
Abstract
Peptides have great potential as therapeutic agents, however, their clinic applications are severely hampered by their instability and short circulation half-life. Zero-order release carriers could not only extend the circulation lifetime of peptides, but also maintain the plasma drug level constant, and thus maximize their therapeutic efficacy and minimize their toxic effect. Here using PEGylated salmon calcitonin (PEG-sCT)/tannic acid (TA) film as an example, we demonstrated that hydrogen-bonded layer-by-layer films of a PEGylated peptide and a polyphenol could be a platform for zero-order peptide release. The films were fabricated under mild conditions. The second component, TA, is a natural product and presents potential therapeutic activities itself. Unlike common carriers, the new carrier releases the peptide via gradual disintegration of the film because of its dynamic nature. The release of PEG-sCT follows a perfect zero-order kinetics without initial burst release. In addition the release rate could be tuned via external stimuli, such as pH and temperature. When implanted in rats, the films could remain the plasma level of PEG-sCT constant over an extended period. Accordingly, the serum calcium level was reduced and maintained constant over the same period, suggesting an improved therapeutic efficacy of the released drug.
Similar content being viewed by others


Introduction
The tremendous advances in biotechnology are introducing more and more peptide therapeutics for the treatment of various diseases, particularly metabolic diseases and cancers1,2,3. Unfortunately, their intrinsic weaknesses, e.g. poor chemical and physical stability and therefore a short circulating plasma half-life, severely limited their clinical applications4. One method to extend circulation lifetimes is via PEGylation5. It was well documented that PEG conjugation could protect the fragile drug from enzymatic digestion, slow its filtration by the kidneys, and thus increase its retention in the circulation6. Another important approach is the using of drug carrier. Encapsulation of peptide therapeutic in a carrier could not only protect the drug against degradation but also allow its sustained release, leading to prolonged systemic exposure to the drug. Up to now, many carriers were designed for peptide release, including biodegradable microparticles (particularly poly(D,L-lactide-co-glycolide) (PLGA) microparticles)7, 8, micelles9, and hydrogels10. PEG conjugations can be regarded as a type of drug carriers too, in which the drug is chemically bound, instead of physically encapsulated11.
Although significant progress has been achieved in the development of drug carriers for peptide release, some challenges still remain. Particularly the carriers usually release the drug in a “fast-then-slow” manner. The rapid release at the initial stage will result in a high plasma drug level, which will pose a serious toxicity threat for the patients8. For example, biodegradable microspheres, which were extensively investigated for peptide release, usually suffer from a significant initial burst release8, 12. Initial burst was also observed in peptide releasing hydrogels both in vitro and in vivo, and is considered a limiting factor for their applications10. At the late stage of release, however, as the release rate slows down gradually, the plasma drug level will finally drop to a level not high enough to achieve therapeutic effects13. As a result, only a small portion of the precious drug will exert its therapeutic effects.
In this context, an ideal carrier should allow for zero-order release, i.e., the drug release rate is constant over the entire release duration14, 15. In this way, the fluctuation of the plasma drug level will be minimized. The plasma drug level will be maintained within the therapeutic window throughout the entire release period, leading to the highest therapeutic efficacy. Despite of intensive efforts in the last decades, however, achieving zero-order release still remains a big challenge16. Although many carriers and devices were designed for zero-order drug release, many of them are complex, expensive, and their manufacture is difficult and time consuming15. New methods for zero-order release are still highly desirable15,16,17,18,19,20,21,22,23,24,25,26. Here we designed a new carrier which allows for zero-order release of peptide drugs. Initial burst release is completely avoided. Preliminary in vivo tests suggest this carrier could maintain the plasma drug level constant over an extended period and thus achieve therapeutic effects for a long time. It is noteworthy that peptides can not only be used as therapeutics, but also widely used to construct drug carriers for drug delivery27,28,29,30.
Results
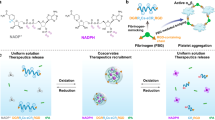
The new carrier designed here is the hydrogen-bonded layer-by-layer (LBL) film of a PEGylated peptide and a polyphenol (Fig. 1 and Figure S1). The peptide drug is first PEGylated and then incorporated into a thin film by layer-by-layer assembly31 with a polyphenol. As a proof of concept, PEGylated salmon calcitonin (PEG-sCT)32, an attractive therapeutic peptide for the treatment of bone disorders, was chosen as a model peptide drug. Tannic acid (TA), a natural hydrolysable tannin, which itself shows antimutagenic, anticarcinogenic, antimicrobial, antioxidant, and antibacterial activities33, was chosen as the polyphenol (Figure 1A). The LBL films were fabricated by dipping quartz slides in aqueous solutions of PEG-sCT and TA alternately. As shown in Fig. 2, the film absorbance increases with increasing dipping cycles, suggesting the successful deposition of PEG-sCT and TA onto the substrate. It is noteworthy that only a small portion of drug in the assembly solution is incorporated into the LBL film. It is expected that loading efficiency can be improved by repeated using of the PEG-sCT and TA solutions. The main driving force for the film buildup is supposed to be the hydrogen bonding between the PEG chain in PEG-sCT and TA (Figure 1B). As an evidence, a shoulder appears at 1733 cm−1 in the FTIR spectra of the LBL film, suggesting some of the intra-molecular hydrogen bonds in TA break as a result of the formation of intermolecular hydrogen bonds between PEG-sCT and TA (Figure 3). The morphology of the films was studied using SEM. Like other LBL films, the PEG-sCT/TA films also present a rough surface34 (Figure S2A). The thickness of an 8-bilayer film was measured to be ~20 nm.

(A) Chemical structure of TA. (B) Hydrogen bonding between TA and PEG-sCT. (C) Release of PEG-sCT from the hydrogen-bonded PEG-sCT/TA film as a result of the gradual disintegration of the film.

UV-vis absorption spectra of PEG-sCT/TA films with various bilayer numbers (0–15). Inset: Plot of absorbance at 216 and 278 nm against the bilayer number. Both bands can be assigned to phenyl groups in TA.

FTIR spectra of PEG-sCT, TA and a PEG-sCT/TA film.
To study the in vitro release of PEG-sCT, the PEG-sCT/TA films were soaked in aqueous solutions and the concentration of PEG-sCT released into the media was analyzed by HPLC (Figure 1C). Figure 4A shows the release profile of PEG-sCT from 4 PEG-sCT/TA films. One can see in all cases the accumulated released amount of PEG-sCT increases linearly with time, with a correlation coefficient >0.99. The result indicates that the films release PEG-sCT at a constant rate. The zero-order kinetics continues even after >90% of PEG-sCT is released. Initial burst release and incomplete release, which are common issues when using biodegradable microparticles8, 12, 35 and hydrogels10 as peptide-releasing carriers, were not observed here. The morphology of the films does not change during the release (Figure S2).

(A,B) Release of PEG-sCT from different bilayer number of PEG-sCT/TA films drawn as percentage release (A) and cumulative released amount (B). (C) Release duration (represented as time for 90% release) as a function of film thickness (represented as the absorbance of the original film at 218 nm). Release media: 50 mM pH7.4 phosphate buffer. T = 37 °C.
The effect of a series of factors on the release of PEG-sCT was studied. As shown in Fig. 5A, an increased pH of the release media results in a faster release of PEG-sCT. Higher temperature also accelerates the release of PEG-sCT from the films, (Fig. 5B) however, an increase in the ionic strength of the release media slows down the peptide release (Figure 5C). These results suggest it will be possible to tune the release rate of the peptide via external stimuli. Note although the release rate was changed, the zero-order kinetics was still followed under all these conditions studied.

Release profiles of PEG-sCT from (PEG-sCT/TA)6 films under various conditions: (A) in 50 mM phosphate buffer of various pH as indicated, T = 37 °C; (B) in 50 mM pH7.4 phosphate buffer at various temperatures; (C) in various concentration of phosphate buffer. pH = 7.4, T = 37 °C.
A suitable carrier for peptide should not alter the structure of the incorporated drug. For this purpose, the secondary structure of PEG-sCT released into the media was studied by circular dichroism (CD). As shown in Fig. 6, the native PEG-sCT presents a typical negative band at ~199 nm and a low magnitude of ellipticity at 222 nm, suggesting a major random coil structure with a small helical content36, 37. The CD spectra of sCT-PEG released from the film were similar to that of the native drug, suggesting the secondary structure of the peptide remains unchanged.

CD spectra of a solution of native PEG-sCT, mixed solution of PEG-sCT and TA, and release media into which PEG-sCT and TA released.
The major biological effect of PEG-sCT is to lower blood Ca2+ level32. To check if the LBL film could generate a hypocalcaemic response, the films were subcutaneously implanted in SD rats. As shown in Fig. 7A, PEG-sCT was detected in the blood soon after the implantation. Particularly the plasma concentration of PEG-sCT remains almost constant at ~10 ng/mL before a sudden drop to zero. The result strongly suggests that the in vivo release of PEG-sCT also follows a zero-order kinetics. The serum calcium level after the film implantation was shown in Fig. 7B. Compared to the group implanted with PEG/TA, a ~20% decrease in serum calcium level was observed for rats implanted with PEG-sCT/TA films, which is typical calcium level reduction achieved with sCT in rats38. The result confirms that the PEG-sCT released from the films remain bioactive in vivo. In addition, the calcium level maintains constant in the releasing period of the corresponding PEG-sCT/TA film.

PEG-sCT levels in rat serum (A) and plasma calcium levels (B) after implantation.
Discussion
Most drug carriers typically release a drug in a fast-then-slow manner. For example, the drug release from a hydrogel carrier usually follows Higuchi’s kinetics, i.e., the release is proportional to the square root of time, because of a diffusion-controlled release39. Quite differently, the release of PEG-sCT from the PEG-sCT/TA films follows a perfect zero-order kinetics. No initial burst release was observed (Figure 4). In addition, the zero-order kinetics is followed for the entire release process (Figure 4A and also Figure S3 in supporting information, in which complete release profiles of a 4-bilayer and 8-bilayer film with data beyond 100% release were shown). In contrast, for many zero-order release carriers reported previously, constant release can only be observed in a certain period of the release process40.
The zero-order release of PEG-sCT from the PEG-sCT/TA films can be explained by the unique release mechanism of these films. Unlike common drug carriers which release drug via diffusion or polymer degradation13, here PEG-sCT was released via the gradual disintegration of the film (Figure 1C). We34, 41, 42 previously observed that hydrogen-bonded LBL films disintegrate gradually in aqueous solutions, because the film materials are bonded with dynamic, reversible hydrogen bonds. It is noteworthy the gradual disintegration of hydrogen-bonded films is different from the pH-induced disintegration of the same films. In the latter case, the films disintegrate as a result of the breakage of hydrogen bonds, which is a consequence of the pH-induced dissociation of the hydrogen-donor, for example PAA (poly(acrylic acid)) in PVPON/PAA (PVPON: poly(vinyl pyrrolidone)) films43. It occurs only at a pH higher than the pKa of the hydrogen-donor. In addition, it usually occurs instantly. In contrast in the former case, most of the hydrogen bonds in the film remain intact. It occurs even at a pH much lower than the pKa of PAA as in the case of PVPON/PAA film41. The gradual disintegration of PEG-sCT/TA film or PEG/TA film in aqueous solutions was monitored using UV-vis, QCM and Fabry-Perot fringes (Figures S4–S6). Gradual disintegration was also observed from LBL films linked with other dynamic bonds44, such as phenylboronate ester bonds45, 46 and Schiff base bonds47.
As shown in Fig. 1C, the film disintegration, i.e., the dissociation of the film material, PEG-sCT/TA complex, results in the release of free PEG-sCT and TA into the media. Meanwhile free PEG-sCT and TA in the media may deposit back onto the film, or form soluble PEG-sCT/TA complex and remain in the solution. The whole process can be described using the following reactions:
The net rate of the film disintegration, or, the release rate of PEG-sCT, can be written as:
where [PEG-sCT/TA] is the concentration of PEG-sCT/TA complex in the film, [PEG-sCT] and [TA] are concentrations of free PEG-sCT and TA in the media, respectively, and k1 and k2 the rate constants. Under the experimental conditions, [PEG-sCT] and [TA] in the media are very low. Therefore the second term, i.e., the rate of free PEG-sCT and TA to deposit back, is negligible. The release rate of PEG-sCT can be re-written as
Equation 3 indicates that the release of PEG-sCT is determined by the dissociation of PEG-sCT/TA complex. As a solid-like material, concentration of PEG-sCT/TA complex in the film, [PEG-sCT/TA], could be regarded as constant, therefore the release of PEG-sCT is determined only by the rate constant k1. For dynamic films from polymers with a wide molecular weight distribution, k1 is larger for species with a lower molecular weight. Consequently those films exhibit a “fast-then-slow” release pattern34, 45,46,47. In contrast here both PEG-sCT and TA have a narrow molecular weight distribution, therefore k1 for all species can be regarded to be the same48, 49. Therefore the PEG-sCT/TA films disintegrate at a constant rate and releases PEG-sCT at a constant rate.
In Fig. 4B, the release profiles of the 4 films were replotted in terms of cumulative released amount of PEG-sCT, instead of percentage release as in Fig. 4A. One can see these films release PEG-sCT at almost the same rate, despite that their thickness is different. Furthermore as shown in Fig. 4C, the duration for a film to continuously release PEG-sCT is proportional to its thickness. This feature makes the new carrier highly predictable. For a film with a known film thickness, one may predict how long the film will continuously release the drug. On the other hand, one could fabricate a film with a particular thickness according to its predetermined release duration.
The new carrier not only allows for zero-order release of PEG-sCT, but tuning the release rate using external stimuli, such as pH and temperature (Fig. 5). The effects of the external stimuli on the release rate of PEG-sCT can also be explained by the unique release mechanism of the new carrier. An elevated pH increases the dissociation of TA and therefore weakens the hydrogen bonding between TA and PEG-sCT. In addition, with an enhanced dissociation, the electrostatic repulsion among the charged TA molecules increases. Both effects are favorable for the film disintegration. Therefore the release rate of PEG-sCT increases with increasing pH as shown in Fig. 5A. The accelerated release of PEG-sCT at higher temperature, as shown in Fig. 5B, could be attributed to the weakened hydrogen bonding between PEG-sCT and TA. It is well-known that hydrogen bonding is sensitive to temperature and heating could partially break the hydrogen-bonds in hydrogen-bonded LBL films50, 51. The retarded release of PEG-sCT at an elevated ionic strength, as shown in Fig. 5C, can be explained by the enhanced screening of the electrostatic repulsion among the charged TA molecules at higher ionic strength.
The key goal of a release system is to maintain the plasma drug concentration within the therapeutic window for a prolonged period of time. For zero-order release carriers, as the drug is released at constant rate, it will be possible to maintain the plasma drug concentration constant. This will avoid a plasma drug concentration which is too high to exert toxic effects, or too low to fail to exert therapeutic effects, and therefore lead to the best outcome. As expected, for rats with implanted PEG-sCT/TA films, the plasma concentration of PEG-sCT remains almost constant (Fig. 7A). When a 20 bilayer film was implanted, the plasma PEG-sCT level can be maintained for 10 days, which is much longer than the half-life of PEG-sCT when administered via injection (~83 min)38. Along with a constant plasma PEG-sCT level, the serum calcium level remains almost constant until the serum PEG-sCT level drops to zero. The result suggested that almost all drugs exert therapeutic effects because the drug concentration was remained in the therapeutic window during the whole releasing period. This property is highly desirable considering the high synthetic and production costs of a peptide drug52. As mentioned above, when using a carrier with a “fast and slow” release pattern, a significant portion of the drug will not exert a therapeutic effect, because the drug concentration becomes lower than the therapeutic level at the later stage.
In conclusion, using PEG-sCT/TA film as an example, we demonstrated that hydrogen-bonded LBL films of PEGylated peptide and polyphenol could be used for the zero-order release of peptide drugs. The films were fabricated under mild conditions without the use of any harmful solvents. The second component, TA, is a natural product and presents potential therapeutic activities itself. The in vitro release profile of the peptide suggests a zero-order kinetics without initial burst release. In addition the release rate could be tuned via external stimuli, such as pH and temperature. In vivo test showed that the implanted LBL films could maintain the plasma level of PEG-sCT constant over an extended period and thus keep a constant, lowered serum calcium level. Further studies using microspheres as substrate for film fabrication, which will allow administration via subcutaneous injection, are under way.
Methods
Materials
Linear salmon calcitonin ((D)Ala-Ser-Asn-Leu-Ser-Thr-Cys-Val-Leu-Gly-Lys-Leu-Ser-Gln-Glu-Leu-His-Lys-Leu-Gln-Thr-Tyr-Pro-Arg-Thr-Asn-Thr-Gly-Ser-(D)Ala-Thr-Pro-NH2, sCT) (purity >95%) was purchased from Hangzhou SINOPEP Pharmaceutical Inc. Methoxy poly(ethylene glycol)-maleimide (mPEG-Mal) (Mw = 5 K) was purchased from Beijing Kaizheng Biothch Development Co. Ltd. Tannic acid (TA) and poly(ethylene glycol) (PEG, Mw = 5 K) were purchased from Sigma-Aldrich. The chemicals were used as received without further purification. Salmon calcitonin ELISA Kit was purchased from Shanghai Enzyme-linked Biotechnology Co., Ltd. Calcium Colorimetric Assay Kit was obtained from Genmed Co., Ltd, Shanghai.
Synthesis of PEGylated salmon calcitonin (PEG-sCT)
PEG-sCT was synthesized by the reaction between the maleimide group at the end of mPEG-Mal and 7-Cys in sCT. Briefly, 2.0 g of sCT was dissolved in 30 mL of 0.2 M NaH2PO4 in 50 mL Erlenmeyer flask. Then 4.10 g of mPEG-Mal was added into the solution. The reaction was allowed to proceed for 1 hour. HPLC was used to monitor the reaction until the reaction finished. The crude product was purified by Biotage SP-1 middle-pressure preparative liquid chromatography (MPLC) to >98% purity with RP-C18 column (SepaFlash HP, 40–60 μm (SP), Santai Technologies, Inc.). The product was collected, concentrated and lyophilized. The yield was 3.15 g. The purity of product was determined by analytical HPLC (SHIMADZU SPD-10A) with RP-C4 column (Phenonmenex, Jupiter 5 u, 4.6 × 250 mm, 300 Å) at a flow-rate of 1.0 mL/min (Figure S7A). The molecular weight of the peptides was confirmed by MALDI-TOF mass spectra (Figure S7B).
Fabrication of LBL films
The PEG-sCT/TA LBL films were fabricated using quartz slides with a size of 44 mm × 10 mm × 1 mm as substrate (Figure S1). Before use the slides were cleaned in boiling piranha solution (3:7 v/v H2O2−H2SO4 mixture) (caution: this solution is extremely corrosive!), rinsed thoroughly with deionized (DI) water, and then dried. PEG-sCT and TA solutions, both with a concentration of 0.5 mg/mL, were prepared using 10−3 M HCl. To fabricate the films, the substrates were immersed in PEG-sCT and TA solutions alternately, each for 5 min, intermediated with twice washing in 10−3 M HCl, each for 1 min. This cycle was repeated until the desired bilayer numbers were reached. PEG/TA films were fabricated similarly. The films were not released from the substrate and used as prepared in the following tests.
In Vitro Release of PEG-sCT
The LBL films were immersed in 20 mL of release media (pH7.4 50 mM phosphate buffer, if not otherwise specified) which were incubated at 37 °C (if not otherwise specified). A rotary table was used to keep the release media homogeneous. Every 20 minutes the release media was completely withdrawn and replaced with same volume of pre-warmed fresh media. The concentration of peptide in the release media was measured by HPLC. To measure the total amount of peptide in the films, they were completely disintegrated by immersion in pH9.0 50 mM phosphate buffer. All experiments were carried out in triplicate. The results were reported as mean ± standard deviation.
In vivo release and hypocalcemic efficacy
In vivo release of PEG-sCT was carried out with male Spraguee Dawley (SD) rats (200–240 g). The SD rats were cared in accordance with international standards on animal welfare. The rats were divided into 4 groups (n = 5 for each group). The control group was treated with PEG/TA film, while the other 3 groups were treated with PEG-sCT/TA film with various bilayer numbers. The size of the films was all 10 mm × 10 mm × 1 mm. The rats were first anesthetized with sodium pentobarbital (65 mg/kg i.p.). The film was then implanted subcutaneously at the back of each rat through a small incision in the neck region. An i.v. catheter was inserted into the jugular vein using standard aseptic surgical procedures. Blood samples were collected from the jugular vein at pre-determined time intervals. All samples were centrifuged at 10,000 rpm for 10 min and serum was collected. They were stored at −10 °C before analysis. Concentrations of PEG-sCT and calcium were determined using Salmon calcitonin ELISA kit and Calcium Colorimetric Assay Kit, respectively. All experimental procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals of the China National Academy of Sciences, and were approved by the Animal Care and Use Committee of the Beijing Institute Pharmacology & Toxicology.
Other characterizations
UV-vis absorption spectra were recorded on a UV-1800 spectrophotometer (SHIMADZU, Japan). Fourier transform infrared (FTIR) spectra were measured on Bio-Rad FTS-6000 spectrometer. CD measurement was performed on a Biologic MOS-450 (Claix, France) at room temperature in a 0.1 cm path cell. The CD signals were recorded from 190 to 260 nm using a bandwidth of 4.0 nm and a scanning rate of 50 nm/min. SEM images were recorded on an FEI NanoSEM 430 Scanning Electron Microscope. Atomic force microscopy (AFM) images were acquired using a Benyuan CSPM5000s scanning probe microscope in tapping mode.
Statistical analysis
All experiments were repeated at least three times. The results were expressed as mean ± SD.
References
Lawrence, S. Billion dollar babies-biotech drugs as blockbusters. Nat. Biotechnol. 25, 380 (2007).
Kovalainen, M. et al. Novel Delivery Systems for Improving the Clinical Use of Peptides. Pharmacol. Rev. 67, 541 (2015).
Fosgerau, K. & Hoffmann, T. Peptide therapeutics: current status and future directions. Drug Discov. Today 20, 122 (2015).
Zhang, J., Desale, S. S. & Bronich, T. K. Polymer-based vehicles for therapeutic peptide delivery. Therapeutic Delivery 6, 1279 (2015).
Alconcel, S. N. S., Baas, A. S. & Maynard, H. D. FDA-approved poly(ethylene glycol)-protein conjugate drugs. Polym. Chem 2, 1442 (2011).
Fishburn, C. S. The pharmacology of PEGylation: Balancing PD with PK to generate novel therapeutics. J. Pharm. Sci. 97, 4167 (2008).
Sophocleous, A. M. et al. The nature of peptide interactions with acid end-group PLGAs and facile aqueous-based microencapsulation of therapeutic peptides. J. Control. Release 172, 662 (2013).
Wang, J., Wang, B. M. & Schwendeman, S. P. Characterization of the initial burst release of a model peptide from poly(d,l-lactide-co-glycolide) microspheres. J. Control. Release 82, 289 (2002).
Hu, Q. et al. A novel approach for the intravenous delivery of leuprolide using core-cross-linked polymeric micelles. J. Control. Release 205, 98 (2015).
Vermonden, T., Censi, R. & Hennink, W. E. Hydrogels for Protein Delivery. Chem. Rev. 112, 2853 (2012).
Du, A. W. & Stenzel, M. H. Drug Carriers for the Delivery of Therapeutic Peptides. Biomacromolecules 15, 1097 (2014).
Ye, M., Kim, S. & Park, K. Issues in long-term protein delivery using biodegradable microparticles. J. Control. Release 146, 241 (2010).
Uhrich, K. E., Cannizzaro, S. M., Langer, R. S. & Shakesheff, K. M. Polymeric Systems for Controlled Drug Release. Chem. Rev. 99, 3181 (1999).
Weidner, J. Drug delivery. Drug Discov. Today 7, 632 (2002).
Gokhale, A. Achieving Zero-Order Release Kinetics Using Multi-Step Diffusion-Based Drug Delivery. Pharmaceutical Technology 38, 46 (2014).
Celia, C. et al. Sustained Zero-Order Release of Intact Ultra-Stable Drug-Loaded Liposomes from an Implantable Nanochannel Delivery System. Adv. Healthc. Mater. 3, 230 (2014).
Ali, M. et al. Zero-order therapeutic release from imprinted hydrogel contact lenses within in vitro physiological ocular tear flow. J. Control. Release 124, 154 (2007).
Lei, L. et al. Zero-order release of 5-fluorouracil from PCL-based films featuring trilayered structures for stent application. Eur. J. Pharm. Biopharm. 78, 49 (2011).
Zhang, H. et al. PEG/lecithin-liquid-crystalline composite hydrogels for quasi-zero-order combined release of hydrophilic and lipophilic drugs. RSC Adv. 3, 22927 (2013).
Fine, D. et al. A robust nanofluidic membrane with tunable zero-order release for implantable dose specific drug delivery. Lab Chip 10, 3074 (2010).
Brazel, C. S. & Peppas, N. A. Modeling of drug release from Swellable polymers. Eur. J. Pharm. Biopharm. 49, 47 (2000).
Lee, P. I. Novel approach to zero-order drug delivery via immobilized nonuniform drug distribution in glassy hydrogels. J. Pharm. Sci. 73, 1344 (1984).
Tahara, K., Yamamoto, K. & Nishihata, T. Overall mechanism behind matrix sustained release (SR) tablets prepared with hydroxypropyl methylcellulose 2910. J. Control. Release 35, 59 (1995).
Wang, Q., Wang, J. X., Lu, Q. H., Detamore, M. S. & Berkland, C. Injectable PLGA based colloidal gels for zero-order dexamethasone release in cranial defects. Biomaterials 31, 4980 (2010).
Ferrati, S. et al. Leveraging nanochannels for universal, zero-order drug delivery in vivo. J. Control. Release 172, 1011 (2013).
Ferrati, S. et al. The Nanochannel Delivery System for Constant Testosterone Replacement Therapy. J. Sex. Med. 12, 1375 (2015).
Xiao, H. et al. Biodegradable polymer − cisplatin(IV) conjugate as a pro-drug of cisplatin(II). Biomaterials 32, 7732 (2011).
Zhou, D. et al. Layer-by-Layer Assembled Polypeptide Capsules for Platinum-Based Pro-Drug Delivery. Bioconjugate Chem. 23, 2335 (2012).
Xiao, H. et al. Photosensitive Pt(IV)–azide prodrug-loaded nanoparticles exhibit controlled drug release and enhanced efficacy in vivo. J. Control. Release 173, 11 (2014).
Han, K. et al. A Tumor Targeted Chimeric Peptide for Synergistic Endosomal Escape and Therapy by Dual-Stage Light Manipulation. Adv. Funct. Mater. 25, 1248 (2015).
Decher, G. Fuzzy Nanoassemblies: Toward Layered Polymeric Multicomposites. Science 277, 1232 (1997).
Youn, Y. S., Na, D. H. & Lee, K. C. High-yield production of biologically active mono-PEGylated salmon calcitonin by site-specific PEGylation. J. Control. Release 117, 371 (2007).
Costa, E., Coelho, M., Ilharco, L. M., Aguiar-Ricardo, A. & Hammond, P. T. Tannic Acid Mediated Suppression of PNIPAAm Microgels Thermoresponsive Behavior. Macromolecules 44, 612 (2011).
van Dijkhuizen-Radersma, R. et al. Stability aspects of salmon calcitonin entrapped in poly(ether-ester) sustained release systems. Int. J. Pharmaceut 248, 229 (2002).
Bhandari, K. H., Newa, M., Chapman, J. & Doschak, M. R. Synthesis, characterization and evaluation of bone targeting salmon calcitonin analogs in normal and osteoporotic rats. J. Control. Release 158, 44 (2012).
Tang, Y. & Singh, J. Thermosensitive Drug Delivery System of Salmon Calcitonin: In Vitro Release, In Vivo Absorption, Bioactivity and Therapeutic Efficacies. Pharm. Res. 27, 272 (2010).
Ryan, S. M. et al. Conjugation of salmon calcitonin to a combed-shaped end functionalized poly(poly(ethylene glycol) methyl ether methacrylate) yields a bioactive stable conjugate. J. Control. Release 135, 51 (2009).
Censi, R. et al. Photopolymerized thermosensitive hydrogels for tailorable diffusion-controlled protein delivery. J. Control. Release 140, 230 (2009).
van de Manakker, F. et al. Protein-Release Behavior of Self-Assembled PEG-beta-Cyclodextrin/PEG-Cholesterol Hydrogels. Adv. Funct. Mater. 19, 2992 (2009).
Guan, Y. et al. Fabry-Perot fringes and their application to study the film growth, chain rearrangement, and erosion of hydrogen-bonded PVPON/PAA films. J. Phys. Chem. B 110, 13484 (2006).
Guan, Y., Zhang, Y. J., Zhou, T. & Zhou, S. Q. Stability of hydrogen-bonded hydroxypropylcellulose/poly(acrylic acid) microcapsules in aqueous solutions. Soft Matter 5, 842 (2009).
Zhou, L., Chen, M., Tian, L., Guan, Y. & Zhang, Y. Release of Polyphenolic Drugs from Dynamically Bonded Layer-by-Layer Films. ACS Appl. Mater. Interfaces 5, 3541 (2013).
Sukhishvili, S. A. & Granick, S. Layered, erasable, ultrathin polymer films. J. Am. Chem. Soc. 122, 9550 (2000).
Guan, Y. & Zhang, Y. Dynamically bonded layer-by-layer films: Dynamic properties and applications. J. Appl. Polym. Sci. 131, 40918 (2014).
Ding, Z. B., Guan, Y., Zhang, Y. & Zhu, X. X. Layer-by-layer multilayer films linked with reversible boronate ester bonds with glucose-sensitivity under physiological conditions. Soft Matter 5, 2302 (2009).
Zhang, X., Guan, Y. & Zhang, Y. Dynamically bonded layer-by-layer films for self-regulated insulin release. J. Mater. Chem. 22, 16299 (2012).
Zhou, L., Chen, M., Guan, Y. & Zhang, Y. Dynamic layer-by-layer films linked with Schiff base bond for sustained drug release. RSC Adv 5, 83914 (2015).
Zhao, Y., Yuan, Q., Li, C., Guan, Y. & Zhang, Y. Dynamic Layer-by-Layer Films: A Platform for Zero-Order Release. Biomacromolecules 16, 2032 (2015).
Zhao, Y., Gu, J., Jia, S., Guan, Y. & Zhang, Y. Zero-order release of polyphenolic drugs from dynamic, hydrogen-bonded LBL films. Soft Matter 12, 1085 (2016).
Zhang, Y. J., Guan, Y., Yang, S. G., Xu, J. & Han, C. C. Fabrication of hollow capsules based on hydrogen bonding. Adv. Mater. 15, 832 (2003).
Yang, S. G., Zhang, Y. J., Yuan, G. C., Zhang, X. L. & Xu, J. A. Porous and nonporous nanocapsules by H-bonding self-assembly. Macromolecules 37, 10059 (2004).
Vlieghe, P., Lisowski, V., Martinez, J. & Khrestchatisky, M. Synthetic therapeutic peptides: science and market. Drug Discov. Today 15, 40 (2010).
Acknowledgements
We thank financial support for this work from the National Natural Science Foundation of China (21274068, 21374048, 51625302, 81202465, and 81573354), Tianjin Committee of Science and Technology (16JCZDJC32900), ICCAS State Key Laboratory of Polymer Physics and Chemistry (PPCL-2014CX-6), PCSIRT program (IRT1257), and the National Key Technologies R & D Program for New Drugs of China (2012ZX09301003). We thank Mr. Zhao Meng, Bin Li and Wei Wang for kindly help in this work.
Author information
Authors and Affiliations
Contributions
Yongjun Zhang, Qingbin Meng and Ying Guan conceived and designed the experiments. Ya-Nan Zhao and Na Wen fabricated the films and performed the in vitro tests. Xiaoyu Xu synthesized the peptide. Xiaoyu Xu and Jiankun Qie purified and characterized the peptide. Ya-Nan Zhao, Rui Song, Danni Cao, Siqi Cheng and Yansheng Dong performed the animal experiments. Yongjun Zhang, Qingbin Meng, Ying Guan and Keliang Liu analyzed the data. Yongjun Zhang and Ying Guan wrote the manuscript. All authors discussed the results.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, YN., Xu, X., Wen, N. et al. A Drug Carrier for Sustained Zero-Order Release of Peptide Therapeutics. Sci Rep 7, 5524 (2017). https://doi.org/10.1038/s41598-017-05898-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05898-6
This article is cited by
-
Loading and release of insulin from chitosan oligosaccharide biopolymeric patch for oromucosal delivery
Chemical Papers (2024)
-
pH-sensitive beads containing curcumin loaded nanostructured lipid carriers for a colon targeted oral delivery system
Journal of Pharmaceutical Investigation (2022)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
