
Abstract
The chronic disruption of circadian rhythms has been implicated in the risk of cancer development in humans and laboratory animals. The gene product CLOCK is a core molecular component of the circadian oscillator, so that mice with a mutated Clock gene (Clk/Clk) exhibit abnormal rhythms in various physiological processes. However, we demonstrated here that Clk/Clk mice resisted chemical carcinogen-induced tumorigenesis by suppressing epidermal growth factor (EGF) receptor-mediated proliferation signals. The repetitive application of 7,12-dimethylbenz[α]anthracene (DMBA) to skin on the back resulted in the significant development of tumors in wild-type mice, whereas chemically-induced tumorigenesis was alleviated in Clk/Clk mice. Although the degree of DMBA-induced DNA damage was not significantly different between wild-type and Clk/Clk mice, EGF receptor-mediated Ras activation was not detected in DMBA-treated Clk/Clk mice. Genetic and biochemical experiments revealed that the suppression of EGF receptor-mediated signal transduction in DMBA-treated Clk/Clk mice was associated with the expression of the cellular senescence factor p16INK4a. These results suggest an uncovered role for CLOCK in the development of chemical carcinogen-induced primary tumors and offers new preventive strategies.
Similar content being viewed by others

Introduction
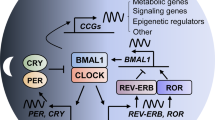
In mammals, circadian rhythms in physiological functions are generated by a molecular oscillator driven by a transcriptional-translational feedback loop consisting of negative and positive regulators1,2,3. The gene products of Clock and Bmal1 (also known as Arntl) form a heterodimer that acts as a positive transcription factor to activate the transcription of the Period (Per) and Cryptochrome (Cry) genes. Once the PER and CRY proteins have reached a critical concentration, they act as negative transcription factors to attenuate CLOCK/BMAL1-mediated transactivation. The alternating activation and repression of the CLOCK/BMAL1-driven positive loop and PER/CRY-controlled negative loop result in a circadian oscillation in the molecular clock4, 5 and also allow organisms to synchronize their physiological and behavioral functions to anticipatory changes in their environment6,7,8. Therefore, disruption of the circadian clock has detrimental effects on health and increases the risk of cancer development9,10,11.
In fact, human night shift workers are at an increased risk of developing breast, prostate, colon, and endometrial cancers as well as non-Hodgkin lymphoma12,13,14,15,16. These epidemiological findings are supported by animal studies in which repetitive changes in the light-dark cycle were found to facilitate the growth of implanted tumors17. Although the genetic ablation of negative or positive components of the circadian clock leads to disrupted rhythms in physiological functions, we previously demonstrated the different contributions of each component of the circadian clock to oncogene-induced cellular transformation18. Mouse embryonic fibroblasts prepared from animals deficient in the negative circadian clock regulators, Per2 or Cry1/2, were susceptible to transformation induced by the co-expression of H-RasV12 and SV40 large T antigen (SV40LT). Consistent with these findings, mice with a mutated Per2 gene (Per2 m/m) were predisposed to spontaneous as well as radiation-induced tumor development19,20,21. On the other hand, cells prepared from mice deficient in the positive circadian clock regulators, Clock or Bmal1, showed resistance to oncogene-induced transformation. However, the tumorigenic phenotype of mice deficient in positive circadian clock regulators currently remains unknown.
Mice with a mutated Clock gene exhibit abnormal rhythms in physiology and behavior22,23,24. We herein demonstrated that Clk/Clk mice resisted chemical carcinogen-induced tumorigenesis by suppressing EGF receptor-mediated proliferation signals. The application of DMBA to the back skin of Clk/Clk mice induced the expression of the cellular senescence factor p16INK4a, which prevents EGF receptor-mediated Ras activation. Collectively, the results of the present study suggest a role for CLOCK in the development of chemically-induced primary tumors and will contribute to new preventive strategies.
Results
Resistance of Clk/Clk mice to the DMBA-induced development of skin tumors
To investigate the role of the Clock gene in tumorigenesis, wild-type and Clk/Clk mice were subjected to a conventional chemically-induced skin tumor protocol with the biweekly application of DMBA for 8 weeks. This protocol was employed because DMBA is known to induce the development of skin tumors25,26,27. In wild-type and Clk/Clk mice, tumors with a diameter of more than 1 mm appeared 5 weeks after the initiation of the DMBA treatment (Fig. 1a and b). The numbers and sizes of tumors gradually increased during the duration of the experiment; however, chemical carcinogen-induced tumorigenesis was significantly alleviated in Clk/Clk mice (F 1,16 = 6.302, P <0.05; Fig. 1a). The results of the histological analysis also revealed that obvious epidermal hyperplasia was induced in wild-type mice 2 weeks after the initiation of the DMBA treatment, during which tumors had not yet been observed on the skin (Fig. 1c). Since the expression of proliferating cell nuclear antigen (PCNA) was significantly increased in the skin of DMBA-treated wild type mice (P < 0.01; Fig. 1d), application of the chemical carcinogen appears to induce the transition of skin cells into proliferative state. In contrast to wild-type mice, DMBA-induced hyperplasia and PCNA expression were attenuated in Clk/Clk mice. These results suggest that Clk/Clk mice resist chemical carcinogen-induced tumorigenesis.

Comparison of DMBA-induced skin tumorigenesis between wild-type and Clk/Clk mice. Animals were treated with 100 μg DMBA twice a week. (a) Time course of DMBA-induced skin tumor formation in wild-type and Clk/Clk mice. Each column indicates the average number of tumors and their size distribution obtained from 6–12 mice per each group (means ± s.e.m.). Tumors with a diameter of more than 1 mm were counted every week. DMBA-induced tumorigenesis was significantly alleviated in Clk/Clk mice (F 1,16 = 6.302, P = 0.023; two-way repeated measures ANOVA). (b) Representative photographs of skin tumor formation in wild-type and Clk/Clk mice 8 weeks after initiation of the DMBA treatment. The yellow arrows indicate the formation of tumors. (c) Histological analysis of the dorsal skin of wild-type and Clk/Clk mice after the DMBA treatment. Mice were treated with DMBA or vehicle (200 μL acetone) for 2 weeks. Skin samples were stained with hematoxylin and eosin. Yellow arrows indicate the epidermis. Scale bar, 100 μm. Histological data were confirmed in more than three mice in each group. (d) The protein levels of proliferating cell nuclear antigen (PCNA) in the skin of wild-type and Clk/Clk mice 2 weeks after initiation of the DMBA treatment. Full-size images are presented in Supplementary Fig. 2. Values show the means ± s.e.m. (n = 3). **P < 0.01 significant difference between two groups (F 3,8 = 9.664, P = 0.005; ANOVA with Tukey-Kramer’s post-hoc test).
DMBA-induced DNA damage to the skin of wild-type and Clk/Clk mice
DMBA is converted to its bioactive form after being metabolized by Cyp1a1 and Cyp1b128, 29 and then causes DNA damage by forming adducts with the nucleobase30, 31. To elucidate the mechanisms responsible for differences in tumorigenicity between wild-type and Clk/Clk mice, we assessed the protein levels of these enzymes in the skin of wild-type and Clk/Clk mice after the DMBA treatment. Consistent with previous findings32, the protein levels of Cyp1a1, but not Cyp1b1 in the skin of wild-type mice were increased 12 hours after a single dose of DMBA (Fig. 2a). The similar induction of Cyp1a1 expression was also detected in Clk/Clk mice, but with no significant differences in the DMBA-induced expression of metabolic enzymes between wild-type and Clk/Clk mice.

Induction of Cyp proteins and formation of DNA adducts in the skin of wild-type and Clk/Clk mice after the DMBA treatment. (a) Protein levels of Cyp1a1 and Cyp1b1 in the skin of wild-type and Clk/Clk mice after the DMBA treatment. Skin homogenates were prepared 12 hours after the treatment with 100 μg DMBA. Plus and minus indicate the treatment with DMBA and vehicle (200 μL acetone), respectively. Full-size images are presented in Supplementary Fig. 3. Western blotting data were confirmed in more than three mice in each group. (b) Amounts of DMBA-DNA adducts in the skin of wild-type and Clk/Clk mice. DMBA was applied twice at 8-hour intervals. DNA was extracted from the skin of mice at 0 or 24 hours after the second DMBA treatment. The fluorescence intensity derived from DMBA-DNA adducts was measured, and intensity was normalized by DNA contents in samples (Means ± s.e.m.; n = 3–4).
We also evaluated DNA damage in the skin of mice after the DMBA treatment by measuring the intensity of fluorescence derived from DMBA-DNA adducts33. Since a single dose of DMBA was insufficient to detect fluorescence intensity, the amount of DMBA-DNA adducts was measured after the application of DMBA twice to mouse skin at 8-hour intervals. In wild-type and Clk/Clk mice, the intensity of fluorescence derived from DMBA-DNA adducts was detected 24 hours after the second application of DMBA, while no significant differences were observed between the genotypes (Fig. 2b). These results suggest that the degree of DMBA-induced DNA damage in Clk/Clk mice was similar to that observed in wild-type mice.
Influence of the Clock gene mutation on cell proliferation signals after the DMBA treatment
Ras, a small GTP-binding protein, is an important component of the signal transduction pathway used by growth factors to initiate cell growth and differentiation34. Although the aberrant activation of Ras by non-hydrolyzable GTP-binding mutations is a major cause of tumorigenesis35, 36, this GTP-binding protein also contributes to the wound healing process37. The repetitive application (twice a week) of DMBA to the back skin of wild-type mice for two weeks caused elevations in the levels of the active form of Ras (P < 0.05; Fig. 3a). Since the total amount of Ras was not markedly affected by the DMBA treatment (Fig. 3b, top panel), elevated levels of the active form of Ras in DMBA-treated wild-type mice were unlikely to be associated with changes in its protein abundance. Furthermore, the activation of Ras in DMBA-treated wild-type mice was significantly suppressed by the selective EGF receptor inhibitor AG1478 (P < 0.05, Fig. 3a), suggesting that Ras activation is due to the stimulation of EGF receptors with its cognate ligands. In fact, we also found the enhancement of phosphorylation of EGF receptor in the skin of DMBA-treated wild-type mice (Fig. 3b, middle panel). In contrast to wild-type mice, the same treatment with DMBA did not induce the activation of Ras in the skin of Clk/Clk mice (Fig. 3a). Since phosphorylation of EGF receptor in Clk/Clk mice was also not induced by DMBA treatment (Fig. 3b, middle panel), the failure to activate Ras in DMBA-treated Clk/Clk mice appeared to be associated with inactivation of EGF receptor. Activated Ras enhances the protein kinase activity of Raf, which subsequently phosphorylates and activates MEK, resulting in the propagation of the signal to downstream effectors, such as MAPK38. Consistent with Ras activation, the phosphorylation state of MEK was also elevated in DMBA-treated wild-type mice (Fig. 3b, bottom panel), but the DMBA-induced MEK phosphorylation was not induced in Clk/Clk mice.

Biochemical analysis of cell proliferation signaling in the skin of wild-type and Clk/Clk mice after the DMBA treatment. DMBA (100 μg) or vehicle (200 μL acetone) was applied biweekly to the back skin of mice for 2 weeks. Plus and minus indicate the treatment with DMBA and vehicle, respectively. (a) Amount of the active form of Ras in the skin of wild-type and Clk/Clk mice 2 weeks after initiation of the DMBA treatment. AG1478 (100 μg) was applied to the dorsal skin of mice 3 hours before sampling. Values of absorbance at 450 nm were obtained and normalized by total protein levels (means ± s.e.m.; n = 5). *P < 0.05 significant difference between the two groups (F 7,23 = 11.945, P < 0.001; ANOVA with Tukey-Kramer’s post-hoc test). (b) The protein levels of all (active and inactive) forms of Ras, phosphorylated EGF receptor (pEGFR), and phosphorylated MEK1 (pMEK1) in the skin of wild-type and Clk/Clk mice 2 weeks after initiation of the DMBA treatment. Full-size images are presented in Supplementary Fig. 4. Western blotting data were confirmed in more than three mice in each group. (c) mRNA levels of endogenous ligands for the EGF receptor in the skin of wild-type and Clk/Clk mice after initiation of the DMBA treatment (Means ± s.e.m.; n = 5). *P < 0.05 significant difference between the two groups (F 3,16 = 5.986, P = 0.006 for Hb-egf, F 3,16 = 7.257, P = 0.003 for Amphiregulin, F 3,16 = 8.365, P = 0.001 for Tgf-α; ANOVA with Tukey-Kramer’s post-hoc test).
After ligand binding, the EGF receptor forms a homodimeric complex and rapidly activates Ras via several adaptor proteins39, 40. EGF, heparin-binding EGF-like growth factor (HB-EGF), transforming growth factor-α (TGF-α), and amphiregulin have been identified as endogenous ligands of EGF receptors41,42,43,44. After the biweekly treatment with DMBA for 2 weeks, the mRNA levels of Hb-egf and Amphiregulin increased in wild-type and Clk/Clk mice (Fig. 3c). The expression of Tgf-α mRNA in wild-type mice was not significantly affected by the DMBA treatment, whereas these mRNA levels were significantly decreased in DMBA-treated Clk/Clk mice (P < 0.05). In contrast to Tgf-α, the mRNA levels of Egf slightly increased in DMBA-treated Clk/Clk mice (P = 0.074). These results suggest that some types of ligands for the EGF receptor are involved in the activation of Ras in wild-type mice during chemical carcinogen-induced tumorigenesis. However, the expression profiles of ligands for the EGF receptor were unable to explain failed Ras activation in DMBA-treated Clk/Clk mice.
Induction of apoptotic cells and cellular senescence in DMBA-treated Clk/Clk mice
During oncogenic transformation, cells acquire genetic mutations that override the normal cell-cycle mechanism, resulting in abnormal proliferation. The failure of cells to override this mechanism often causes reversible cell-cycle arrest, apoptotic cell death, or cellular senescence45. Therefore, we also investigated cell death events and senescence in the skin of mice after the DMBA treatment. The results of the histochemical analysis using the terminal deoxynucleotidyltransferase (TdT)-mediated dUTP-digoxigenin nick-end labeling (TUNEL) assay revealed that the number of apoptotic cells in DMBA-treated Clk/Clk mice was less than that in wild-type mice (Fig. 4a). Consistent with this result, the protein levels of cleaved caspase-3 were also lower in Clk/Clk mice than in wild-type mice (Fig. 4b).

DMBA-induced apoptotic cell death and cellular senescence in the skin of wild-type and Clk/Clk mice. Animals were treated with 100 μg DMBA or vehicle (200 μL acetone) twice a week. (a) Immunofluorescence staining of apoptotic cells in the skin of wild-type and Clk/Clk mice 2 weeks after initiation of the DMBA treatment. Apoptotic cells were detected by the TUNEL assay (Green). DAPI was used to stain nuclei (Blue). White arrows indicate TUNEL-positive cells. Dashed lines denote the epidermis-dermis border. Scale bar, 100 μm. (b) The expression of cleaved caspase-3 and p16INK4a proteins in the skin of wild-type and Clk/Clk mice 2 weeks after initiation of the DMBA treatment. Plus and minus indicate the treatment with DMBA and vehicle (200 μL acetone), respectively. Full-size images are presented in Supplementary Fig. 5. Data shown in panel a and b were confirmed in more than three mice in each group. (c) mRNA levels of inflammatory cytokines in the skin of wild-type and Clk/Clk mice 2 weeks after initiation of the DMBA treatment. (Means ± s.e.m.; n = 5). *P < 0.05 significant difference between the two groups (F 3,14 = 6.412, P = 0.006 for IL-6, F 3,16 = 5.211, P = 0.011 for Ccl2, F 3,16 = 3.682, P = 0.034 for Tnf-α; ANOVA with Tukey-Kramer’s post-hoc test). Plus and minus indicate the treatment with DMBA and vehicle (200 μL acetone), respectively.
The tumor suppressor gene Cdkn2a acts as a canonical inducer of cellular senescence46. Cdkn2a generates the different transcript variants, p16Ink4a and p19Arf (also known as p14ARF in humans), by using different first exons and alternate polyadenylation sites47. The p16Ink4a variants encode structurally related protein isoforms that inhibit CDK4 kinase48. The CDK4 inhibitor prevents the phosphorylation of pRB by disrupting the activity of the CDK4-cyclin D complex. On the other hand, p19ARF stabilizes p53 by sequestering MDM249. The protein levels of p16INK4a in the skin of Clk/Clk mice were significantly increased by the DMBA treatment, whereas no obvious band derived from p16INK4a was detected in wild-type mice (Fig. 4b). We also attempted to assess the protein levels of p19ARF in DMBA-treated mice; however, this protein was not detected in the skin of mice.
Since senescent cells show the senescence-associated secretory phenotype (SASP) reflecting the production of inflammatory cytokines and chemokines50, 51, we investigated the expression of typical inflammatory cytokines and chemokines in the skin of DMBA-treated mice. In wild-type and Clk/Clk mice, the mRNA levels of Tgf-β1 increased after the DMBA treatment, while expression levels were not significantly different between the genotypes (Fig. 4c). On the other hand, the expression of IL-6, Ccl2, and Tnf-α was significantly increased only in DMBA-treated Clk/Clk mice (P < 0.05, respectively), suggesting that the treatment of Clk/Clk mice with DMBA induces cellular senescence rather than neoplastic transformation.
The expression of p16INK4a prevents EGF receptor-mediated Ras activation
In contrast to reversible cell-cycle arrest, cellular senescence is defined by the irreversible loss of proliferative potential52, 53. In the final set of experiments, we used cultured human keratinocyte HaCaT cells to investigate whether elevation of p16INK4a protein levels prevents EGF receptor-mediated Ras activation. The infection of HaCaT cells with retroviral vectors expressing flag-tagged human p16INK4a had a negligible effect on the protein levels of Ras and the EGF receptor (Fig. 5a). The mRNA levels of IL-6, Ccl2, and Tnf-α were significantly increased in p16INK4a-expressing cells (P < 0.01, respectively; Fig. 5b). The treatment of control HaCaT cells with EGF significantly induced the phosphorylation of EGF receptor as well as activation of Ras (P < 0.05; Fig. 5c), but neither EGF receptor phosphorylation nor Ras activation were detected in EGF-treated p16INK4a-expressing cells. These results indicate that the expression of p16INK4a leads to the failure of EGF receptor-mediated Ras activation. This may be the mechanism responsible for the resisting phenotype of Clk/Clk mice to chemical carcinogen-induced tumorigenesis.

Effects of p16INK4a on EGF receptor-mediated Ras activation in human keratinocytes. HaCaT cells were infected with retroviral vectors expressing Flag-tagged p16INK4a. (a) Protein levels of Flag-tagged p16INK4a, total Ras, and EGF receptor in HaCaT cells. Plus and minus indicate the infection with p16INK4a-Flag expressing vectors. Full-size images are presented in Supplementary Fig. 6. Data shown were confirmed in more than three independent experiments. (b) mRNA levels of inflammatory cytokines in cells infected or not infected with the p16INK4a-Flag expressing vectors. (Means ± s.e.m.; n = 6). **P < 0.01 significant difference control cells (t 10 = −8.393, P < 0.001 for IL-6, t 10 = −4.161, P = 0.002 for Ccl2, t 10 = −3.975, P = 0.034 for Tnf-α; unpaired t-test, two-sided). (c) Protein levels of phosphorylated EGFR (right) and the amount of the active form of Ras (left) in cells infected or not infected with the p16INK4a-Flag expressing vectors. For right panel, western blotting data were confirmed in three mice in each group. Full-size images are presented in Supplementary Fig. 7. For left panel, cells were incubated in serum-starved media for 24 hours and then stimulated with 200 ng/ml of EGF for 1 minute. Plus and minus indicate the introduction of the p16INK4a-Flag vector and the EGF treatment, respectively. Values of absorbance at 450 nm were obtained and normalized by total protein levels (Means ± s.e.m.; n = 4). *P < 0.05 significant difference between the two groups (F 3,12 = 29.245, P < 0.001; ANOVA with Tukey-Kramer’s post-hoc test).
Discussion
Consistent with our previous findings obtained using an oncogene-induced cellular transformation model18, the present results also revealed that the genetic ablation of the circadian gene Clock in mice resulted in resistance to chemically-induced tumorigenesis. During the development of primary cancers, chemical carcinogens induce cellular damage, apoptotic cell death, and inflammation. Consequently, cell growth and repair factors are released in the damaged and inflamed area for the restoration of tissues. The abnormal proliferation of epidermal cells occurred in the skin of DMBA-treated wild-type mice, and this appears to have been caused by EGF receptor-mediated Ras activation. Abnormal cell proliferation signals override the normal cell-cycle mechanism, resulting in tumor development in wild-type mice. On the other hand, epidermal cells in the skin of Clk/Clk mice may have difficulties tolerating excessive cell proliferation signals. The failure of epidermal cells in Clk/Clk cells to override the normal cell-cycle mechanism appeared to induce p16INK4a expression, thereby showing the SASP. Therefore, Clk/Clk mice exhibit the phenotype that resists chemically-induced tumorigenesis.
Tumorigenesis is known to proceed in three steps: initiation, promotion, and progression54,55,56. We previously reported a decrease in the expression of enzymes responsible for producing the active metabolite of diethylnitrosamine in the livers of Clk/Clk mice57. This led to the alleviation of DNA damage in the hepatocytes of Clk/Clk mice, even when they were exposed to chemical carcinogens. These facts indicate that a dysfunction in CLOCK influences the initiation step of chemically-induced tumorigenesis. However, no significant differences were observed in the DMBA-induced dermal expression of Cyp1a1 and 1b1 between wild-type and Clk/Clk mice. Furthermore, the amount of DMBA-DNA adducts in the skin of Clk/Clk mice was similar to that in wild-type mice. These findings suggest that the resistance of Clk/Clk mice to DMBA-induced tumorigenesis is not attributable to the initiation step.
Regarding the promotion step, a marked difference was observed in the activation of Ras between wild-type and Clk/Clk mice. The EGF receptor is a member of the ligand-activated receptor and tyrosine kinase family of transmembrane signaling proteins, which are fundamentally important regulators of cell growth and differentiation58. Previous studies reported that the EGF receptor is strongly expressed in epidermal cells, and its ligands are often up-regulated in the damaged or inflamed area of skin59, 60. Therefore, EGF receptor-mediated Ras activation in DMBA-treated mice appeared to have occurred in the epidermal layer. This notion is also supported by previous findings that DMBA-induced tumor development in the skin of mice is associated with the malignant transformation of epidermal cells61, 62. Although the DMBA treatment induced the expression of several types of ligands for the EGF receptor in wild-type and Clk/Clk mice, Ras activation was only observed in DMBA-treated wild-type mice. The genotype-dependent difference in Ras activation may also be reflected in the disparity in the degree of epidermal hyperplasia between wild-type and Clk/Clk mice.
The hallmark of cellular senescence is an inability to progress through the cell cycle. Senescent cells arrest growth irreversibly, but remain metabolically active63. Once arrested, cells fail to proliferate in response to adequate growth stimuli and often become resistant to cell death signals64, 65. The number of apoptotic cells in the skin of DMBA-treated Clk/Clk mice was smaller than that in wild-type mice; however, the treatment with DMBA induced the expression of p16INK4a only in Clk/Clk mice. We previously demonstrated that ATF4 is a potent repressor of the expression of p16INK4a66. Elevated levels of ATF4 in oncogene-introduced wild-type cells are sufficient to suppress the expression of p16INK4a and drive oncogenic transformation. On the other hand, CLOCK acts as a transcriptional activator of the Atf4 gene18; thus, the expression levels of ATF4 in Clk/Clk cells remain low even when they receive oncogenic growth signals. Therefore, unresponsiveness of the Atf4 gene to oncogenic stimuli allows the constitutive expression of p16INK4a in Clk/Clk cells, resulting in the induction of SASP reflecting the expression of Ccl2, IL-6 and Tnfα 18. We also confirmed that ATF4 expression was not significantly induced in the skin of DMBA-treated Clk/Clk mice (Supplementary Fig. 1), which likely allow the expression of p16INK4a. Furthermore, transgenic expression of p16INK4a in human keratinocytes induced the SASP as well as prevented EGF receptor-mediated Ras activation. Therefore, the induction of p16INK4a expression appears to cause the failure of Ras activation in DMBA-treated Clk/Clk mice and their resisting phenotype to chemically carcinogen-induced primary cancer.
Since the basal mechanism of the circadian clock is well conserved in many mammalian species, CLOCK is assumed to function in almost the same manner in human cells. The present results suggest an uncovered role for CLOCK in the development of chemically-induced primary tumors and will contribute to new preventive strategies.
Methods
Treatment of animals
Female Clk/Clk mice with an ICR background and female wild-type mice of the same strain were housed in groups (from 3 to 6 per cage) in a light-controlled room (lights on from 7:00 to 19:00) at 24 ± 1 °C and humidity of 60 ± 10% with food and water ad libitum. Mice were used for experiments between the ages of seven and nine weeks. All protocols using mice were reviewed and approved by the Animal Care and Use Committee, Kyushu University. All methods were performed in accordance with the relevant guidelines and regulations.
Cell culture and treatment
HaCaT cells were maintained in Dulbecco’s modified Eagle’s medium (Sigma-Aldrich, St. Louis, MO) supplemented with 10% fetal bovine serum (Thermo Fisher Scientific, Waltham, MA) and antibiotics at 37 °C in a humidified 5% CO2 atmosphere. Cells were infected with retroviral vectors expressing Flag-tagged p16INK4a (Addgene #24934). After growing cells to semi-confluence, serum was starved for 24 hours. EGF (Thermo Fisher Scientific) was added to media at a final concentration of 200 ng mL−1. One minute later, media were aspirated and cells were frozen by liquid nitrogen. Cells were used in the assessment of the active form of Ras.
Chemically-induced skin tumor model
A chemically-induced tumor formation analysis was performed according to a previously described protocol with some modifications25,26,27. The dorsal skin of mice was shaved and, 1 day later, 100 μg of DMBA (Sigma-Aldrich) dissolved in 200 μL of acetone (Nacalai Tesque, Kyoto, Japan) was applied biweekly to the shaved area for 8 weeks. The diameters of tumors were measured using a caliper (Shinwa Rules Co., Ltd., Niigata, Japan) and the number of tumors with a diameter of more than 1 mm was counted once a week.
Histological analysis
The dorsal skin of mice was shaved and 100 μg DMBA or vehicle (200 μL acetone) were applied biweekly for 2 weeks. Skin tissues were collected and fixed in 4% buffered formalin. Fixed skin tissues were transferred to 15% sucrose in PBS for 24 hours and then 30% sucrose in PBS for 24 hours. These samples were frozen in −80 °C and serially sectioned on a cryostat at a thickness of 14 μm. Sections were observed by microscopy after staining with hematoxylin (Muto Pure Chemicals Co., Ltd., Tokyo, Japan) and eosin (Muto Pure Chemicals Co., Ltd.).
Western blotting
Skin tissues were homogenized with CelLyticTM MT Cell Lysis Reagent (Sigma-Aldrich). Ten micrograms of the protein lysate was resolved by SDS-PAGE, transferred to a PVDF membrane, and probed with antibodies against PCNA (1:1,000; ab29, Abcam), Cyp1a1 (1:1,000; #871, Daiichi Pure Chemical Co., Ltd., Tokyo, Japan), Cyp1b1 (1:1,000; ab185954, Abcam, Cambridge, UK), Actin (1:1,000; sc1616-HRP, Santa Cruz Biotechnology, Santa Cruz, CA), Pan-Ras (1:1000; Component of STA-440, Cell Biolabs, Inc., San Diego, CA), EGF receptor (1:1000; #2232, Cell Signaling Technology, Danvers, MA), phosphorylated (Y1086) EGF receptor (1:1,000; ab32086, Abcam), MEK1 (1:1,000; ab32091, Abcam), phosphorylated (E342) MEK1 (1:1,000; ab396379, Abcam), Cleaved Caspase-3 (1:5000; #9661, Cell Signaling Technology), Flag-M2 (1:1000; F3165, Sigma-Aldrich), and p16INK4a (1:500; sc1207, Santa Cruz Biotechnology). Specific antigen-antibody complexes were visualized using horseradish peroxidase-conjugated secondary antibodies and Chemi-Lumi One (Nacalai Tesque) or ImmunoStar reagent (Wako, Osaka, Japan).
Measurement of DMBA-DNA adduct formation
The dorsal skin of mice was shaved and, 1 day later, 300 μg of DMBA dissolved in 200 μL of acetone was applied twice at 8-hour intervals. DNA was extracted from skin with the Genomic DNA Purification Kit (Promega, Madison, WI) 0 or 24 hours after the second application of DMBA. The fluorescence intensity of DMBA-DNA adducts was measured using a FlexStation3 (Molecular Devices, Sunnyvale, CA) with a 1/2 AreaPlate-96 (Black; Perkin Elmer, Waltham, MA). Emission data at 445 nm were obtained with excitation at 390 nm based on the spectrum previously reported33, and the intensity of fluorescence was normalized by DNA concentrations in the samples. DNA concentrations were measured using a nucleic acid analyzer (DW-K2800; Drawell International Technology Co., Ltd., PuDong, Shanghai, China).
Measurement of the active form of Ras
The dorsal skin of mice was shaved and 100 μg DMBA or vehicle (200 μL acetone) was applied twice a week. Skin tissues were collected 2 weeks after the initiation of the DMBA treatment. One hundred micrograms of the EGF receptor antagonist AG1478 (Wako) was applied to the dorsal skin of mice 3 hours before sampling. The amount of the active form of Ras was measured using an ELISA Kit (STA-440; Cell Biolabs, Inc.). The values of absorbance at 450 nm were normalized by total protein concentrations in the samples. Protein concentrations were measured using the BCA assay (Thermo Fisher Scientific).
Quantitative RT-PCR analysis
RNA was extracted from the skin of mice by RNAiso reagent (Takara Bio Inc., Osaka, Japan). In order to quantify the mRNA levels of each factor, cDNA was synthesized by reverse transcription using a ReverTra Ace quantitative real-time PCR kit (Toyobo, Osaka, Japan). Diluted cDNA samples were analyzed by PCR using a THUNDERBIRDSYBR qPCR Mix (Toyobo) and LightCycler® 96 System (Roche, Basel, Switzerland). Data were normalized using 18 s mRNA as a control. PCR primer sequences are described in Supplementary Table 1.
TUNEL staining
Skin sections were prepared using the same procedure described above. Apoptosis in skin sections was evaluated using a DeadEnd™ Fluorometric TUNEL System (Promega).
Statistical and data analyses
The values presented are expressed as Means ± s.e.m.. The significance of differences in the total number of tumors between wild-type and Clk/Clk mice was tested by a two-way repeated measures ANOVA. The significance of differences among groups was analyzed by a one-way ANOVA followed by Tukey-Kramer’s post-hoc test. Equal variances were not formally tested. P < 0.05 was considered to be significant.
Data availability
All data supporting the results of the present study are included in the article, either in the main figures or supplementary information files.
References
Gekakis, N. et al. Role of the CLOCK protein in the mammalian circadian mechanism. Science 280, 1564–1569 (1998).
Darlington, T. K. et al. Closing the circadian loop: CLOCK-induced transcription of its own inhibitors per and tim. Science 280, 1599–1603 (1998).
Kume, K. et al. mCRY1 and mCRY2 are essential components of the negative limb of the circadian clock feedback loop. Cell 98, 193–205 (1999).
Jin, X. et al. A molecular mechanism regulating rhythmic output from the suprachiasmatic circadian clock. Cell 96, 57–68 (1999).
Cheng, M. Y. et al. Prokineticin 2 transmits the behavioural circadian rhythm of the suprachiasmatic nucleus. Nature 417, 405–410 (2002).
Krieger, D. T., Allen, W., Rizzo, F. & Krieger, H. P. Characterization of the normal temporal pattern of plasma corticosteroid levels. J. Clin. Endocrinol. Metab. 32, 266–284 (1971).
Dimitrov, S. et al. Cortisol and epinephrine control opposing circadian rhythms in T cell subsets. Blood 113, 5134–5143 (2009).
Daan, S., Beersma, D. G. & Borbely, A. A. Timing of human sleep: recovery process gated by a circadian pacemaker. Am. J. Physiol. 246, R161–83 (1984).
Morris, C. J., Purvis, T. E., Hu, K. & Scheer, F. A. Circadian misalignment increases cardiovascular disease risk factors in humans. Proc. Natl. Acad. Sci. USA 113, E1402–11 (2016).
Magrini, A. et al. Shift work and autoimmune thyroid disorders. Int. J. Immunopathol. Pharmacol. 19, 31–36 (2006).
Vallieres, A., Azaiez, A., Moreau, V., LeBlanc, M. & Morin, C. M. Insomnia in shift work. Sleep Med. 15, 1440–1448 (2014).
Schernhammer, E. S. et al. Rotating night shifts and risk of breast cancer in women participating in the nurses’ health study. J. Natl. Cancer Inst. 93, 1563–1568 (2001).
Rao, D., Yu, H., Bai, Y., Zheng, X. & Xie, L. Does night-shift work increase the risk of prostate cancer? a systematic review and meta-analysis. Onco Targets Ther. 8, 2817–2826 (2015).
Schernhammer, E. S. et al. Night-shift work and risk of colorectal cancer in the nurses’ health study. J. Natl. Cancer Inst. 95, 825–828 (2003).
Viswanathan, A. N., Hankinson, S. E. & Schernhammer, E. S. Night shift work and the risk of endometrial cancer. Cancer Res. 67, 10618–10622 (2007).
Lahti, T. A., Partonen, T., Kyyronen, P., Kauppinen, T. & Pukkala, E. Night-time work predisposes to non-Hodgkin lymphoma. Int. J. Cancer 123, 2148–2151 (2008).
Filipski, E. et al. Effects of chronic jet lag on tumor progression in mice. Cancer Res. 64, 7879–7885 (2004).
Katamune, C. et al. Different Roles of Negative and Positive Components of the Circadian Clock in Oncogene-induced Neoplastic Transformation. J. Biol. Chem. 291, 10541–10550 (2016).
Wood, P. A. et al. Period 2 mutation accelerates ApcMin/+ tumorigenesis. Mol. Cancer. Res. 6, 1786–1793 (2008).
Fu, L., Pelicano, H., Liu, J., Huang, P. & Lee, C. The circadian gene Period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 111, 41–50 (2002).
Lee, S., Donehower, L. A., Herron, A. J., Moore, D. D. & Fu, L. Disrupting circadian homeostasis of sympathetic signaling promotes tumor development in mice. PLoS One 5, e10995 (2010).
Vitaterna, M. H. et al. Mutagenesis and mapping of a mouse gene, Clock, essential for circadian behavior. Science 264, 719–725 (1994).
Miller, B. H. et al. Circadian clock mutation disrupts estrous cyclicity and maintenance of pregnancy. Curr. Biol. 14, 1367–1373 (2004).
Wakatsuki, Y., Kudo, T. & Shibata, S. Constant light housing during nursing causes human DSPS (delayed sleep phase syndrome) behaviour in Clock-mutant mice. Eur. J. Neurosci. 25, 2413–2424 (2007).
Hara, T., Matsumura, S., Hakuno, F., Takahashi, S. & Chida, K. PKCalpha suppresses 7,12-dimethylbenz[a]anthracene-induced skin tumor formation. Anticancer Res. 32, 3097–3101 (2012).
Moore, R. J. et al. Mice deficient in tumor necrosis factor-alpha are resistant to skin carcinogenesis. Nat. Med. 5, 828–831 (1999).
Murphy, J. E., Morales, R. E., Scott, J. & Kupper, T. S. IL-1 alpha, innate immunity, and skin carcinogenesis: the effect of constitutive expression of IL-1 alpha in epidermis on chemical carcinogenesis. J. Immunol. 170, 5697–5703 (2003).
Buters, J. T. et al. Cytochrome P450 CYP1B1 determines susceptibility to 7, 12-dimethylbenz[a]anthracene-induced lymphomas. Proc. Natl. Acad. Sci. USA 96, 1977–1982 (1999).
Kleiner, H. E. et al. Role of cytochrome p450 family members in the metabolic activation of polycyclic aromatic hydrocarbons in mouse epidermis. Chem. Res. Toxicol. 17, 1667–1674 (2004).
Devanesan, P. D. et al. Identification and quantitation of 7,12-dimethylbenz[a]anthracene-DNA adducts formed in mouse skin. Chem. Res. Toxicol. 6, 364–371 (1993).
Izzotti, A. et al. Patterns of DNA adduct formation in liver and mammary epithelial cells of rats treated with 7,12-dimethylbenz(a)anthracene, and selective effects of chemopreventive agents. Cancer Res. 59, 4285–4290 (1999).
Ide, F. et al. Skin and salivary gland carcinogenicity of 7,12-dimethylbenz[a]anthracene is equivalent in the presence or absence of aryl hydrocarbon receptor. Cancer Lett. 214, 35–41 (2004).
Ivanovic, V. et al. Cell and microsome mediated binding of 7,12-dimethylbenz(a)anthracene to DNA studied by fluorescence spectroscopy. Cancer Lett. 4, 131–140 (1978).
Margolis, B. & Skolnik, E. Y. Activation of Ras by receptor tyrosine kinases. J. Am. Soc. Nephrol. 5, 1288–1299 (1994).
Pylayeva-Gupta, Y., Grabocka, E. & Bar-Sagi, D. RAS oncogenes: weaving a tumorigenic web. Nat. Rev. Cancer. 11, 761–774 (2011).
Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer. 3, 11–22 (2003).
Doma, E., Rupp, C. & Baccarini, M. EGFR-ras-raf signaling in epidermal stem cells: roles in hair follicle development, regeneration, tissue remodeling and epidermal cancers. Int. J. Mol. Sci. 14, 19361–19384 (2013).
Roberts, P. J. & Der, C. J. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 26, 3291–3310 (2007).
Lowenstein, E. J. et al. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 70, 431–442 (1992).
Hallberg, B. & Rayter, S. I. & Downward, J. Interaction of Ras and Raf in intact mammalian cells upon extracellular stimulation. J. Biol. Chem. 269, 3913–3916 (1994).
COHEN, S. & ELLIOTT, G. A. The stimulation of epidermal keratinization by a protein isolated from the submaxillary gland of the mouse. J. Invest. Dermatol. 40, 1–5 (1963).
Higashiyama, S., Abraham, J. A., Miller, J., Fiddes, J. C. & Klagsbrun, M. A heparin-binding growth factor secreted by macrophage-like cells that is related to EGF. Science 251, 936–939 (1991).
de Larco, J. E. & Todaro, G. J. Growth factors from murine sarcoma virus-transformed cells. Proc. Natl. Acad. Sci. USA 75, 4001–4005 (1978).
Shoyab, M., McDonald, V. L., Bradley, J. G. & Todaro, G. J. Amphiregulin: a bifunctional growth-modulating glycoprotein produced by the phorbol 12-myristate 13-acetate-treated human breast adenocarcinoma cell line MCF-7. Proc. Natl. Acad. Sci. USA 85, 6528–6532 (1988).
Hanahan, D. & Weinberg, R. A. Hallmarks of cancer: the next generation. Cell 144, 646–674 (2011).
Collado, M. & Serrano, M. Senescence in tumours: evidence from mice and humans. Nat. Rev. Cancer. 10, 51–57 (2010).
Quelle, D. E., Zindy, F., Ashmun, R. A. & Sherr, C. J. Alternative reading frames of the INK4a tumor suppressor gene encode two unrelated proteins capable of inducing cell cycle arrest. Cell 83, 993–1000 (1995).
Serrano, M., Hannon, G. J. & Beach, D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature 366, 704–707 (1993).
Honda, R. & Yasuda, H. Association of p19(ARF) with Mdm2 inhibits ubiquitin ligase activity of Mdm2 for tumor suppressor p53. EMBO J. 18, 22–27 (1999).
Acosta, J. C. et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 15, 978–990 (2013).
Andriani, G. A. et al. Whole chromosome Instability induces senescence and promotes SASP. Sci. Rep. 6, 35218 (2016).
Hayflick, L. & Moorhead, P. S. The serial cultivation of human diploid cell strains. Exp. Cell Res. 25, 585–621 (1961).
Collado, M., Blasco, M. A. & Serrano, M. Cellular senescence in cancer and aging. Cell 130, 223–233 (2007).
Pitot, H. C. The molecular biology of carcinogenesis. Cancer 72, 962–970 (1993).
Barrett, J. C. Mechanisms of multistep carcinogenesis and carcinogen risk assessment. Environ. Health Perspect. 100, 9–20 (1993).
Grivennikov, S. I., Greten, F. R. & Karin, M. Immunity, inflammation, and cancer. Cell 140, 883–899 (2010).
Matsunaga, N. et al. Influence of CLOCK on cytotoxicity induced by diethylnitrosamine in mouse primary hepatocytes. Toxicology 280, 144–151 (2011).
Sibilia, M. & Wagner, E. F. Strain-dependent epithelial defects in mice lacking the EGF receptor. Science 269, 234–238 (1995).
Liou, A., Elias, P. M., Grunfeld, C., Feingold, K. R. & Wood, L. C. Amphiregulin and nerve growth factor expression are regulated by barrier status in murine epidermis. J. Invest. Dermatol. 108, 73–77 (1997).
Stoll, S., Garner, W. & Elder, J. Heparin-binding ligands mediate autocrine epidermal growth factor receptor activation In skin organ culture. J. Clin. Invest. 100, 1271–1281 (1997).
Indra, A. K. et al. Malignant transformation of DMBA/TPA-induced papillomas and nevi in the skin of mice selectively lacking retinoid-X-receptor alpha in epidermal keratinocytes. J. Invest. Dermatol. 127, 1250–1260 (2007).
Capozza, F. et al. Absence of caveolin-1 sensitizes mouse skin to carcinogen-induced epidermal hyperplasia and tumor formation. Am. J. Pathol. 162, 2029–2039 (2003).
Salama, R., Sadaie, M., Hoare, M. & Narita, M. Cellular senescence and its effector programs. Genes Dev. 28, 99–114 (2014).
Hampel, B., Malisan, F., Niederegger, H., Testi, R. & Jansen-Durr, P. Differential regulation of apoptotic cell death in senescent human cells. Exp. Gerontol. 39, 1713–1721 (2004).
Wang, E. Senescent human fibroblasts resist programmed cell death, and failure to suppress bcl2 is involved. Cancer Res. 55, 2284-2292 (1995).
Horiguchi, M. et al. Stress-regulated transcription factor ATF4 promotes neoplastic transformation by suppressing expression of the INK4a/ARF cell senescence factors. Cancer Res. 72, 395–401 (2012).
Acknowledgements
We are grateful for the technical support provided by the Research Support Center, Graduate School of Medical Sciences, Kyushu University. This work was supported in part of the Japan Society for the Promotion of Science (JSPS) KAKENHI Grants FCG6670317 (to S. K.) and 16H02636 (to S. O.).
Author information
Authors and Affiliations
Contributions
K.H., C.K., S.K., and S.O. designed the research; K.H., C.K., S.K., and S.O. acquired the data; C.K., N.M., and S.K., supplied the experimental materials; K.H., N.K., S.K., and S.O. drafted the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hashikawa, Ki., Katamune, C., Kusunose, N. et al. Dysfunction of the circadian transcriptional factor CLOCK in mice resists chemical carcinogen-induced tumorigenesis. Sci Rep 7, 9995 (2017). https://doi.org/10.1038/s41598-017-10599-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-10599-1
This article is cited by
-
Construction of a CCL20-centered circadian-signature based prognostic model in cervical cancer
Cancer Cell International (2023)
-
Alteration of circadian machinery in monocytes underlies chronic kidney disease-associated cardiac inflammation and fibrosis
Nature Communications (2021)
-
Diurnal expression of MRP4 in bone marrow cells underlies the dosing-time dependent changes in the oxaliplatin-induced myelotoxicity
Scientific Reports (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
