction, between the lowest singlet states Chl Qy and Car S1, \({J}_{{Q}_{y}{S}_{1}}\), was evaluated using the transition density cube (TDC) method32 (for precise definitions see Methods and Supporting Information). It was calculated under the rotation of Chl around the axis perpendicular to the chlorin ring and originating at the Mg atom (z axis, Fig. 1f). Since all the Cars and Chls assume slightly different molecular conformations enforced by the binding pockets we used planar (vacuum-optimized) structures superimposed onto the prototype Lut 620–Chl a612 and Lut 621–Chl a603 pairs from LHCII (nomenclature according to Liu et al.7).
The resulting coupling dependence is shown in Fig. 2. The origin corresponds to the configuration of the planar molecules superimposed on the originals from the crystal structure. Results for both L1 and L2 pairs are shown to represent the effect of slight differences in the placement and initial orientation. The couplings are an order of magnitude smaller than their typical Chl–Chl counterparts (20–120 cm−1)33, which reflects the “dark” nature of the S1 state. The positive and negative peaks are reminiscent of the dipole–dipole interaction (sign is arbitrary). Most importantly, we notice that the actual orientation from the crystal structure corresponds to the minimal coupling (2.7 cm−1) between the two pigments. Furthermore, at this orientation the dependence demonstrates a shallow plateau as opposed to the steep dependence at the other J = 0 orientation.

Chl–Lut resonance interaction \({{\boldsymbol{J}}}_{{{\boldsymbol{Q}}}_{{\boldsymbol{y}}}{{\boldsymbol{S}}}_{1}}\) as a function of the Chl rotation around the z axis. Results for L1 and L2 sites of a single Lhcb1 monomer (red and blue, accordingly) are qualitatively identical, except for a shallow local minimum for L1 site at −15°. Dots correspond to the calculated values, lines correspond to the spline interpolated values. The inset shows the dependence in the immediate vicinity of the original orientation at which the couplings are 2.6 cm−1 and 2.7 cm−1 for L1 and L2 sites, accordingly.
Flexibility of Chl–Car pairs in fluctuating environment
Having revealed the regularity of the Chl–Car orientational motif, a natural question arises: is such a configuration preserved in vivo, and if so, does it have physiological significance? Furthermore, it is important to know how stable and rigid such a configuration is. In order to study the extent of the configuration-space available to Cars with respect to the Chls we performed a Molecular Dynamics (MD) simulation of an LHCII trimer (Fig. 1d) within a lipid membrane. A 1 μs trajectory was generated and 1000 snapshots taken at every nanosecond are considered. Planar, vacuum-optimized Chl a and Lut were superimposed onto the extracted L1/L2 pairs (result for a random snapshot shown in Supplementary Fig. 3). This was done in order to have well quantum-chemically optimized molecules for coupling calculation. For details of the simulation and subsequent coupling calculations see the Methods and Supplementary Information.
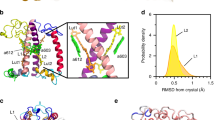
The MD trajectory reveals that both Luts are rigidly fixed position-wise relative to their Chl partners: the fluctuations are of the order of 0.5 Å or smaller (conf., Fig. 3a). Specifically, the central C16 = C37 bond is situated at (1.04 ± 0.52; −5.53 ± 0.41; −2.87 ± 0.30) Å for L1 and at (1.88 ± 0.45; −5.19 ± 0.34; −3.29 ± 0.29) Å for L2, which means that Lut 620 is slightly closer to the chlorin ring of its partner Chl a612 (z coordinate). At the same time both sites appear to be flexible enough to yield considerable orientational fluctuations. To quantify these fluctuations we introduce angles \({\phi }_{yx}\) and \({\phi }_{zx}\) (Fig. 3b,c). The former corresponds to the inclination of the backbone projection in the xy plane towards the x axis and partially relates to the rotation investigated in Fig. 2. The latter corresponds to the projection in the xz plane and describes the inclination of the Car towards its Chl. For the L1 site the mean values are \({\phi }_{yx}={(11.1\pm 4.2)}^{^\circ }\) and \({\phi }_{zx}={(6.8\pm 3.1)}^{^\circ }\). For the L2 site the mean values are \({\phi }_{yx}={(15.1\pm 3.5)}^{^\circ }\) and \({\phi }_{zx}={(3.0\pm 3.9)}^{^\circ }\). The corresponding distributions within the full trimer are shown in Fig. 3d,e. While Lut 621 is more inclined towards y axis than Lut 620, the opposite is true for the inclination towards the z axis. Interestingly, the latter trend is not observed directly within the crystal structure (see arrows in Fig. 3e), while Lut 621 in the MD trajectory relaxes toward markedly different orientation (Supplementary Fig. 4).

L1 and L2 pairs’ statistics from Molecular Dynamics simulation. (a) Distribution of the position of the central C16 = C37 bond of the two Luts within one monomer showing the distinction between L1 (red) and L2 (blue) sites. All coordinates and directions in this work are represented in the Chl a reference frame given in Fig. 1e,f. (b,c) Definition of the characteristic angles \({\phi }_{yx}\) and \({\phi }_{zx}\) that the Lut backbone (C9–C30 axis) projections form with the x coordinate axis: \({\phi }_{yx}\) angle corresponds to the projection of the backbone axis in the xy plane (b), likewise, \({\phi }_{zx}\) angle corresponds to the projection of the backbone axis in the xz plane (c). (d,e) The distribution of the \({\phi }_{yx}\,\,\)and \({\phi }_{zx}\) angles for L1 (d) and L2 sites (e) within the full trimer. Distributions of \({\phi }_{yx}\) and \({\phi }_{zx}\) are shown in red and blue, accordingly. The arrows indicate the corresponding average values directly from the crystal structure PDB: 1RWT7 (averaged over all three trimers captured in the structure). The envelopes show the normal distribution fit.
Orientational dependence of Chl–Car coupling from probing configuration phase-space
We note that the rotation angle in Fig. 2, in its immediate vicinity around 0, effectively corresponds to the angle \(\,{\phi }_{yx}\). The variation of \({\phi }_{yx}\) therefore means that modulation of the coupling is to be expected. However, we also note that comparable variation is present in the angle \({\phi }_{zx}\) as well. Lastly, the orientation of a rigid-body is fully described by three angles: in our case the rotation around the backbone axis of a Car is the third degree of freedom. All this points to the need to evaluate multiple Interaction Energy Surfaces (IES) in such a configuration phase-space. We calculated the IES for both sites (which represents the variation of coordinate in itself) varying the angles \({\phi }_{yx}\) and \({\phi }_{zx}\) and also the direction of the plane vector of the Luts. The latter dependence was found to be marginal (Supplementary Fig. 2).
The IES \({J}_{{\rm{L}}1/{\rm{L}}2}({\phi }_{yx},{\phi }_{zx})\) are shown in Fig. 4. Only absolute values of the couplings are shown, since the transfer rates are insensitive to the sign. The interpolated values of the couplings are shown along with the actual orientations from the MD trajectory (circles). The main feature of both surfaces is the decrease of the interaction energy going from smaller to larger angles, where the dependence passes the minimal J = 0 boundary. There are several minor local features too, especially at the L1 site. These features represent the fact that at such small distances the coupling is sensitive to even minute atomic differences (see Supporting Information). Interaction energy along \({\phi }_{yx}\) varies rather moderately and in line with the plateau-like dependence of Fig. 2. The variation in the \({\phi }_{zx}\) direction is stronger, and taken together they yield a substantial change in the interaction for relatively small deflections. The physiologically probable deflections can be inferred from the MD values of the orientation. While the average couplings are \({J}_{{\rm{L}}1}=3.6\pm 1.3\) cm−1 and \({J}_{{\rm{L}}2}=3.1\pm 0.9\) cm−1, they can increase almost twice or vanish within one trajectory. The angular dependence of the interaction energies for different displacements are shown and discussed in the Supplementary Information.

The Chl Qy and Car S1 state interaction energy surfaces. (a,b) The IES (absolute values) are shown for the L1 and L2 sites, respectively. The white circles represent the actual values of the two angles from the MD trajectory. The dashed lines show the J = 3 cm−1 values for guidance. For the interpolation of the 41 TDC coupling values a 100 × 100 point grid was used.
Chl-to-Car excitation transfer, energy quenching and the functional role of the process
The calculated couplings provide insight into possible energy transfer processes in their own right, however, in order to appreciate their significance a full model of excitation relaxation is needed. Therefore, we firstly calculated the transfer rate of the excitation from the Qy to the S1 state as a function of the angles \({\phi }_{yx}\) and \(\,{\phi }_{zx}\). We used Förster resonance energy transfer (FRET) theory34, which is justified by the smallness of the coupling and the relatively large energy gap between the states35. Lut S1 energy was set to 14220 cm−1 as determined from the two-photon absorption data36 (see Supplementary Information), and the typical energy of Chl a Qy state (14900 cm−1)33 was used. The angular dependence of the transfer rates on the coupling in Fig. 2 is shown to be very strong because of the \(|J{|}^{2}\) factor (Supplementary Fig. 5). By contrast, the transfer rates mostly negligibly depend on the energy gap between Qy and S1. This shows, that the uncertainty of the S1 energy is a minor factor in the details of such energy transfer.
The calculated transfer rates demonstrate how the protein scaffold operates on the level of individual Chl–Car pairs, avoiding orientations that would facilitate excitation quenching via S1 state (Supplementary Fig. 5). Now we look at the role of such transfer in the overall biological functioning of the entire LHC sub-unit. In order to illustrate how transfer to S1 affects the excitation evolution within one Lhcb/Lhca complex under the conditions of closed RCs, we consider a coarse-grained, purely kinetic model, summarized in Fig. 5a. We partition the Chl a sub-population within a single LHC into the pool (6 Chl a’s; green block in Fig. 5a) and separate Chl a612 and Chl a603 (L1 and L2 sites, accordingly). All the Chl b’s are assumed to instantly populate the Chl a pool, which is lower in energy (80% of excitation transferred in less than 1 ps16). The pool transfers the excitation towards Chls a612/603 with the rate \({k}_{{{\rm{pQ}}}_{y}}\). The excitation leaves the singled-out Chls either back to the pool (rate \({k}_{{{\rm{Q}}}_{y}{\rm{p}}}\)) or to their Car partners (the calculated FRET rates \({k}_{{{\rm{Q}}}_{y}{{\rm{S}}}_{1}}\)). All Chls have the lifetime of 4 ns37, while the lifetime of Lut S1 state is considered 14 ps22. The robustness of this model with respect to parameter variation is discussed in the Supplementary Information.

Lifetime of the Chl pool in the presence of Luts. (a) A pool of 6 Chl a’s (together with Chl a612 and Chl a603 representing the Chl a subsystem of an Lhca/Lhcb unit) is equilibrating with Chl a612/Chl a603 which in turn transfer excitation to their Car partners Lut 620/Lut 621. (b) The lifetime of the whole complex based on the L1/L2 pigment pair structures from the MD simulation. The inset shows the lifetime distribution based on the MD snapshots. (c) The lifetime of the complex as a function of the Car tilting angles, \({\tau }_{{\rm{complex}}}({\phi }_{yx},{\phi }_{zx})\), calculated for the coarse-grained model a using the L2 site IES, \({J}_{{\rm{L}}2}({\phi }_{yx},{\phi }_{zx})\), for both sites because of the smoothness of this particular IES. The dashed lines indicate the typical time-scales of the light-harvesting regime in the membrane (2 ns37) and the quenched regime under high-light conditions (~400 ps38). The circles represent the actual tilt angles for Lut 621 from the MD trajectory.
Having calculated the IES, we map the coupling values onto particular instances of the L1/L2 pair orientations from the MD trajectory. The obtained coupling trajectory is then converted into the rates, which in turn are used within the coarse-grained model. The resultant values of the net lifetime of the whole complex, \({\tau }_{{\rm{complex}}}\), are shown in Fig. 5b. The transient dynamics (first ~200 ns) can be observed, where the initial lifetime of 1.5 ns drops to the subsequent average value of 0.86 ns. Such a sub-nanosecond lifetime has been observed for quenched LHCII crystals39, which is a sign that the MD trajectory is within a local minimum not too far from the crystal structure used as the starting point.
We further employ the coarse-grained model to see the effect of alternative Chl–Car orientations on the lifetime \({\tau }_{{\rm{complex}}}\). To that end we use the full IES of the L2 site (Fig. 4c) for both sites, because of the smoothness of this particular IES as opposed to the detailed IES of the L1 site (Fig. 4b). The resulting \({\tau }_{{\rm{complex}}}\) map is shown in Fig. 5c. Even though a number of approximations have been invoked, the result provides us with several valuable insights. Naturally, the diagonal dependence of the interaction energy translates into the diagonal dependence of the net lifetime. The strong coupling at small angles translates into short lifetime of the complex (lower left corner) and vice versa for the large angles (upper right corner). Again the strong dependence of FRET rates on the coupling leads to profound change of the lifetime upon slight changes in the angles. The red stripe, corresponding to the intrinsic Chla lifetime of 4 ns, is separated from the light-harvesting configuration in the membrane (\({\tau }_{{\rm{complex}}}\approx 2\,{\rm{ns}}\) 37) by just 5°−10°. A tilt reducing the angles by \( \sim {5}^{^\circ }\) can bring the lifetime to the domain of values typical for the LHCII crystals (~1 ns), as mentioned previously. A further tilt of 5°−10° could bring the lifetime to as low as 400 ps, a characteristic value of LHCII lifetime under the NPQ conditions38. This means that, at least in principle, the re-orientation of the transition densities of Luts could be one of the key ingredients in switching from the light-harvesting state into the NPQ mode of operation within LHCs.
Discussion
In principle, there are 6 explicit degrees of freedom in placing a Car molecule with respect to Chl: 3 coordinates to displace the molecular center and 3 angles to orient the molecule at a given position. Additionally, there are the “intrinsic” degrees of freedom that describe the actual molecular shape governed both by the chemical structure and the coordinating residues of the apoprotein. As it turns out, in such a vast phase-space of possible configurations, nature appears to be consistently restrictive (Fig. 1e,f). The benefit of close co-facial pairing of certain Chls and Cars has been rationalized in terms of efficient triplet quenching40,41. However, that alone does not explain the restrictive binding and its repetition throughout the variety of LHCs. Furthermore, having a pigment of such a short lifetime as Cars22 poses a threat to the excitation energy storage within the antenna before any subsequent transfer to the RCs can take place. Our coupling calculations, spanning the orientation phase-space simultaneously in several directions, revealed that the specific mutual orientation actually corresponds to the minimal resonance interaction between the lowest-lying singlet states of the two pigments (Fig. 2). This enables the photosystems to benefit from all the functionality of Cars (structural, light-harvesting and protective roles22,42) without depleting the system of energy.
On the other hand, the fact that Cars induce observable energy dissipation, even with the suppressed interaction, cannot be ignored (Fig. 5c). Even the modest couplings yield excitation quenching, the only question being whether the Luts in LHCII are only minor quenchers, or are they important enough to be the major agents of the NPQ mechanism? The \({|J|}^{2}\) factor within the FRET rate implies a very strong LHC lifetime dependence on the Chl–Car coupling, making the ensuing dynamics extremely susceptible to both the precise inter-pigment configuration and the intra-pigment transition density distribution. Increasingly, the sensitivity of this pathway is being discussed in the context of NPQ mechanism. Several recent studies cite the Car S1 state as the quencher of excess energy proposing that this pathway is modulated by some relative movement of the Chls and Cars43,44,45. Our MD simulations, in conjunction with the coupling calculations, provide insight into how specific variations of these configurational degrees of freedom play a physiological role. There are three major aspects regarding what can be learnt from the presented results.
Position
There are arguments regarding Cars’ (non)involvement in NPQ that relate to the mutual Chl–Car positioning. One group of such arguments claims that no translational movement is possible within as tightly bound a scaffold as a protein, meaning there is simply not enough room for Cars to act as NPQ “switches”46. There is, however, another group of arguments anticipating very specific movements as switches45,47. The MD simulation favors the former arguments, because the position fluctuations are confined within ~1 Å. Of course, the simulated dynamics represent thermal fluctuations, not abrupt conformational changes, yet the coupling dependence on distance (see Supplementary Information) points to drastic displacements required for actual switching.
Orientation
As opposed to the distance, a wide angular distribution of the Chl with respect to the Car conjugated backbone is supported by the protein (Fig. 2d,e). More importantly, even moderate tilts are sufficient for a substantial change in the interaction, hence the sensitivity of the LHC excitation lifetime to the fluctuations of the mutual Chl–Car configuration within the MD trajectory. Such sensitivity in turn implies that the functional state of an LHC unit can be easily shifted towards a state that markedly decreases the excitation lifetime (Fig. 5c), supporting some earlier proposed NPQ mechanisms48. The physical orientations capable of inducing or preventing the dissipative regime in the whole LHC are shown in Fig. 6.

Representative structures of the boundary regimes of the excitation density control. A tilt of Luts with respect to Chls by less than 20° is sufficient to switch from the excitation energy preserving/light-harvesting mode (full orange structures) to the highly dissipative/quenching mode (transparent structures). The shown Lut structures correspond to the marginal angular values of Fig. 5c: the lower left corner (5.7°−9.2°, transparent) and the upper right corner \((({26.4}^{^\circ },{15.1}^{^\circ });\,J\approx 0;\,{\rm{full}}\,{\rm{color}})\).
Distortions
One common argument is that the Car S1 state may become “more allowed” in the face of protein-induced distortions away from planarity. This is an aspect disregarded in our coupling calculations (conf., Supplementary Fig. 3). While some deformations are present within the MD results, they are beyond the current computational capabilities due to complicated character of the S1 state combined with the system size25. However, even under such circumstances several reasonable and important observations can be made. There are two types of quantitative changes possibly induced by the deformation: a shift in energy and a change in the dipole moment. The former change appears to be irrelevant in the overall transfer because of smallness of the resonance interaction and strong influence of the environment (see discussion in the Supplementary Information). This additionally rules out any gearshift-type mechanism of NPQ which relies on closing the Chl–Car energy gap as a switch23. Interestingly, this indirectly points to the non-uniqueness of L1 site as a quencher in LHCII and CP29, which it was proposed to be based on the association with the lowest energy Chl cluster (Chl a610/611/612)33. The possible change in the dipole moment is more difficult to account for, but the trend can be named with certainty: due to deformation and the ensuing admixture of one-electron configurations into the S1 state (effective mixing with dipole-allowed state S2) its dipole moment can only increase49, thus increasing the coupling. At the same time such increase must be very well bound, for there is no spectroscopic evidence of the S1 state opening up upon deformation (e.g., heavily bent Neo in the N1 pocket does not produce S1 optical signal). But we agree that the geometry aspect is of paramount importance when dealing with the Car S1 state and needs further clarification, which would come with the development of more suitable quantum chemical methods. Lastly, there are qualitative geometrical changes associated with the head-group rotation, however, their quenching role is precluded by the virtual absence of transition density in these groups (Supplementary Fig. 1b,c).
In conclusion, we have demonstrated that the positioning of Cars within the green lineage eukaryotic antenna complexes is governed by the principle of minimal resonance interaction between Car S1 and spatially closest Chl Qy states. This explains the remarkably regular Car binding maintained within homologous sites. Despite being minimal, the coupling is sensitive to even slight deviations of mutual pigment orientation. We demonstrate that a small increase in coupling translates into significant excitation quenching of the whole LHC unit. Therefore minor adjustments of Car orientation toward Chl sustained by the protein scaffold are sufficient for the transitions between light-harvesting and photoprotective (quenched) global states. The repetition of the Chl–Car configuration also implies that multiple NPQ sites are possible, not just within the major antenna. The obtained slow transfer-to-trap rates ensure that quenching does not compete with the open RCs and that the quencher is only significant once the RCs close. Such a concept of quenching has been termed “economic photoprotection”50. Lastly, the results show that further steered-MD studies provide a viable path for pinpointing the precise transitions between the harvesting/quenched configurations under in vivo conditions.
Methods
Electronic structure and coupling calculations
Ground state geometries of the molecules were optimized using density-functional theory (B3LYP functional) as implemented in Gaussian09 package51. The lowest excited singlet states of the vacuum-optimized pigments were calculated by a full Configuration Interaction calculation within a Complete Active Space using the semi-empirical AM1 Hamiltonian (AM1-CAS-CI) as implemented in the package MOPAC201652. This methodology was benchmarked for Cars by Kusumoto et al.53 Using the obtained wave-functions we calculated the transition densities (custom code54) within the TDC famework32:
where \({\Psi }_{g}\) and \({\Psi }_{e}^{\ast }\) are the ground and excited state wave-functions, and \(\delta x,\delta y,\delta z\) define the grid size of the cube. The electronic coupling (Coulombic part only) was calculated as \(J=\frac{{e}^{2}}{4\pi \varepsilon {\varepsilon }_{0}}\sum _{i,j}{M}_{m}(i){M}_{n}(j)/|\mathop{{r}_{i}}\limits^{\rightharpoonup }-\mathop{{r}_{j}}\limits^{\rightharpoonup }|,\) where Chl and Lut transition dipole moments were re-scaled to 4.49 D55 and 0.767 D25, respectively.
Molecular dynamics simulation
The high resolution X-ray crystal structure of LHCII from spinach7 (PDB: 1RWT) was used for MD simulation. We selected the trimer of chains C, H, and E. A DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) bilayer membrane was generated by the CHARMM-GUI56 with 450 lipids in each layer. The membrane was generated in a rectangular box with upper and lower water layers containing 37 water molecules per lipid molecule. LHCII was pre-equilibrated before placing into the lipid bilayer: a LHCII shape cavity was generated at the center of the membrane by removing the lipids that were closer than 1.5 Å from the complex. Then, the LHCII complex was inserted into the pore according to the suggested orientation of protein membrane database57. The MD of LHCII embedded in the membrane was performed following the protocol described by Ogata and coworkers58. MD runs were performed with the Amber14 suite59. The Amber ff14SB force field was used to describe the protein60. All carotenoids were modelled by an ad hoc force field described in Prandi et al.61 Chls a were modelled with the set of parameters reported in Ceccarelli et al.62 and modified by Zhang and coauthors63; Chls b were described with the same set of parameters of Chls a except for the aldehyde group on porphyrin ring, taking parameters from the General Amber Force Field64. The DOPC membrane was described with the Lipid14 forcefield65. Since this force field for lipid does not contain parameters for the internal DPPG molecule, the previous version of the force field (Lipid1166) was used for it. Water molecules were described through the TIP3P model67, and ionic parameters were taken from Joung and Cheatham68. The full protocol is given in the Supporting Information.
Resonance transfer rates
The FRET rates between pigments m and n are given by69
where \({J}_{mn}\) is the inter-pigment coupling and \(A(t),F(t)\) are the acceptor absorption and donor fluorescence time-domain response functions, related to the corresponding spectra via the Fourier transform. Spectral information for Chl a is reported by Renger et al.70 while the corresponding parameters for Lut were extracted from the two-photon absorption data by Walla et al.36 as detailed in the Supporting Information.
Coarse-grained model simulation
The evolution of the coarse-grained model of five sites (pool and four pigments) is governed by a Master equation, which in turn is fully described by the matrix of inter-site transfer and on-site decay rates. The pool-to-Chl a612/603 and the reverse rates, \({k}_{{{\rm{pQ}}}_{y}}\) and \({k}_{{{\rm{Q}}}_{y}{\rm{p}}}\), are related by the entropic factor: \({k}_{{{\rm{pQ}}}_{y}}={k}_{{{\rm{Q}}}_{y}{\rm{p}}}/6\equiv k/6\), which simply accounts for the fact that transfer from n sites to one particular site is n time less likely than the reverse; we used a characteristic value \({k}^{-1}=1\,{\rm{ps}}\). The lifetime of the complex \({\tau }_{{\rm{complx}}.}\) is then directly related to the eigenvalues of the matrix, as given in the Supporting Information along with the discussion of the possible parameter variation.
Data availability
All the couplings, rates, simulation data and pigment pair structures are available upon request to the corresponding author.
References
Blankenship, R. E. Molecular Mechanisms of Photosynthesis (BlackwellScience, Oxford, 2002).
Ruban, A.V. The Photosynthetic Membrane: Molecular Mechanisms and Biophysics of Light Harvesting (John Wiley & Sons Ltd., 2013).
van Amerongen, H. & Croce, R. Light harvesting in photosystem II. Photosynthesis Res. 116 (2013).
Amunts, A. & Nelson, N. Plant photosystem I design in the light of evolution. Structure 17, 637–650 (2009).
Croce, R. & van Amerongen, H. Natural strategies for photosynthetic light harvesting. Nat. Chem. Biol. 10, 492–501 (2014).
van Amerongen, H., Valkunas, L. & van Grondelle, R. Photosynthetic Excitons (World Scientific, Singapore, 2000).
Liu, Z. F. et al. Crystal structure of spinach major light-harvesting complex at 2.72 A resolution. Nature 428, 287–292 (2004).
Qin, X., Suga, M., Kuang, T. & Shen, J.-R. Structural basis for energy transfer pathways in the plant PSI-LHCI supercomplex. Science 348, 989–995 (2015).
Wei, X. et al. Structure of spinach photosystem II-LHCII supercomplex at 3.2 A resolution. Nature 534, 69–87 (2016).
Su, X. et al. Structure and assembly mechanism of plant C2S2M2-type PSII-LHCII supercomplex. Science 357, 815–820 (2017).
Bennett, D. I. G., Amarnath, K. & Fleming, G. R. A Structure-Based Model of Energy Transfer Reveals the Principles of Light Harvesting in Photosystem II Supercomplexes. J. Am. Chem. Soc. 135, 9164–9173 (2013).
Renger, T. & Müh, F. Understanding photosynthetic light-harvesting: a bottom up theoretical approach. Phys. Chem. Chem. Phys. 15, 3348–3371 (2013).
Romero, E., Novoderezhkin, V. I. & van Grondelle, R. Quantum design of photosynthesis for bio-inspired solar-energy conversion. Nature 543, 355–365 (2017).
Ritz, T., Damjanović, A. & Schulten, K. The quantum physics of photosynthesis. Chem Phys Chem 3, 243–248 (2002).
Frank, H. A. & Cogdell, R. J. Carotenoids in photosynthesis. Photochem. Photobiol. 63, 257–264 (1996).
van Amerongen, H. & van Grondelle, R. Understanding the energy transfer function of LHCII, the major light-harvesting complex of green plants. J. Phys. Chem. B 105, 604–617 (2001).
Ballottari, M., Mozzo, M., Girardon, J., Hienerwadel, R. & Bassi, R. Chlorophyll triplet quenching and photoprotection in the higher plant monomeric antenna protein Lhcb5. J. Phys. Chem. B 117, 11337–11348 (2013).
Pospíšil, P. Production of reactive oxygen species by photosystem II as a response to light and temperature stress. Frontiers in plant science 7 (2016).
Foyer, C.H. & Harbinson, J. Relationships between antioxidant metabolism and carotenoids in the regulation of photosynthesis. In The photochemistry of carotenoids 305–325 (Springer, 1999).
Müller, P., Li, X.-P. & Niyogi, K. K. Non-photochemical quenching. A response to excess light energy. Plant Physiol. 125, 1558–1566 (2001).
Ruban, A. V., Johnson, M. P. & Duffy, C. D. P. The photoprotective molecular switch in the photosystem II antenna. Biochim. Biophys. Acta - Bioenerg. 1817, 167–181 (2012).
Polivka, T. & Sundstrom, V. Ultrafast dynamics of carotenoid excited states - From solution to natural and artificial systems. Chem. Rev. 104, 2021–2071 (2004).
Liao, P.-N. et al. On the role of excitonic interactions in carotenoid–phthalocyanine dyads and implications for photosynthetic regulation. Photosynthesis Res. 111, 237–243 (2012).
Schulten, K. & Karplus, M. On the origin of a low-lying forbidden transition in polyenes and related molecules. Chem. Phys. Lett. 14, 305–309 (1972).
Andreussi, O., Knecht, S., Marian, C. M., Kongsted, J. & Mennucci, B. Carotenoids and Light-Harvesting: From DFT/MRCI to the Tamm-Dancoff Approximation. J. Chem. Theory Comput. 11, 655–666 (2015).
Chmeliov, J. et al. An ‘all pigment’ model of excitation quenching in LHCII. Phys. Chem. Chem. Phys. 17, 15857–15867 (2015).
Fox, K. F. et al. The Carotenoid pathway: What is important for excitation quenching in plant antenna complexes? Phys. Chem. Chem. Phys. 19, 22957–22968 (2017).
Dreuw, A., Fleming, G. & Head-Gordon, M. Role of electron-transfer quenching of chlorophyll fluorescence by carotenoids in non-photochemical quenching of green plants. Biochem. Soc. Trans. 33, 858–862 (2005).
Kühlbrandt, W., Wang, D. N. & Fujiyoshi, Y. Atomic model of plant light-harvesting complex by electron crystallography. Nature 367, 614–621 (1994).
Croce, R., Müller, M. G., Bassi, R. & Holzwarth, A. R. Carotenoid-to-chlorophyll energy transfer in recombinant major light-harvesting complex (LHCII) of higher plants. I. Femtosecond transient absorption measurements. Biophys. J. 80, 901–915 (2001).
Humphrey, W., Dalke, A. & Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graphics 14, 33–38 (1996).
Krueger, B. P., Scholes, G. D. & Fleming, G. R. Calculation of couplings and energy-transfer pathways between the pigments of LH2 by the ab initio transition density cube method. J. Phys. Chem. B 102, 5378–5386 (1998).
Müh, F., Madjet, M. E. A. & Renger, T. Structure-Based Identification of Energy Sinks in Plant Light-Harvesting Complex II. J.Phys. Chem. B 114, 13517–13535 (2010).
Förster, T. Zwischenmolekulare energiewanderung und fluoreszenz. Annalen der physik 437, 55–75 (1948).
Balevičius, V., Gelzinis, A., Abramavicius, D., Mančal, T. & Valkunas, L. Excitation dynamics and relaxation in a molecular heterodimer. Chem. Phys. 404, 94–102 (2012).
Walla, P. J., Linden, P. A., Ohta, K. & Fleming, G. R. Excited-state kinetics of the carotenoid S1 state in LHC II and two-photon excitation spectra of lutein and β-carotene in solution: efficient Car S1 → Chl electronic energy transfer via hot S1 states? J. Phys. Chem. A. 106, 1909–1916 (2002).
Belgio, E., Johnson, M. P., Juric, S. & Ruban, A. V. Higher Plant Photosystem II Light-Harvesting Antenna, Not the Reaction Center, Determines the Excited-State Lifetime-Both the Maximum and the Nonphotochemically Quenched. Biophys. J. 102, 2761–2771 (2012).
Miloslavina, Y. et al. Far‐red fluorescence: A direct spectroscopic marker for LHCII oligomer formation in non‐photochemical quenching. FEBS Lett. 582, 3625–3631 (2008).
Pascal, A. A. et al. Molecular basis of photoprotection and control of photosynthetic light-harvesting. Nature 436, 134–137 (2005).
Mozzo, M., Dall’Osto, L., Hienerwadel, R., Bassi, R. & Croce, R. Photoprotection in the antenna complexes of photosystem II Role of individual xanthophylls in chlorophyll triplet quenching. J. Biol. Chem. 283, 6184–6192 (2008).
Cupellini, L., Jurinovich, S., Prandi, I. G., Caprasecca, S. & Mennucci, B. Photoprotection and triplet energy transfer in higher plants: the role of electronic and nuclear fluctuations. Phys. Chem. Chem. Phys. 18, 11288–11296 (2016).
Landrum, J. T. Carotenoids: physical, chemical, and biological functions and properties, (CRC Press, 2009).
Ruban, A. V. et al. Identification of a mechanism of photoprotective energy dissipation in higher plants. Nature 450, 575–U22 (2007).
Liguori, N., Periole, X., Marrink, S. J. & Croce, R. From light-harvesting to photoprotection: structural basis of the dynamic switch of the major antenna complex of plants (LHCII). Scientific Reports 5, 10 (2015).
Kondo, T., Chen, W. J. & Schlau-Cohen, G. S. Single-Molecule Fluorescence Spectroscopy of Photosynthetic Systems. Chem. Rev. 117, 860–898 (2017).
Barros, T., Royant, A., Standfuss, J., Dreuw, A. & Kühlbrandt, W. Crystal structure of plant light-harvesting complex shows the active, energy-transmitting state. EMBO J. 28, 298–306 (2009).
Schlau-Cohen, G. S. et al. Single-Molecule Identification of Quenched and Unquenched States of LHCII. J. Phys. Chem. Lett. 6, 860–867 (2015).
Wentworth, M., Ruban, A. V. & Horton, P. Thermodynamic investigation into the mechanism of the chlorophyll fluorescence quenching in isolated photosystem II light-harvesting complexes. J. Biol. Chem. 278, 21845–21850 (2003).
Knecht, S., Marian, C. M., Kongsted, J. & Mennucci, B. On the photophysics of carotenoids: a multireference DFT study of peridinin. J. Phys. Chem. B 117, 13808–13815 (2013).
Belgio, E. et al. Economic photoprotection in photosystem II that retains a complete light-harvesting system with slow energy traps. Nature Communications 5, 8 (2014).
Frisch, M. J. et al. Gaussian 09. (Gaussian, Inc., Wallingford, CT, USA, 2009).
Stewart, J. J. P. MOPAC2016. 16.175W edn (Stewart Computational Chemistry, 2016).
Kusumoto, T. et al. Femtosecond Transient Absorption Spectroscopic Study of a Carbonyl-Containing Carotenoid Analogue, 2-(all-trans-Retinylidene)-indan-1,3-dione. J. Phys. Chem. A 115, 2110–2119 (2011).
Bricker, W. P. & Lo, C. S. Efficient Pathways of Excitation Energy Transfer from Delocalized S2 Excitons in the Peridinin-Chlorophyll a-Protein Complex. J. Phys. Chem. B 119, 5755–5764 (2015).
Knox, R. S. & Spring, B. Q. Dipole strengths in the chlorophylls. Photochem. Photobiol. 77, 497–501 (2003).
Jo, S., Kim, T., Iyer, V. G. & Im, W. Software News and Updates CHARMM-GUI: A Web-Based Graphical User Interface for CHARMM. J. Comput. Chem. 29, 1859–1865 (2008).
Lomize, M. A., Lomize, A. L., Pogozheva, I. D. & Mosberg, H. I. OPM: Orientations of proteins in membranes database. Bioinformatics 22, 623–625 (2006).
Ogata, K., Yuki, T., Hatakeyama, M., Uchida, W. & Nakamura, S. All-atom molecular dynamics simulation of photosystem II embedded in thylakoid membrane. J. Am. Chem. Soc. 135, 15670–15673 (2013).
Case, D. A. et al. {Amber 14}, (2014).
Maier, J. A. et al. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput. 11, 3696–3713 (2015).
Prandi, I. G., Viani, L., Andreussi, O. & Mennucci, B. Combining Classical Molecular Dynamics and Quantum Mechanical Methods for the Description of Electronic Excitations: The Case of Carotenoids. J. Comput. Chem. 37, 981–991 (2016).
Ceccarelli, M., Procacci, P. & Marchi, M. An ab initio force field for the cofactors of bacterial photosynthesis. J. Comput. Chem. 24, 129–142 (2003).
Zhang, L., Silva, D. A., Yan, Y. & Huang, X. Force field development for cofactors in the photosystem II. J. Comput. Chem. 33, 1969–1980 (2012).
Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A. & Case, D. A. Development and testing of a general amber force field. J. Comput. Chem. 25, 1157–1174 (2004).
Dickson, C. J. et al. Lipid14: the amber lipid force field. J. Chem. Theory Comput. 10, 865–879 (2014).
Skjevik, Å. A., Madej, B. D., Walker, R. C. & Teigen, K. LIPID11: a modular framework for lipid simulations using amber. J. Phys. Chem. B 116, 11124–11136 (2012).
Jorgensen, W. L., Chandrasekhar, J., Madura, J. D., Impey, R. W. & Klein, M. L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 (1983).
Joung, I. S. & Cheatham, T. E. III. Molecular dynamics simulations of the dynamic and energetic properties of alkali and halide ions using water-model-specific ion parameters. J. Phys. Chem. B 113, 13279–13290 (2009).
Valkunas, L., Abramavicius, D. & Mancal, T. Molecular excitation dynamics and relaxation: quantum theory and spectroscopy, (John Wiley & Sons, 2013).
Renger, T. & Marcus, R. A. On the relation of protein dynamics and exciton relaxation in pigment-protein complexes: An estimation of the spectral density and a theory for the calculation of optical spectra. J. Chem. Phys. 116, 9997–10019 (2002).
Acknowledgements
V.B. and C.D.P.D. acknowledge the support from the Leverhulme Trust RPG-2015-337. This research utilized Queen Mary’s MidPlus computational facilities, supported by QMUL Research-IT and funded by EPSRC grant EP/K000128/1. W.P.B acknowledges support from the Photosynthetic Antenna Research Center (PARC), an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Award DE-SC0001035 for initial development of the TDC calculation code, as well as support from Army Research Office (ARO-MURI) Award W911NF1210420 for further development.
Author information
Authors and Affiliations
Contributions
V.B. and K.F.F. performed quantum chemical calculations under supervision of C.D.P.D.; V.B. performed transfer rate and complex lifetime calculations; W.P.B. developed TDC calculation software; S.J. and I.G.P. developed the force fields and performed MD simulation under supervision of B.M.; V.B. and C.D.P.D. designed the study and wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Balevičius, V., Fox, K.F., Bricker, W.P. et al. Fine control of chlorophyll-carotenoid interactions defines the functionality of light-harvesting proteins in plants. Sci Rep 7, 13956 (2017). https://doi.org/10.1038/s41598-017-13720-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-13720-6
This article is cited by
-
Ultrafast excited-state dynamics of Luteins in the major light-harvesting complex LHCII
Photochemical & Photobiological Sciences (2024)
-
A perspective on the major light-harvesting complex dynamics under the effect of pH, salts, and the photoprotective PsbS protein
Photosynthesis Research (2023)
-
Towards the description of charge transfer states in solubilised LHCII using subsystem DFT
Photosynthesis Research (2023)
-
Structure of the stress-related LHCSR1 complex determined by an integrated computational strategy
Communications Biology (2022)
-
A different perspective for nonphotochemical quenching in plant antenna complexes
Nature Communications (2021)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
