
Abstract
Current therapies for multiple sclerosis (MS) reduce both relapses and relapse-associated worsening of disability, which is assumed to be mainly associated with transient infiltration of peripheral immune cells into the central nervous system (CNS). However, approved therapies are less effective at slowing disability accumulation in patients with MS, in part owing to their lack of relevant effects on CNS-compartmentalized inflammation, which has been proposed to drive disability. Bruton tyrosine kinase (BTK) is an intracellular signalling molecule involved in the regulation of maturation, survival, migration and activation of B cells and microglia. As CNS-compartmentalized B cells and microglia are considered central to the immunopathogenesis of progressive MS, treatment with CNS-penetrant BTK inhibitors might curtail disease progression by targeting immune cells on both sides of the blood–brain barrier. Five BTK inhibitors that differ in selectivity, strength of inhibition, binding mechanisms and ability to modulate immune cells within the CNS are currently under investigation in clinical trials as a treatment for MS. This Review describes the role of BTK in various immune cells implicated in MS, provides an overview of preclinical data on BTK inhibitors and discusses the (largely preliminary) data from clinical trials.
Key points
-
Bruton tyrosine kinase (BTK) is an important intracellular signalling molecule involved in regulating the maturation, proliferation, survival and activation of B cells and myeloid cells.
-
BTK inhibitors might concurrently target adaptive and innate immune mechanisms in the periphery and central nervous system (CNS).
-
This ability makes BTK inhibitors a promising therapeutic approach for relapsing and progressive forms of multiple sclerosis (MS).
-
Preclinical studies show that BTK inhibition can suppress key pathological features of MS, including B cell activation, CNS lymphocyte infiltration, leptomeningeal inflammation, pro-inflammatory microglial activation and demyelination.
-
The efficacy and safety of five BTK inhibitors are currently being evaluated in clinical trials in patients with relapsing and progressive MS.
-
The BTK inhibitors under investigation in clinical trials differ in their selectivity, mode of binding, target occupancy, inhibitory potency and CNS penetrance.
Similar content being viewed by others

Introduction
Multiple sclerosis (MS) is a central nervous system (CNS) disorder characterized by inflammation, demyelination, gliosis and neuroaxonal degeneration1,2,3,4. Although MS has traditionally been viewed as primarily mediated by T cells, B cells and nearly all cell types of the innate immune system (Box 1) also seem to have important roles in both the initiation and propagation of MS3,5,6,7. Peripheral immune cells that cross the blood–brain barrier (BBB) drive relapses and the formation of focal demyelinating plaques8. These infiltrating peripheral immune cells, as well as endothelial cells and innate immune cells such as microglia, macrophages and astrocytes, are thought to contribute to CNS-compartmentalized neuroinflammation and neurodegeneration in MS through the production of cytokines, chemokines, excitotoxins, free radicals, complement components and secondary messengers7,9,10.
Although substantial progress has been made in the development of highly effective treatments for relapsing MS (RMS) in the past 25 years, limited therapeutic options are available for patients with non-relapsing progressive MS (PMS)2,11,12,13. Salient pathological features of PMS include widespread activation of microglia and macrophages, astrogliosis, persistent lymphocyte activity in meningeal aggregates and dispersed in the CNS parenchyma, increased cortical demyelination, deficient remyelination, chronic active lesions, diffuse neuroaxonal damage and atrophy of both white and grey matter2,11,13,14,15.
Chronic active lesions were first described in histopathological studies as white matter lesions with a demyelinated, hypocellular core, surrounded by activated microglia and macrophages at the lesion rim16,17,18,19. Advances in MRI made it possible to visualize two subsets of chronic active lesions in vivo: paramagnetic rim lesions (PRLs) or iron rim lesions and slowly expanding lesions (SELs)20,21. PRLs and iron rim lesions are detected with iron-sensitive MRI techniques, such as susceptibility-weighted imaging or T2*-weighted imaging20, owing to the presence of magnetic susceptibility in the rim of the lesion, which correspond in histopathological studies to iron-laden activated pro-inflammatory macrophages or microglia and (to a much lesser extent) reactive astrocytes21,22,23. SELs are foci of gradual and concentric expansion within existing T2 lesions that can be identified in serial conventional MRI scans of the same patient24. PRLs and SELs are more frequently seen in patients with PMS and are associated with more aggressive disease and more rapid disease progression21,25,26,27,28, although such lesions might represent only a portion of the total burden of chronic active lesions29.
Currently available disease-modifying therapies for MS modulate the peripheral immune response but have only a limited effects on CNS-compartmentalized inflammation, including that mediated by the CNS-resident immune cells that are thought to drive the accumulation of disability7,30. This circumstance explains why therapies that are effective in preventing relapses and relapse-associated worsening driven by transient infiltration of peripheral immune cells in patients with RMS might not necessarily offer a benefit in patients with established PMS7. Despite successful clinical trial results for B cell-depleting antibody therapies such as rituximab, ocrelizumab and ofatumumab31,32,33,34,35, these agents only modestly limit disease progression36, probably owing to poor brain penetration of monoclonal antibodies, which limits their ability to act on the compartmentalized inflammation in PMS37,38,39,40. Furthermore, continued use of these non-selective therapies causes chronic B cell depletion41, which can potentially increase the risk of serious infections and possibly malignancy36.
To overcome the limitations of B cell-depleting antibody therapies for MS, research has focused on Bruton tyrosine kinase (BTK) inhibitors. BTK is an intracellular signalling molecule that is produced in B cells and cells of most haematopoietic lineages (including monocytes, macrophages, microglia, mast cells and neutrophils) except for natural killer cells and fully differentiated plasma cells42. BTK is an essential proximal component of several signalling pathways downstream of the B cell receptor (BCR) that control B cell maturation, survival and activation, as well as production of cytokines and antigen-dependent stimulation of T cells43,44,45,46. As a result, BTK has emerged as a treatment target in B cell malignancies and a growing number of autoimmune disorders45,47 (Supplementary Table 1). Moreover, BTK also has orthologous roles in macrophages, microglia and mast cells: BTK controls their activation, the liberation of histamine through degranulation in mast cells and the secretion of pro-inflammatory cytokines as well as inflammasome activation45,48,49,50. BTK is also produced in several cells of neural origin, including neurons51 and astrocytes52,53, whereas oligodendrocytes do not produce BTK52. Accordingly, the development of CNS-penetrant BTK inhibitors brings hope for a new era of MS treatments that simultaneously target both peripheral immune cells, which drive MS relapses, and CNS-compartmentalized inflammation, which is thought to be involved in disability accumulation.
This Review highlights the role of BTK in signalling pathways of B lymphocytes and other immune cells that are considered important in MS. We also provide an update on the growing body of data supporting the scientific rationale for use of BTK inhibitors to treat MS and discuss the promising preliminary results of clinical trials.
An overview of BTK inhibitors
In 1952, the paediatrician Ogden C. Bruton described an 8-year-old boy with ‘complete absence of gamma globulin with otherwise normal serum proteins and recurrent pneumococcal sepsis’54. The inherited immunodeficiency described by Bruton was termed X-linked agammaglobulinaemia (XLA). In the early 1990s, the BTK gene (encoding BTK) was identified as the key mutation site in patients with XLA, and BTK was shown to participate in the BCR signalling pathway after antigen engagement46,55,56.
After the discovery of BTK, a large number of BTK inhibitors were developed for the treatment of B cell malignancies, the survival of which requires BCR-mediated BTK signalling57. LFM-A13 was the first (non-selective) BTK inhibitor to be developed58, although this agent also potently inhibits Janus kinase 2, another non-receptor tyrosine kinase59,60. A search for selective BTK inhibitors led in 2007 to the synthesis of ibrutinib61,62. To date, ibrutinib is approved for the treatment of chronic lymphocytic leukaemia (CLL), small lymphocytic lymphoma, mantle-cell lymphoma (MCL), Waldenström macroglobulinaemia, marginal zone lymphoma and chronic graft-versus-host disease63. In addition, acalabrutinib is approved for the treatment of CLL, small lymphocytic lymphoma and MCL64 and zanubrutinib for MCL65. BTK inhibitors are also under investigation for the treatment of several other haematological disorders (including malignancies and other lymphoproliferative disorders) and non-haematological malignancies (including melanoma, breast neoplasms, prostate cancer, urogenital carcinoma, gastrointestinal adenocarcinoma, adenocarcinoma of the lung, pancreas and kidney and glioblastoma), as well as severe acute respiratory syndrome coronavirus-2 infection, which causes coronavirus disease 2019 (ref. 66).
Moreover, BTK inhibitors have been tested or are actively being investigated as potential treatments for autoimmune diseases, including rheumatoid arthritis, Sjögren syndrome, systemic lupus erythematosus, pemphigus vulgaris, chronic spontaneous urticaria, asthma and MS67. The development of BTK inhibitor treatments for autoimmune diseases was fuelled by the observation that B cell-specific overexpression of BTK induces systemic autoimmunity in mice68,69 and the finding that BTK levels are upregulated in B cells from people with rheumatoid arthritis or Sjögren syndrome70. BTK inhibitors have shown variable efficacy in autoimmune conditions, ranging from no benefit (in Sjögren syndrome and systemic lupus erythematosus) to favourable efficacy in RMS67. At the time of writing (September 2022), 330 clinical trials of BTK inhibitors, including in patients with autoimmune disorders, are registered at ClinicalTrials.gov. Table 1 includes the five BTK inhibitors in late-stage (phases II–III) development for MS. Supplementary Table 1 lists notable trials of the same agents for other indications.
BTK in immune cell signalling
BTK — along with IL-2-inducible T cell kinase, resting lymphocyte kinase, tyrosine kinase expressed in hepatocellular carcinoma (TEC) and bone-marrow-expressed kinase70 — comprises the Tec family of non-receptor protein tyrosine kinases, which participate in the signalling pathways of B cells and myeloid cells, a lineage that includes microglia, macrophages, monocytes, mast cells, basophils, neutrophils and dendritic cells. The presence of a pleckstrin homology (PH) domain is the most characteristic conserved motif of Tec family proteins. This PH domain enables Tec kinases to bind to phosphoinositides, such as phosphatidylinositol (3,4,5)-trisphosphate (PtdIns(3,4,5)P3), in the cytoplasmic leaflet of the plasma membrane. Thus, receptor-mediated increases in membrane phosphoinositide concentration lead to recruitment of Tec kinases to the plasma membrane, where they phosphorylate downstream signalling molecules71. As such, the PH domain of Tec proteins is essential for their roles in controlling downstream effectors that regulate diverse cell processes, including growth, differentiation, metabolism, survival and apoptosis72,73,74,75,76.
BTK is crucial for B cell development and maturation in the bone marrow, secondary lymphoid tissues and in the periphery. For example, patients with XLA, which is caused by mutations distributed across the five structural domains of BTK77,78, have a near-complete lack of circulating B cells (<2% of total lymphocytes in peripheral blood) and plasma cells77. By contrast, in mice with X-linked immunodeficiency (XID)79, a genetic model of XLA, missense mutations in the Btk region encoding the PH domain of BTK cause only partial B cell depletion. Circulating B cell levels are less severely affected in mice with XID than in patients with XLA owing to the presence of compensatory mechanisms that maximize pre-B cell survival79,80,81.
In the early stages of B cell development in the bone marrow, BTK controls the progression of pre-B cells to immature B cells by regulating the IL-7-induced proliferation of large (cycling) pre-B cells and their transition to small (resting) pre-B cells and by regulating immunoglobulin light chain gene expression41,44,82. BTK also controls the negative selection of autoreactive immature B cells69,83. Outside the bone marrow, BTK participates in the follicular migration (via control of integrin gene expression) and maturation of B cells; BTK also regulates the activation and terminal differentiation of B cells into memory B cells or plasma cells41,44,82,83,84,85.
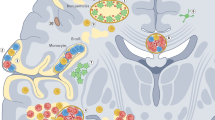
In addition to its involvement in B cell signalling, BTK has important signalling roles mediated by the IgG-specific Fc receptor (FcγR) in macrophages and microglia (Fig. 1) and by the IgE-specific Fc receptor (FcεR) in mast cells. Through its interactions with these Fc receptors, BTK regulates degranulation, the release of histamine and pro-inflammatory cytokines, reactive oxygen species (ROS) production and inflammasome activation in myeloid cells43,47,86.

Bruton tyrosine kinase (BTK) mediates signalling from the B cell receptor (BCR) in B cells and IgG Fc receptor III (FcγRIII, also known as CD16) in macrophages and microglia to downstream elements crucial to immune cell function. a, BCR activation leads to the formation and activation of a signalling complex in which BTK is linked with 1-phosphatidylinositol 4,5-bisphosphate phosphodiesterase-γ2 (PLCγ2) through the scaffold protein SLP65 (Src homology 2 domain-containing leukocyte protein of 65 kDa). Phosphorylation of PLCγ2 by BTK triggers several downstream signalling pathways, including inositol 1,4,5-triphosphate (IP3) receptor (IP3R)-dependent calcium mobilization (green), cytoskeletal remodelling (light blue) and activation of nuclear factor of activated T cells (NFAT; green), nuclear factor (NF)-κB (olive green) and RAS GTPase pathways (red). b, FcγRIII activation recruits and activates Lck/Yes-related novel protein tyrosine kinase (LYN) and spleen tyrosine kinase (SYK), which phosphorylate linker for the activation of T cells family member 1 (LAT) and establish a signalling complex comprising BTK and PLCγ2. In turn, PLCγ2 is responsible for generating diacylglycerol and IP3, two secondary messengers that control cellular effector responses via protein kinase Cβ (PKC-β) and intracellular Ca2+ release, respectively. Diacylglycerol interacts with a scaffold complex comprising B cell leukaemia and/or lymphoma 10 (BCL10), B cell adaptor for phosphoinositide-3-kinase (BCAP) and caspase recruitment domain-containing protein 11 (CARD11). ER, endoplasmic reticulum; ERK, extracellular signal-regulated protein kinase; ITAM, immunoreceptor tyrosine-based activation motif; MALT1, mucosa-associated lymphoid tissue lymphoma translocation protein 1; PI3K, phosphoinositide 3-kinase; PtdIns(4,5)P2, phosphatidylinositol 4,5-bisphosphate; PtdIns(3,4,5)P3, phosphatidylinositol (3,4,5)-trisphosphate; TRAF6, tumour necrosis factor receptor-associated factor 6; WASP, Wiskott–Aldrich syndrome protein. Adapted from refs. 57,210, Springer Nature.
BTK in B cell signalling
The BCR consists of a membrane-bound immunoglobulin comprising two heavy chains, two light chains and a heterodimer of Igα and Igβ (also known as CD79a and CD79b, respectively). Both Igα and Igβ have cytoplasmic tails containing immunoreceptor tyrosine-based activation motifs (ITAMs), which are necessary for intracellular signal transduction downstream of the BCR. Antigen binding to the BCR leads to phosphorylation (usually by tyrosine-protein kinase LYN) of tyrosine residues located on ITAMs in the BCR87 and in the intracellular tail of the BCR co-receptor CD19 (ref. 88). These phosphorylation events result in the creation of docking sites for spleen tyrosine kinase (SYK)89,90 and the recruitment of phosphoinositide 3-kinase (PI3K), respectively57,91. PI3K can also be activated independently of CD19 by B cell phosphoinositide 3-kinase adapter protein 192.
Upon BCR stimulation, BTK (which is ordinarily located in the cytoplasm) is recruited to the plasma membrane, where PtdIns(3,4,5)P3 interacts with the PH domain of BTK43,93. Moreover, CD40 activates BTK via SYK, both independently and in synergy with the BCR94,95,96. Upon full activation of BTK in association with B cell linker protein (also known as Src homology 2 domain-containing leukocyte protein of 65 kDa (SLP65)), BTK phosphorylates phospholipase C γ2 (PLCγ2)97, which generates the secondary messengers inositol 1,4,5-triphosphate (IP3) and diacylglycerol98 (Fig. 1a). IP3, via its receptor, mobilizes calcium influx from intracellular stores99, which stimulates the calmodulin–calcineurin pathway leading to activation of nuclear factor of activated T cells (NFAT)43,57. Diacylglycerol, together with the increased calcium influx, in turn activates protein kinase Cβ98. This enzyme interacts with a complex that comprises multiple signalling cofactors (including tumour necrosis factor (TNF) receptor-associated factor 6 (TRAF6)) to activate the IκB kinase complex. Activation of the IκB kinase complex results in the degradation of IκB, which enables nuclear factor-κB (NF-κB) to enter the nucleus43,100,101,102 (Fig. 1a). NFAT and NF-κB transcription factors control the expression of genes that are essential for B cell survival, proliferation, tolerance, immunoglobulin class switching and the release of chemokines and cytokines103,104,105,106,107,108.
In addition to activating the NF-κB pathway, protein kinase Cβ and diacylglycerol stimulate mitogen-activated protein kinase signalling, leading to activation and nuclear translocation of extracellular signal-regulated protein kinases (ERKs) 1 and 2 (ref. 109) (Fig. 1a). Important targets of ERK1 and ERK2 include the transcription factors ELK1 and MYC, which are involved in pre-B cell survival and proliferation110.
BTK is also essential for the cytoskeletal remodelling that occurs following BCR activation111,112,113. This process involves VAV proteins, which are activated by CD19 and linked to BTK via SLP65 (ref. 114) (Fig. 1a). BTK activation is negatively regulated by SH2-domain-containing inositol polyphosphate 5ʹphosphatase-1 (SHIP1), which is recruited to immune tyrosine inhibitory motifs on low-affinity IgG Fc region receptor IIb FcγRIIB, an inhibitory receptor on B cells. In a process that involves LYN-mediated phosphorylation of immune tyrosine inhibitory motifs, SHIP1 hydrolyses PtdIns(3,4,5)P3, causing the release of BTK from the plasma membrane115,116.
Beyond BCR signal transduction, BTK is essential in Toll-like receptor (TLR) signalling in B cells. BTK interacts with the Toll and IL-1 receptor (TIR) domain of TLRs117,118,119, as well as with adapter proteins that couple TLRs to downstream protein kinases120,121. For instance, BTK interacts with the adapter protein myeloid differentiation factor 88 (MyD88)117 to trigger TRAF6-dependent activation of NF-κB and cytokine production via recruitment of IL-1 receptor-associated kinases43,122. As TRAF6 is also recruited by the BCR to activate NF-κB101, these two signalling pathways are proposed to represent a key mechanism through which BTK contributes to synergistic B cell responses to dual TLR and BCR stimulation43,123,124.
BTK in innate immune cell signalling
BTK is also involved in signalling networks that contribute to innate immunity, including FcεR-mediated signal transduction in mast cells and basophils48,49, 125. Antigen binding to IgE molecules associated with FcεR induces phosphorylation of ITAMs in FcεR β and γ chains48. As in BCR signal transduction, phosphorylation of FcεR ITAMs recruits and activates first LYN and then SYK126,127. These two kinases phosphorylate multiple tyrosine residues in linker for the activation of T cells family member 1 (LAT) and establish a signalling complex that consists of BTK, PLCγ1 and PLCγ2 (ref. 128). BTK and SYK, via phosphorylation of PLCγ, initiate the generation of secondary messengers IP3 and diacylglycerol128,129. The IP3-induced release of intracellular calcium and diacylglycerol-mediated activation of PKC isoforms both result in degranulation and activation of several transcription factors, including members of the NFAT family and activator protein-1 complex, that control cytokine production48,129.
In contrast to the considerable understanding of BTK in BCR signalling in B cells, little is known about the function of BTK in individual myeloid cell populations43. The evidence accrued to date suggests that BTK, via Fc receptor signalling, participates in myeloid cell activation, maturation, migration, survival, phagocytosis and pro-inflammatory cytokine production (Fig. 1b). Among the cell surface Fc receptors expressed by innate immune cells and specific for different immunoglobulin isotypes, only FcγRs are known to signal via BTK: in cultured macrophages130,131,132, BTK inhibition blocked FcγR-induced phagocytosis130 as well as production of IL-1β, IL-6 and TNF133. The role of BTK in FcγR-induced cytokine production was attributed to FcγRIII, an activating FcγR133. FcγRIII contains two ITAMs on the intracellular portion of its Fcγ chain that are phosphorylated upon IgG binding. As in the BCR signalling pathway described earlier, phosphorylation of these ITAMs results in cellular activation via SYK, BTK and PI3K-mediated PtdIns(3,4,5)P3 generation43,134 (Fig. 1b).
The involvement of BTK in innate immune cell responses is also mediated by TLR signalling, including TLR regulation by major histocompatibility complex class II molecules. BTK interacts with MyD88 and TIR-domain-containing adapter molecule 1 (TICAM1, also known as TRIF) in innate immune cells to facilitate TLR signalling pathways that lead to NF-κB activation and interferon-β production117,119,135,136. The interaction of BTK with MyD88 and TICAM1 is essential for the upregulation of TLR signalling by intracellular major histocompatibility complex class II molecules in macrophages and dendritic cells137.
Evidence for a role of BTK in MS
A series of observations in human tissue has provided evidence for an important role of BTK in MS, which might be particularly relevant in progressive forms of the disease. In memory B cells isolated from the blood of patients with relapsing-remitting MS (RRMS) or secondary progressive MS (SPMS), the ratio of phosphorylated BTK to unmodified BTK protein (which indicates the level of BTK activation) was increased compared with that in healthy control individuals138. In brain autopsy samples from patients with SPMS, the levels of BTK+ cells were increased around the rim of lesions (results available only in abstract form)139. Immunostaining of SPMS brain tissue revealed that the BTK protein was present in microglia, with a marked increase in the concentration of BTK+CD68+ cells in lesions compared with that in normal-appearing white matter (results available only in abstract form)139,140. When expression of the BTK gene was assessed in brain samples from patients with MS, BTK mRNA levels were notably increased in primary progressive MS lesions and SPMS lesions compared with surrounding healthy tissue (P = 0.02). Finally, single-nucleus RNA sequencing of microglia in brain samples from patients with PMS and healthy control individuals showed a high level of enrichment for both BTK mRNA and a BTK activation signature in patient-derived microglia. This BTK activation signature included upregulation of SPP1 and RGS1 genes and downregulation of CX3CR1 and P2RY12 genes (results available only in abstract form)139.
CX3CR1 is a receptor for chemokine ligand CX3CL1, which has both pro-inflammatory and anti-inflammatory properties141. Thus, the altered expression of CX3CR1 in disease-associated microglia from patients with PMS indicates an altered chemokine profile. CXCL13 is another chemokine that is highly expressed in microglia and has been implicated in the pathogenesis of MS, specifically in recruiting B cells into the CNS142. Microglia have a key role in the recruitment of adaptive immune cells into the CNS143,144,145. This trafficking of lymphocytes into the CNS represents a disease-associated mechanism that could be plausibly targeted with BTK inhibitors. Of note, in an in vitro study, the BTK inhibitor evobrutinib attenuated the migration of brain-homing (CXCR3+ switched) B cells derived from healthy blood samples through monolayers of human brain endothelial cells138. CXCL13 and several cytokines produced by microglia, such as TNF and IL-1β146, are thought to attract and maintain B cells within the CNS147. Indeed, CNS-compartmentalized B cells are now considered to be one of the main drivers in the immunopathogenesis of PMS148,149. These cells might directly damage the CNS through the secretion of exosomes and pathogenic microvesicles (which have been shown to induce neuron and oligodendrocyte death in vitro) or through the release of antibodies that promote T cell reactivation in CNS barriers and facilitate their entry into the parenchyma150, as well as increasing complement deposition on myelin, which in turn could lead to enhanced myelin phagocytosis, membrane attack complex formation and B cell activation148.
In patients with MS, B cells might indirectly promote CNS injury by driving the polarization of autoreactive T helper type 1 (TH1) and TH17 cells both in the periphery and within the CNS, and by their release of various pro-inflammatory cytokines that activate macrophages and microglia and might also promote the formation of follicle-like structures. In MS brain tissue, the formation of such structures is associated with pronounced cortical demyelination and thinning, microglial activation and CNS infiltration of T cells, macrophages and some plasma cells148. Importantly, the CNS-resident B cells in patients with MS are presumed to be unaffected by anti-CD20 B cell-depleting therapies. Hence, the potential of BTK inhibitors to modulate these compartmentalized B cells and thereby attenuate their presumed contributions to CNS-compartmentalized inflammation and injury could provide a clear benefit in patients with MS.
Preclinical work in models of MS
In vitro and in vivo preclinical studies have demonstrated the therapeutic potential of BTK inhibitors in models of MS and have also provided insights into their putative mechanisms of action (Fig. 2). Most of the findings have emerged from studies of evobrutinib and tolebrutinib, both of which are covalent BTK inhibitors151,152. However, it is important to note that many of the preliminary data discussed here have not been peer-reviewed for full publication and should therefore be interpreted with caution.

Bruton tyrosine kinase (BTK) inhibitors directly modulate the functions of B cells and myeloid cells (including macrophages and microglia) and therefore target both adaptive and innate mechanisms that contribute to the immunopathology of multiple sclerosis on both sides of the blood–brain barrier (BBB)41. Although BTK inhibitors do not modulate T cell function directly, they can interfere with deleterious B cell–T cell interactions (not shown)163. CNS, central nervous system.
Effects on B cells
B cells are implicated in both relapse and progression of MS39,148,153,154. BTK inhibitors alter B cell function but, in contrast to B cell depletion therapies, preserve B cell viability and survival11,44,155. In a phase I clinical trial, administration of a BTK inhibitor to healthy individuals was followed by a transient increase in circulating B cell counts156. Such changes are a class effect of drugs that target BCR signalling and are offset over time by diminished maturation of B lymphocytes; CD19+ cell counts are reduced during chronic exposure to such agents85,157,158. The mechanism by which treatment with ibrutinib produces transient lymphocytosis and a sustained reduction in lymphadenopathy158 was investigated in primary cultures of malignant cells from patients with CLL157. BTK inhibitor treatment resolved the pathologically heightened (BCR-driven and chemokine-driven) signalling processes that control integrin-mediated retention and homing of malignant B cells in lymph node and bone marrow microenvironments157.
Further insight into the functional consequences of BTK inhibition was gained from preclinical (in vitro) and phase I (in vivo) pharmacodynamic studies of the BTK inhibitor BIIB091 (ref. 156). BTK inhibition suppressed several early signalling events and downstream cellular effector functions in B cells and myeloid cells: BCR-induced phosphorylation of PLCγ2 and antigen presentation to T cells; activation, proliferation and differentiation of B cells; and Fc receptor-triggered ROS generation by neutrophils, TNF secretion by monocytes and basophil degranulation156. Broad suppression of BTK-dependent inflammatory actions in multiple immune cell types, including B cells, basophils, neutrophils, monocytes and microglia, was also reported in an early in vitro study of tolebrutinib (results available only in abstract form)159.
Studies in mice with experimental autoimmune encephalomyelitis (EAE), a model of MS, suggest that B cells could be an essential therapeutic target of BTK inhibitors in MS. Both evobrutinib and tolebrutinib ameliorate disease severity in a dose-dependent manner in B cell-dependent and T cell-dependent EAE models (results available only as an abstract)44,159,160,161. In mice with B cell-dependent EAE, BTK inhibitor-induced reductions in disease severity occurred alongside marked changes in B cell function, including suppression of antigen-driven B cell activation and maturation, suppression of BCR-triggered pro-inflammatory cytokine secretion, reduced infiltration of B cells into the CNS and attenuation of B cell-dependent antigen presentation, which is required for the development of encephalitogenic T cells44.
The mechanism underlying BTK inhibitor-induced improvements in T cell-dependent EAE is less fully understood, although some evidence suggests that B cells are involved. Evobrutinib treatment of mice with T cell-dependent EAE resulted in reduced leptomeningeal inflammation, which was visualized with post-contrast, ultrahigh field MRI; however, histological and immunohistochemical examination of the inflamed leptomeninges revealed that the proportions of B cells were more consistently reduced by evobrutinib treatment than were the proportions of T cells, whereas those of myeloid cells were unchanged162. This finding is consistent with cell culture experiments, in which BTK inhibitors suppressed antigen-specific T cell proliferative and pro-inflammatory responses only in the presence of B cells. Thus, BTK inhibitors probably target interactions between B cells and T cells163.
Macrophages
Microglia and macrophages are thought to have key roles in the pathogenesis of MS, especially in promoting immune infiltration, demyelination and neurodegeneration and thereby driving disease progression7,164 (Box 2). Early data from an in vitro study of macrophages from mice with XID demonstrated that these cells had reduced pro-inflammatory function coupled with an increased tendency to undergo apoptosis. The potential effect of BTK depletion on macrophages was further supported in XID mice with TH1-dependent EAE. Slower disease induction and reduced disease severity were observed in these mice compared with wild-type mice165. To further investigate possible B cell-independent mechanisms of action for evobrutinib, the effects of this agent were assessed in an in vitro study of monocytes and macrophages. Evobrutinib promoted apoptosis of pro-inflammatory macrophages that had been caused to differentiate by treatment with granulocyte-macrophage colony-stimulating factor. Furthermore, when treated simultaneously with granulocyte-macrophage colony-stimulating factor and evobrutinib, these macrophages became skewed towards an anti-inflammatory phenotype characterized by increased uptake of apoptotic cells (results available only in abstract form)166.
Microglia
Evidence from mouse studies also provides support for important in vivo effects of BTK inhibition on microglia. In a study of BtkE41K knock-in mice, which produce a constitutively active variant of BTK, the amplification of microglial proliferation in these mice was inhibited by treatment with a small-molecule BTK inhibitor167. Another study reported reduced EAE severity in mice with conditional (tamoxifen-inducible) inactivation of BTK pointing to a role of BTK in long-lived, tissue-resident myeloid cells, including microglia (results available only in non-peer-reviewed form so far)160.
In mice, fenebrutinib administration starting 2 days before EAE induction by immunization with myelin oligodendrocyte glycoprotein (MOG35–55) led to reduced disease severity scores compared with those in EAE mice that did not receive fenebrutinib. This benefit was associated with reduced microglial activation, as assessed by immunohistochemical staining for the myeloid marker IBA1 in the spinal cord (results available only in abstract form)168. In mice with cuprizone-induced demyelination, immunohistochemistry studies revealed that BTK colocalized with IBA1 in brain tissue. Treatment of these mice with the tolebrutinib tool compound PRN2675 (a derivative of tolebrutinib with a single fluorine substituted for a proton on the reactive moiety) revealed a BTK-dependent transcriptional signature in which interferon signalling was among the most profoundly altered pathways (results available only in abstract form)169. In mice with MOG35–55-induced EAE, several genes linked to disease-associated microglia were upregulated after EAE induction. Treatment with PRN2675 led to reduced expression of these genes as well as reduced disease scores (results available only in abstract form)170. Furthermore, in mice with EAE induced by passive transfer of encephalitogenic T cells, pre-treatment with evobrutinib reversed the upregulation of several disease-associated molecules involved in activation and antigen presentation by microglia (results available only in abstract form)171.
Decreased microglial activation was also observed in evobrutinib-treated primary mouse microglia exposed in vitro to cytokine and TLR stimulation171. In another in vitro study, a BTK-dependent transcriptional signature was revealed in IgG-stimulated primary mouse microglia139. Treatment of these cells with tolebrutinib altered microglial gene expression and was associated with the downregulation of pro-inflammatory genes (results available only in abstract form)139,140.
A transcriptomic study identified a BTK-dependent mRNA expression signature in cultured human-induced pluripotent stem cell-derived microglia and in a tri-culture system consisting of human neurons, astrocytes and microglia140. After pro-inflammatory signalling was activated in the induced pluripotent stem cell-derived microglia by FcγR stimulation, treatment with tolebrutinib modulated the expression of several genes encoding pro-inflammatory cytokines and chemokines and led to a dose-dependent inhibition of Fc receptor-driven TNF production140. Inflammatory signalling was also reduced by BTK inhibition in the tri-culture system140. Taken together, the RNA sequencing data indicate that modulation of BTK activity in these cells affects several neuroinflammatory pathways relevant to MS140. However, it is important to consider that these results have not yet been peer-reviewed. Similarities and differences in findings between the microglial monoculture and tri-culture systems are currently being investigated.
Protecting and restoring myelin
In addition to the effects of BTK inhibition on the immune system, evidence is beginning to emerge that this approach has potential beneficial effects on tissue protection and repair. In a mouse model of cortical demyelination induced by administration of recombinant antibodies derived from patients with MS and human complement components, pretreatment with the tolebrutinib-like compound PRN2675 inhibited myelin degradation and the migration of microglia to sites of demyelination and also prevented the loss of myelin and oligodendrocytes (results available only in abstract form)172. Treatment with another experimental BTK inhibitor, BTKi-1 (which has properties similar to evobrutinib), promoted myelin repair in ex vivo mouse cerebellar organotypic slice cultures as well as in vivo in transgenic MBP-GFP-NTR Xenopus tadpoles, two complementary experimental models of demyelination and remyelination that lack adaptive immune cells52. Importantly, evobrutinib achieved these effects in systems in which the BBB is not entirely intact. Therefore, whether the BTK inhibitor concentrations used in these models (ranging between 50 nM and 1 μM)52 can be achieved in the brains of patients with MS remains unclear.
Clinical trials of BTK inhibitors in MS
Currently, the safety and efficacy of five BTK inhibitors are being evaluated in patients with RMS or PMS: evobrutinib, fenebrutinib, remibrutinib and tolebrutinib in phase III trials and orelabrutinib in a phase II trial (Table 1).
Pharmacokinetics and pharmacodynamics
BTK inhibitors are small-molecule agents that have two key therapeutic advantages compared with large molecules: they can be taken orally and have good intracellular access47. Molecules being actively pursued in current phase II and phase III trials for MS include agents with irreversible (evobrutinib, orelabrutinib, remibrutinib and tolebrutinib) and reversible (fenebrutinib) modes of binding173,174. Irreversible BTK inhibitors establish a covalent bond with cysteine 481, a residue in the ATP-binding site of BTK. Reversible inhibitors non-covalently and weakly bind to a specific pocket in the SH3 domain of BTK through hydrogen bonds, ionic bonds or hydrophobic interactions, thereby causing BTK to take up an inactive conformation173. In general, irreversible covalent BTK inhibitors achieve higher binding affinities and have longer durations of action than do reversible inhibitors, which might increase their potency and potentially lower the required dose level and/or frequency175. However, irreversible inhibitors can pose safety challenges related to the potential immunogenicity of covalently bound drug–receptor complexes and their degradation products175. Second-generation BTK inhibitors offer the advantages of high selectivity for a small number of kinases, which improves their toxicity profiles.
Table 2 summarizes the pharmacokinetic and pharmacodynamic data for BTK inhibitors under investigation in clinical trials, as well as important differences in their selectivity, mode of binding, target occupancy and CNS penetrance. Although the covalent BTK inhibitors evobrutinib and tolebrutinib irreversibly inactivate BTK, signalling is restored in 5–7 days as the inactivated enzyme is gradually replaced through normal protein turnover151,152. Fenebrutinib inhibits BTK for a relatively short period of time (time to dissociation 18 h in vitro) compared with other reversible BTK inhibitors176.
The efficacy of different BTK inhibitors in patients with MS is likely to be influenced by pharmacodynamic and pharmacokinetic variation (Table 2). Current data indicate that the potency of BTK inhibitors varies considerably: for example, evobrutinib requires higher doses to reach half-maximal inhibitory concentration (IC50), a quantitative index of inhibitory strength, than do tolebrutinib and fenebrutinib67. However, it is important to keep in mind that IC50 values are affected by the biochemical and cellular assays used to derive them and do not always predict the potency of a compound in a cellular context66,174. Use of an alternative measure, the half-maximal effective concentration (EC50) required to suppress B cell and basophil activation in fresh whole-blood samples from healthy individuals, shows that the potency of fenebrutinib was slightly higher than that of tolebrutinib (EC50 15 nM versus 80 nM, respectively) and far greater than that of evobrutinib (EC50 1,271 nM)177. One caveat to consider when comparing IC50 and EC50 values for reversible (fenebrutinib) and irreversible (evobrutinib and tolebrutinib) BTK inhibitors is that the irreversible inhibitors have non-equilibrium binding kinetics, which makes comparisons of the two types of inhibitors problematic174.
In the context of MS, brain penetrance is another key property of BTK inhibitors. Early evidence indicates that BTK inhibitors have dissimilar CNS penetrance. Tolebrutinib shows increased CNS penetrance, higher potency and faster reaction rates of BTK inhibition compared with both evobrutinib and fenebrutinib178. For evobrutinib, cerebrospinal fluid (CSF) concentrations measured at 2–3 h post-dose were lower than the IC50 of the compound, suggesting that therapeutic concentrations were not reached (although efficacy studies would be necessary to confirm this)179. By contrast, CSF concentrations of tolebrutinib exceeded the IC90 2 h after administration of a single 120-mg dose in healthy human volunteers in a phase I double-blind, placebo-controlled study151,180. Moreover, the CSF concentration of tolebrutinib might not yet have peaked at the 2-h post-dose time point because this agent reaches its maximum plasma concentration in ~1 h and has a half-life of 90–120 min. Thus, full characterization of the CSF pharmacokinetics of tolebrutinib requires additional measurements taken over a longer duration151. The reader should nonetheless keep in mind that these results were reported only at conferences, and a full peer-reviewed publication is not yet available as of the time of writing.
Safety and tolerability
The toxicity profile of BTK inhibitors is influenced by whether and to which other kinases they bind in addition to BTK (‘off-target’ effects), as well as ‘on-target’ unwanted effects resulting from modulation of the immune system (such as the increased risk of fungal infections previously reported in patients with CLL receiving ibrutinib and acalabrutinib)181,182. Compared with first-generation BTK inhibitors such as ibrutinib (which also bind to other TEC family kinases and other tyrosine kinases), the second-generation BTK inhibitors currently being investigated as treatments for MS bind to only a small number of other kinases (Table 2). This improved selectivity is anticipated to result in fewer off-target effects and a lower risk of cardiotoxicity and commonly observed adverse events, such as cardiac arrhythmias, hypertension, haemorrhage, diarrhoea, arthralgias and rash, that might necessitate treatment discontinuation66,67,173,183,184. The improved safety profile of second-generation BTK inhibitors is thought to reflect decreased off-target inhibition of molecules such as Janus kinase 3, epidermal growth factor receptor and potentially other TEC family members67,184. Of note, tolebrutinib has lower selectivity for BTK than either fenebrutinib or orelabrutinib66.
Phase II safety and efficacy data have been published only for evobrutinib and tolebrutinib185,186. In a phase II, double-blind trial of evobrutinib, 267 adults with active RRMS or SPMS with superimposed relapses were randomly allocated to one of the five treatment groups: evobrutinib 25 mg once daily, evobrutinib 75 mg once daily, evobrutinib 75 mg twice daily, placebo or open-label dimethyl fumarate 240 mg twice daily for 24 weeks185. At week 25, the patients who had received placebo were switched to evobrutinib 25 mg once daily for an additional 24 weeks, whereas those already receiving evobrutinib or dimethyl fumarate continued to receive the originally assigned treatment.
The most common adverse events related to evobrutinib treatment included nasopharyngitis and increased blood levels of liver enzymes and lipase185. By week 52, the groups receiving evobrutinib 75 mg once daily (66%) and 75 mg twice daily (63%) had experienced higher rates of adverse events than did the groups receiving evobrutinib 25 mg (54%) or placebo (56%). Rates of grade 3–4 adverse events were lowest in the evobrutinib 25 mg group (2%) and were similar among all other groups (11–15%). The highest rate of serious adverse events occurred in patients treated with evobrutinib 75 mg twice daily (7% versus 4% in all other groups)185. Five (9%) patients in the placebo group and two (4%) in the dimethyl fumarate group discontinued the study owing to an adverse event. Most of the 16 treatment discontinuations in the evobrutinib groups were due to asymptomatic and reversible increases in blood liver enzyme levels or to hepatobiliary disorders185.
Patients who completed the double-blind phase IIb study were eligible to enrol in an open-label extension from week 48 onwards, in which they initially received evobrutinib 75 mg once daily before switching to evobrutinib 75 mg twice daily after a mean of 49.8 ± 6.17 weeks (results available only in abstract form)187. Overall, 80% of phase IIb participants entered the open-label extension, of whom 77% completed ≥132 weeks of open-label evobrutinib treatment. Mean CD19+ B cell concentrations decreased (from 0.218 × 106 cells/ml at initiation of open-label evobrutinib to 0.122 × 106 cells/ml at week 96 of this treatment) in all participants in the open-label extension who had been randomly allocated to evobrutinib treatment during the phase IIb study (results available only in abstract form)188. The data for the first 2.5 years of the open-label extension indicated no additional concerns regarding elevations in blood levels of either liver enzymes or lipase. Elevated levels of alanine aminotransferase and aspartate aminotransferase over this period were reported only in patients who had received dimethyl fumarate or evobrutinib 25 mg once daily in the double-blind study, and they were all recorded within the first 12 weeks of the open-label extension (that is, while receiving evobrutinib 75 mg once daily). The incidence of elevated blood levels of lipase over the first 2.5 years of the open-label extension (11%) was similar to that during the double-blind period for patients receiving evobrutinib 75 mg (9% in both the once-daily and twice-daily treatment groups)185, and no clinical signs or symptoms were reported among individuals in the open-label extension (results available only in abstract form)188.
The safety of tolebrutinib was evaluated in a phase IIb, double-blind, crossover, dose-finding study in 130 adults with RRMS or relapsing SPMS186. These patients were randomly allocated to receive tolebrutinib once daily at doses of 5, 15, 30 or 60 mg for 12 weeks. This treatment phase was either preceded or followed by a 4-week placebo phase. Across all groups, 54% of the patients experienced adverse events, but their occurrence was not dose-dependent. The most frequent adverse events during tolebrutinib treatment were headache (7%), upper respiratory tract infection (5%) and nasopharyngitis (4%). Three patients had elevated liver enzyme levels, including two (one in the 30-mg tolebrutinib group and the other in the 60-mg tolebrutinib group) with alanine aminotransferase concentrations more than threefold the upper limit of normal. One serious adverse event (severe MS relapse) was reported in a patient receiving 60 mg tolebrutinib. This treatment was not interrupted, and the patient recovered and completed the study. No patients discontinued treatment or withdrew from the study owing to an adverse event186.
Patients who completed the double-blind phase of the phase IIb study were eligible to enrol in an ongoing long-term safety extension study. These patients initially continued the same tolebrutinib dose that they had been receiving in the dose-finding study in a double-blind manner (part A). Subsequently, all patients received open-label tolebrutinib 60 mg daily (part B). After 18 months, 94% of the patients enrolled in the long-term safety study remained included, and the safety data continued to show favourable tolerability without the emergence of any new safety signals (results available only in abstract form)189.
Efficacy
In the 24-week randomized, double-blind phase II study of evobrutinib, only the patients who received 75 mg of evobrutinib daily showed substantially fewer gadolinium-enhancing lesions during weeks 12–24 compared with those who received placebo. The rate ratios (adjusted for the number of lesions of patients at baseline) versus placebo for the total numbers of lesions assessed through week 24 were 1.45 (95% CI 0.72–2.91, P = 0.32) with evobrutinib 25 mg daily, 0.30 (0.14–0.63, P = 0.005) with evobrutinib 75 mg daily and 0.44 (0.21–0.93, P = 0.06) with evobrutinib 75 mg twice daily185.
The annualized relapse rate did not differ between patients in the evobrutinib and placebo groups: at week 24, 77% of the placebo-treated patients remained relapse-free, compared with 74% of those in the evobrutinib 25 mg group; 88% of those receiving evobrutinib 75 mg once daily; 87% of those receiving evobrutinib 75 mg twice daily; and 89% of those receiving dimethyl fumarate. Median Expanded Disability Status Scale (EDSS) scores also remained unchanged at weeks 24 and 48 in all groups. In the open-label extension study, however, switching from evobrutinib 75 mg once daily to 75 mg twice daily was associated with a reduction in the annualized relapse rate regardless of the treatment received during the double-blinded phase of the study. For the pooled patient population, overall EDSS scores and the mean number of gadolinium-enhancing lesions remained low from week 0 to week 192 (results available only in abstract form)187. Moreover, evobrutinib reduced neurofilament light chain levels in a dose-dependent manner during the double-blinded phase of the study, and these reduced levels were maintained up to week 144 (results available only as conference presentation or abstract)190.
Results of the phase IIb study of tolebrutinib showed a dose-dependent decrease in the number of gadolinium-enhancing lesions. The 60-mg tolebrutinib dose provided an 85% (95% CI 28–97, P = 0.03) reduction in new lesion formation versus placebo; 90% of the patients in the 60-mg tolebrutinib group had no new lesions after 12 weeks of treatment, whereas 75% of the patients had no new lesions after 4 weeks of placebo. A dose–response relationship was also found for the number of new or enlarging T2 lesions: the 60-mg dose provided an 89% (95% CI 68–96, P < 0.01) reduction in the number of new or enlarging T2 lesions versus placebo186. Both efficacy and safety findings in the 12 weeks of the core study were consistent between the overall population and the subgroup of patients who had highly active disease191. In the overall population, new gadolinium-enhancing lesion counts remained low in the 60-mg tolebrutinib group and decreased in the groups receiving other doses of tolebrutinib by week 48 of the 72-week long-term safety study (by which time all patients had transitioned to 60 mg tolebrutinib). At week 72, 84.7% of the patients remained relapse-free, and the annualized relapse rate was low (0.17; 95% CI 0.11–0.27). EDSS scores also remained stable over the first 18 months of the long-term safety study (results available only as conference presentation or abstract)189.
Conclusions
A growing body of mechanistic data indicates that, in contrast to currently approved disease-modifying therapies for MS, BTK inhibitors have the potential to target progression in patients with RMS and PMS. BTK inhibitors also satisfy several criteria that are considered to be important in the treatment of PMS11: BTK inhibitors are small molecules that can cross the BBB, thereby enabling these agents to access cells of the adaptive and innate immune system concurrently in the periphery and CNS. BTK inhibitors might dampen the pro-inflammatory pathogenic potential of innate immune cells by shifting them towards a more immunoregulatory phenotype and/or might promote the protection and repair of myelin. Additionally, BTK inhibitors could have potential advantages over B cell-depleting monoclonal antibodies because they modulate myeloid cell function44,45,192,193. However, whether the efficacy of BTK inhibitors exceeds that of B cell-depleting monoclonal antibodies is yet to be determined193. Unfortunately, many of the preclinical data describing the effects of BTK inhibitors on microglia have been presented only in abstract form and not yet formally peer-reviewed. These results should, therefore, be considered with caution. Moreover, it remains unclear whether the high concentrations of BTK inhibitors used in vitro are achievable in vivo in the brains of patients with MS.
Another possible drawback of B cell-depleting therapy is that patients receiving these agents are chronically deprived of regulatory B cells, which have potential benefits in MS194. Whether BTK inhibitor therapy can address this issue is uncertain, as ibrutinib treatment reduces the production of (immunosuppressive) adenosine in regulatory B cells from patients with cancer195. Although the reversibility of the effects of BTK inhibitor treatment holds much promise, these agents are not expected to be able to eradicate antigen-experienced (including autoreactive) adaptive immune cells. However, this apparent limitation potentially provides an elegant opportunity to maintain the effects of immune cell-depleting therapies when BTK inhibitors are administered during the repopulation phase after cessation of cell-depleting therapy196,197.
To date, two phase II trials have shown that BTK inhibitors limit disease activity in patients with RMS and have favourable tolerability and safety profiles185,186. Although the severe adverse effects of BTK inhibitors seen in patients with haematological malignancies have not yet been reported in individuals with MS, the small sample sizes and short observation periods of the available trials do not enable a comprehensive evaluation of these aspects. The safety profiles of BTK inhibitors might also vary between MS and other diseases targeted with these agents owing to the presence of differing comorbidities and pathophysiological processes198. Several clinical trials are underway that are expected to provide more robust data on the long-term efficacy and safety of BTK inhibitors in patients with MS. We anticipate that differences in their pharmacological properties (such as pharmacodynamics, selectivity and CNS penetration) could lead to differences in their efficacy and safety profiles in phase III trials as well as in clinical practice66,184.
Future mechanistic studies are needed to further investigate whether BTK inhibitors offer any additional benefits relevant to patients with MS, in particular the alleviation of oxidative stress, scavenging of iron and promotion of remyelination11. Studies to determine the effect of the different attributes of the various types of BTK inhibitors will also be of interest; for example, studies to investigate whether the less-frequent dosing required for covalent BTK inhibitors translates to improved medication adherence199.
A further possibility that could be explored in future clinical trials is a sequential approach, in which initial treatment with one of the currently approved antibody therapies is followed by treatment with a BTK inhibitor47. To determine which order of administration is the most effective, clinical trials will be necessary. Of note, in patients with malignancies, BTK inhibitor treatment triggers large-scale egress of lymphocytes from the spleen and lymph nodes to the peripheral blood and impairs their tissue homing. The resultant extensive pro-B cell lymphocytosis enables anti-CD20 therapies to kill neoplastic cells that would otherwise remain inaccessible (also termed ‘occult’) within secondary lymphoid organs200. Thus, the oncological approach of induction with a combination of an anti-CD20 agent plus a BTK inhibitor could provide additional benefit derived from the BTK inhibitor-induced egress of pathogenic B cells, which exposes them to anti-CD20-mediated cytolysis and thereby culls these clones from the bone marrow and/or secondary lymphoid niche. Another important consideration is that although B cells and microglia are important therapeutic targets of BTK inhibitors, the contributions of these cells to the pathophysiology of MS have not been fully elucidated. For instance, the processes that drive microglia to adopt either detrimental or beneficial phenotypes in MS require further investigation163,201. Current data show that microglia are involved in regulating tissue homeostasis under physiological conditions and can have pro-inflammatory, anti-inflammatory and potentially regenerative roles in the injured CNS30 (Box 2).
The interaction between B cells and T cells is another process implicated in the pathophysiology of MS that can be therapeutically targeted by BTK inhibitors6,163. Besides their pathogenic properties, B cells (or specific B cell subpopulations) are probably able to execute immunologically counterbalancing functions that restrict tissue inflammation and the pro-inflammatory activation of other immune cells194. A prominent challenge of future research will be to further unravel the response patterns of B cells and other immune cells, particularly microglia, in different contexts; for instance, microglial activation under BTK inhibition could be imaged in vivo with positron emission tomography and radioligands202. Such insights could enable the beneficial properties of the immune system to be harnessed while at the same time inhibiting harmful reactions201.
References
Reich, D. S., Lucchinetti, C. F. & Calabresi, P. A. Multiple sclerosis. N. Engl. J. Med. 378, 169–180 (2018).
Faissner, S., Plemel, J. R., Gold, R. & Yong, V. W. Progressive multiple sclerosis: from pathophysiology to therapeutic strategies. Nat. Rev. Drug Discov. 18, 905–922 (2019).
Dendrou, C. A., Fugger, L. & Friese, M. A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 15, 545–558 (2015).
Krämer, J. et al. Imaging in mice and men: pathophysiological insights into multiple sclerosis from conventional and advanced MRI techniques. Prog. Neurobiol. 182, 101663 (2019).
Baecher-Allan, C., Kaskow, B. J. & Weiner, H. L. Multiple sclerosis: mechanisms and immunotherapy. Neuron 97, 742–768 (2018).
Bar-Or, A. & Li, R. Cellular immunology of relapsing multiple sclerosis: interactions, checks, and balances. Lancet Neurol. 20, 470–483 (2021).
Healy, L. M., Stratton, J. A., Kuhlmann, T. & Antel, J. The role of glial cells in multiple sclerosis disease progression. Nat. Rev. Neurol. 18, 237–248 (2022).
Ruiz, F., Vigne, S. & Pot, C. Resolution of inflammation during multiple sclerosis. Semin. Immunopathol. 41, 711–726 (2019).
DiSabato, D. J., Quan, N. & Godbout, J. P. Neuroinflammation: the devil is in the details. J. Neurochem. 139, 136–153 (2016).
Minagar, A., Maghzi, A. H., McGee, J. C. & Alexander, J. S. Emerging roles of endothelial cells in multiple sclerosis pathophysiology and therapy. Neurol. Res. 34, 738–745 (2012).
Yong, H. Y. F. & Yong, V. W. Mechanism-based criteria to improve therapeutic outcomes in progressive multiple sclerosis. Nat. Rev. Neurol. 18, 40–55 (2022).
Dangond, F. et al. Facing the urgency of therapies for progressive MS — a progressive MS alliance proposal. Nat. Rev. Neurol. 17, 185–192 (2021).
Lassmann, H. Pathogenic mechanisms associated with different clinical courses of multiple sclerosis. Front. Immunol. 9, 3116 (2018).
Magliozzi, R. et al. Meningeal B-cell follicles in secondary progressive multiple sclerosis associate with early onset of disease and severe cortical pathology. Brain 130, 1089–1104 (2007).
Choi, S. R. et al. Meningeal inflammation plays a role in the pathology of primary progressive multiple sclerosis. Brain 135, 2925–2937 (2012).
Absinta, M. et al. Persistent 7-tesla phase rim predicts poor outcome in new multiple sclerosis patient lesions. J. Clin. Invest. 126, 2597–2609 (2016).
Kuhlmann, T. et al. An updated histological classification system for multiple sclerosis lesions. Acta Neuropathol. 133, 13–24 (2017).
Lassmann, H. The contribution of neuropathology to multiple sclerosis research. Eur. J. Neurol. 29, 2869–2877 (2022).
Lassmann, H., Raine, C. S., Antel, J. & Prineas, J. W. Immunopathology of multiple sclerosis: report on an international meeting held at the Institute of Neurology of the University of Vienna. J. Neuroimmunol. 86, 213–217 (1998).
Calvi, A. et al. In vivo imaging of chronic active lesions in multiple sclerosis. Mult. Scler. 28, 683–690 (2022).
Dal-Bianco, A. et al. Slow expansion of multiple sclerosis iron rim lesions: pathology and 7 T magnetic resonance imaging. Acta Neuropathol. 133, 25–42 (2017).
Absinta, M. et al. A lymphocyte–microglia–astrocyte axis in chronic active multiple sclerosis. Nature 597, 709–714 (2021).
Jackle, K. et al. Molecular signature of slowly expanding lesions in progressive multiple sclerosis. Brain 143, 2073–2088 (2020).
Elliott, C. et al. Slowly expanding/evolving lesions as a magnetic resonance imaging marker of chronic active multiple sclerosis lesions. Mult. Scler. 25, 1915–1925 (2019).
Preziosa, P. et al. Slowly expanding lesions predict 9-year multiple sclerosis disease progression. Neurol. Neuroimmunol. Neuroinflamm. 9, e1139 (2022).
Luchetti, S. et al. Progressive multiple sclerosis patients show substantial lesion activity that correlates with clinical disease severity and sex: a retrospective autopsy cohort analysis. Acta Neuropathol. 135, 511–528 (2018).
Frischer, J. M. et al. Clinical and pathological insights into the dynamic nature of the white matter multiple sclerosis plaque. Ann. Neurol. 78, 710–721 (2015).
Absinta, M. & Dal-Bianco, A. Slowly expanding lesions are a marker of progressive MS — yes. Mult. Scler. 27, 1679–1681 (2021).
Simmons, S. B. & Ontaneda, D. Slowly expanding lesions: a new target for progressive multiple sclerosis trials. Neurology 98, 699–700 (2022).
Guerrero, B. L. & Sicotte, N. L. Microglia in multiple sclerosis: friend or foe? Front. Immunol. 11, 374 (2020).
Hauser, S. L. et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N. Engl. J. Med. 376, 221–234 (2017).
Montalban, X. et al. Ocrelizumab versus placebo in primary progressive multiple sclerosis. N. Engl. J. Med. 376, 209–220 (2017).
Hauser, S. L. et al. Ofatumumab versus teriflunomide in multiple sclerosis. N. Engl. J. Med. 383, 546–557 (2020).
Hauser, S. L. et al. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 358, 676–688 (2008).
Hawker, K. et al. Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann. Neurol. 66, 460–471 (2009).
Gelfand, J. M., Cree, B. A. C. & Hauser, S. L. Ocrelizumab and other CD20(+) B-cell-depleting therapies in multiple sclerosis. Neurotherapeutics 14, 835–841 (2017).
Bhargava, P. et al. Trial of intrathecal rituximab in progressive multiple sclerosis patients with evidence of leptomeningeal contrast enhancement. Mult. Scler. Relat. Disord. 30, 136–140 (2019).
Komori, M. et al. Insufficient disease inhibition by intrathecal rituximab in progressive multiple sclerosis. Ann. Clin. Transl. Neurol. 3, 166–179 (2016).
Li, R., Patterson, K. R. & Bar-Or, A. Reassessing B cell contributions in multiple sclerosis. Nat. Immunol. 19, 696–707 (2018).
Bonnan, M. et al. No early effect of intrathecal rituximab in progressive multiple sclerosis (EFFRITE Clinical Trial). Mult. Scler. Int. 2021, 8813498–707 (2021).
Torke, S. & Weber, M. S. Inhibition of Bruton’s tyrosine kinase as a novel therapeutic approach in multiple sclerosis. Exp. Opin. Investig. Drugs 29, 1143–1150 (2020).
Brunner, C., Müller, B. & Wirth, T. Bruton’s tyrosine kinase is involved in innate and adaptive immunity. Histol. Histopathol. 20, 945–955 (2005).
Rip, J., Van Der Ploeg, E. K., Hendriks, R. W. & Corneth, O. B. J. The role of Bruton’s tyrosine kinase in immune cell signaling and systemic autoimmunity. Crit. Rev. Immunol. 38, 17–62 (2018).
Torke, S. et al. Inhibition of Bruton’s tyrosine kinase interferes with pathogenic B-cell development in inflammatory CNS demyelinating disease. Acta Neuropathol. 140, 535–548 (2020).
Carnero Contentti, E. & Correale, J. Bruton’s tyrosine kinase inhibitors: a promising emerging treatment option for multiple sclerosis. Exp. Opin. Emerg. Drugs 25, 377–381 (2020).
Smith, C. I. et al. Expression of Bruton’s agammaglobulinemia tyrosine kinase gene, BTK, is selectively down-regulated in T lymphocytes and plasma cells. J. Immunol. 152, 557–565 (1994).
Garcia-Merino, A. Bruton’s tyrosine kinase inhibitors: a new generation of promising agents for multiple sclerosis therapy. Cells 10, 2560 (2021).
Ellmeier, W., Abramova, A. & Schebesta, A. Tec family kinases: regulation of FcεRI-mediated mast-cell activation. FEBS J. 278, 1990–2000 (2011).
Hata, D. et al. Involvement of Bruton’s tyrosine kinase in FcεRI-dependent mast cell degranulation and cytokine production. J. Exp. Med. 187, 1235–1247 (1998).
Iwaki, S. et al. Btk plays a crucial role in the amplification of FcεRI-mediated mast cell activation by kit. J. Biol. Chem. 280, 40261–40270 (2005).
Yang, E. J., Yoon, J. H. & Chung, K. C. Bruton’s tyrosine kinase phosphorylates cAMP-responsive element-binding protein at serine 133 during neuronal differentiation in immortalized hippocampal progenitor cells. J. Biol. Chem. 279, 1827–1837 (2004).
Martin, E. et al. Bruton’s tyrosine kinase inhibition promotes myelin repair. Brain Plast. 5, 123–133 (2020).
Yu, C. G. et al. Inhibition of Bruton tyrosine kinase reduces neuroimmune cascade and promotes recovery after spinal cord injury. Int. J. Mol. Sci. https://doi.org/10.3390/ijms23010355 (2021).
Bruton, O. C. Agammaglobulinemia. Pediatrics 9, 722–728 (1952).
Vetrie, D. et al. The gene involved in X-linked agammaglobulinaemia is a member of the src family of protein-tyrosine kinases. Nature 361, 226–233 (1993).
Tsukada, S. et al. Deficient expression of a B cell cytoplasmic tyrosine kinase in human X-linked agammaglobulinemia. Cell 72, 279–290 (1993).
Hendriks, R. W., Yuvaraj, S. & Kil, L. P. Targeting Bruton’s tyrosine kinase in B cell malignancies. Nat. Rev. Cancer 14, 219–232 (2014).
Mahajan, S. et al. Rational design and synthesis of a novel anti-leukemic agent targeting Bruton’s tyrosine kinase (BTK), LFM-A13 [alpha-cyano-beta-hydroxy-beta-methyl-N-(2,5-dibromophenyl)propenamide]. J. Biol. Chem. 274, 9587–9599 (1999).
Kiss, R. et al. Identification of a novel inhibitor of JAK2 tyrosine kinase by structure-based virtual screening. Bioorg. Med. Chem. Lett. 19, 3598–3601 (2009).
van den Akker, E. et al. The Btk inhibitor LFM-A13 is a potent inhibitor of Jak2 kinase activity. Biol. Chem. 385, 409–413 (2004).
Pan, Z. et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem 2, 58–61 (2007).
Wen, T., Wang, J., Shi, Y., Qian, H. & Liu, P. Inhibitors targeting Bruton’s tyrosine kinase in cancers: drug development advances. Leukemia 35, 312–332 (2021).
Jansset Biotech, Inc. Imbruvica (Ibrutinib) Capsules and Tablets [Prescribing Information] (Jansset Biotech, Inc., 2020).
AstraZeneca. Calquence (Acalabrutinib) Capsules [Prescribing Information] (AstraZeneca, 2019).
BeiGene USA, Inc. Brukinsa (Zanubrutinib) Capsules [Prescribing Information] (BeiGene USA, Inc., 2019).
Estupinan, H. Y., Berglof, A., Zain, R. & Smith, C. I. E. Comparative analysis of BTK inhibitors and mechanisms underlying adverse effects. Front. Cell Dev. Biol. 9, 630942 (2021).
Ringheim, G. E., Wampole, M. & Oberoi, K. Bruton’s tyrosine kinase (BTK) inhibitors and autoimmune diseases: making sense of BTK inhibitor specificity profiles and recent clinical trial successes and failures. Front. Immunol. 12, 662223 (2021).
Corneth, O. B. et al. Enhanced expression of Bruton’s tyrosine kinase in B cells drives systemic autoimmunity by disrupting T cell homeostasis. J. Immunol. 197, 58–67 (2016).
Kil, L. P. et al. Btk levels set the threshold for B-cell activation and negative selection of autoreactive B cells in mice. Blood 119, 3744–3756 (2012).
Mano, H. Tec family of protein-tyrosine kinases: an overview of their structure and function. Cytokine Growth Factor Rev. 10, 267–280 (1999).
Dickson, E. J. & Hille, B. Understanding phosphoinositides: rare, dynamic, and essential membrane phospholipids. Biochem. J. 476, 1–23 (2019).
Solvason, N. et al. Transgene expression of bcl-xL permits anti-immunoglobulin (Ig)-induced proliferation in xid B cells. J. Exp. Med. 187, 1081–1091 (1998).
Uckun, F. M. et al. BTK as a mediator of radiation-induced apoptosis in DT-40 lymphoma B cells. Science 273, 1096–1100 (1996).
Smith, C. I. et al. The Tec family of cytoplasmic tyrosine kinases: mammalian Btk, Bmx, Itk, Tec, Txk and homologs in other species. Bioessays 23, 436–446 (2001).
Ekman, N. et al. The BMX tyrosine kinase is activated by IL-3 and G-CSF in a PI-3K dependent manner. Oncogene 19, 4151–4158 (2000).
Schaeffer, E. M. et al. Requirement for Tec kinases Rlk and Itk in T cell receptor signaling and immunity. Science 284, 638–641 (1999).
Basile, N. et al. Clinical and molecular analysis of 49 patients with X-linked agammaglobulinemia from a single center in Argentina. J. Clin. Immunol. 29, 123–129 (2009).
Väliaho, J., Smith, C. I. & Vihinen, M. BTKbase: the mutation database for X-linked agammaglobulinemia. Hum. Mutat. 27, 1209–1217 (2006).
Rawlings, D. J. et al. Mutation of unique region of Bruton’s tyrosine kinase in immunodeficient XID mice. Science 261, 358–361 (1993).
Cancro, M. P. et al. xid mice reveal the interplay of homeostasis and Bruton’s tyrosine kinase-mediated selection at multiple stages of B cell development. Int. Immunol. 13, 1501–1514 (2001).
Thomas, J. D. et al. Colocalization of X-linked agammaglobulinemia and X-linked immunodeficiency genes. Science 261, 355–358 (1993).
Middendorp, S., Dingjan, G. M. & Hendriks, R. W. Impaired precursor B cell differentiation in Bruton’s tyrosine kinase-deficient mice. J. Immunol. 168, 2695–2703 (2002).
Ng, Y. S., Wardemann, H., Chelnis, J., Cunningham-Rundles, C. & Meffre, E. Bruton’s tyrosine kinase is essential for human B cell tolerance. J. Exp. Med. 200, 927–934 (2004).
Cariappa, A. et al. The follicular versus marginal zone B lymphocyte cell fate decision is regulated by Aiolos, BTK, and CD21. Immunity 14, 603–615 (2001).
Spaargaren, M. et al. The B cell antigen receptor controls integrin activity through Btk and PLCgamma2. J. Exp. Med. 198, 1539–1550 (2003).
Lunemann, J. D., Malhotra, S., Shinohara, M. L., Montalban, X. & Comabella, M. Targeting inflammasomes to treat neurological diseases. Ann. Neurol. 90, 177–188 (2021).
Schmitz, R., Baumann, G. & Gram, H. Catalytic specificity of phosphotyrosine kinases Blk, Lyn, c-Src and Syk as assessed by phage display. J. Mol. Biol. 260, 664–677 (1996).
Fujimoto, M. et al. CD19 regulates Src family protein tyrosine kinase activation in B lymphocytes through processive amplification. Immunity 13, 47–57 (2000).
Fütterer, K., Wong, J., Grucza, R. A., Chan, A. C. & Waksman, G. Structural basis for Syk tyrosine kinase ubiquity in signal transduction pathways revealed by the crystal structure of its regulatory SH2 domains bound to a dually phosphorylated ITAM peptide. J. Mol. Biol. 281, 523–537 (1998).
Rowley, R. B., Burkhardt, A. L., Chao, H. G., Matsueda, G. R. & Bolen, J. B. Syk protein-tyrosine kinase is regulated by tyrosine-phosphorylated Ig alpha/Ig beta immunoreceptor tyrosine activation motif binding and autophosphorylation. J. Biol. Chem. 270, 11590–11594 (1995).
Xu, Y., Fairfax, K., Light, A., Huntington, N. D. & Tarlinton, D. M. CD19 differentially regulates BCR signalling through the recruitment of PI3K. Autoimmunity 47, 430–437 (2014).
Okada, T., Maeda, A., Iwamatsu, A., Gotoh, K. & Kurosaki, T. BCAP: the tyrosine kinase substrate that connects B cell receptor to phosphoinositide 3-kinase activation. Immunity 13, 817–827 (2000).
Saito, K., Scharenberg, A. M. & Kinet, J. P. Interaction between the Btk PH domain and phosphatidylinositol-3,4,5-trisphosphate directly regulates BTK. J. Biol. Chem. 276, 16201–16206 (2001).
Haxhinasto, S. A. & Bishop, G. A. Synergistic B cell activation by CD40 and the B cell antigen receptor: role of B lymphocyte antigen receptor-mediated kinase activation and tumor necrosis factor receptor-associated factor regulation. J. Biol. Chem. 279, 2575–2582 (2004).
Ying, H., Li, Z., Yang, L. & Zhang, J. Syk mediates BCR- and CD40-signaling integration during B cell activation. Immunobiology 216, 566–570 (2011).
Tanwar, S. et al. Mediation of transitional B cell maturation in the absence of functional Bruton’s tyrosine kinase. Sci. Rep. 7, 46029 (2017).
Su, Y. W. et al. Interaction of SLP adaptors with the SH2 domain of Tec family kinases. Eur. J. Immunol. 29, 3702–3711 (1999).
Liu, W. S. & Heckman, C. A. The sevenfold way of PKC regulation. Cell. Signal. 10, 529–542 (1998).
Taylor, C. W. & Tovey, S. C. IP(3) receptors: toward understanding their activation. Cold Spring Harb. Perspect. Biol. 2, a004010 (2010).
Lin, X. & Wang, D. The roles of CARMA1, Bcl10, and MALT1 in antigen receptor signaling. Semin. Immunol. 16, 429–435 (2004).
David, L. et al. Assembly mechanism of the CARMA1-BCL10-MALT1-TRAF6 signalosome. Proc. Natl Acad. Sci. USA 115, 1499–1504 (2018).
Shinohara, H., Maeda, S., Watarai, H. & Kurosaki, T. IkappaB kinase beta-induced phosphorylation of CARMA1 contributes to CARMA1 Bcl10 MALT1 complex formation in B cells. J. Exp. Med. 204, 3285–3293 (2007).
Mohamed, A. J. et al. Bruton’s tyrosine kinase (BTK): function, regulation, and transformation with special emphasis on the PH domain. Immunol. Rev. 228, 58–73 (2009).
Berland, R. & Wortis, H. H. Normal B-1a cell development requires B cell-intrinsic NFATc1 activity. Proc. Natl Acad. Sci. USA 100, 13459–13464 (2003).
Peng, S. L., Gerth, A. J., Ranger, A. M. & Glimcher, L. H. NFATc1 and NFATc2 together control both T and B cell activation and differentiation. Immunity 14, 13–20 (2001).
Bhattacharyya, S. et al. NFATc1 affects mouse splenic B cell function by controlling the calcineurin–NFAT signaling network. J. Exp. Med. 208, 823–839 (2011).
Barrington, R. A., Borde, M., Rao, A. & Carroll, M. C. Involvement of NFAT1 in B cell self-tolerance. J. Immunol. 177, 1510–1515 (2006).
Gerondakis, S. & Siebenlist, U. Roles of the NF-kappaB pathway in lymphocyte development and function. Cold Spring Harb. Perspect. Biol. 2, a000182 (2010).
Teixeira, C., Stang, S. L., Zheng, Y., Beswick, N. S. & Stone, J. C. Integration of DAG signaling systems mediated by PKC-dependent phosphorylation of RasGRP3. Blood 102, 1414–1420 (2003).
Yasuda, T. et al. Erk kinases link pre-B cell receptor signaling to transcriptional events required for early B cell expansion. Immunity 28, 499–508 (2008).
Westerberg, L., Greicius, G., Snapper, S. B., Aspenström, P. & Severinson, E. Cdc42, Rac1, and the Wiskott–Aldrich syndrome protein are involved in the cytoskeletal regulation of B lymphocytes. Blood 98, 1086–1094 (2001).
O’Rourke, L. M. et al. CD19 as a membrane-anchored adaptor protein of B lymphocytes: costimulation of lipid and protein kinases by recruitment of Vav. Immunity 8, 635–645 (1998).
Eden, S., Rohatgi, R., Podtelejnikov, A. V., Mann, M. & Kirschner, M. W. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature 418, 790–793 (2002).
Chiu, C. W., Dalton, M., Ishiai, M., Kurosaki, T. & Chan, A. C. BLNK: molecular scaffolding through ‘cis’-mediated organization of signaling proteins. EMBO J. 21, 6461–6472 (2002).
Muta, T. et al. A 13-amino-acid motif in the cytoplasmic domain of Fc gamma RIIB modulates B-cell receptor signalling. Nature 368, 70–73 (1994).
Nishizumi, H., Horikawa, K., Mlinaric-Rascan, I. & Yamamoto, T. A double-edged kinase Lyn: a positive and negative regulator for antigen receptor-mediated signals. J. Exp. Med. 187, 1343–1348 (1998).
Jefferies, C. A. et al. Bruton’s tyrosine kinase is a Toll/interleukin-1 receptor domain-binding protein that participates in nuclear factor kappaB activation by Toll-like receptor 4. J. Biol. Chem. 278, 26258–26264 (2003).
Gray, P. et al. MyD88 adapter-like (Mal) is phosphorylated by Bruton’s tyrosine kinase during TLR2 and TLR4 signal transduction. J. Biol. Chem. 281, 10489–10495 (2006).
Lee, K. G. et al. Bruton’s tyrosine kinase phosphorylates Toll-like receptor 3 to initiate antiviral response. Proc. Natl Acad. Sci. USA 109, 5791–5796 (2012).
O’Neill, L. A. & Bowie, A. G. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat. Rev. Immunol. 7, 353–364 (2007).
Botos, I., Segal, D. M. & Davies, D. R. The structural biology of Toll-like receptors. Structure 19, 447–459 (2011).
Muroi, M. & Tanamoto, K. TRAF6 distinctively mediates MyD88- and IRAK-1-induced activation of NF-kappaB. J. Leukoc. Biol. 83, 702–707 (2008).
Rip, J. et al. Toll-like receptor signaling drives BTK-mediated autoimmune disease. Front. Immunol. 10, 95 (2019).
Kenny, E. F. et al. Bruton’s tyrosine kinase mediates the synergistic signalling between TLR9 and the B cell receptor by regulating calcium and calmodulin. PLoS ONE 8, e74103 (2013).
Kawakami, Y. et al. Tyrosine phosphorylation and activation of Bruton tyrosine kinase upon Fc epsilon RI cross-linking. Mol. Cell Biol. 14, 5108–5113 (1994).
Furumoto, Y., Nunomura, S., Terada, T., Rivera, J. & Ra, C. The FcϵRIβ immunoreceptor tyrosine-based activation motif exerts inhibitory control on MAPK and IκB kinase phosphorylation and mast cell cytokine production. J. Biol. Chem. 279, 49177–49187 (2004).
Simon, M., Vanes, L., Geahlen, R. L. & Tybulewicz, V. L. Distinct roles for the linker region tyrosines of Syk in FcepsilonRI signaling in primary mast cells. J. Biol. Chem. 280, 4510–4517 (2005).
Saitoh, S. et al. LAT is essential for Fc(epsilon)RI-mediated mast cell activation. Immunity 12, 525–535 (2000).
Gilfillan, A. M. & Tkaczyk, C. Integrated signalling pathways for mast-cell activation. Nat. Rev. Immunol. 6, 218–230 (2006).
Jongstra-Bilen, J. et al. Dual functions of Bruton’s tyrosine kinase and Tec kinase during Fcgamma receptor-induced signaling and phagocytosis. J. Immunol. 181, 288–298 (2008).
Liang, C. et al. The development of Bruton’s tyrosine kinase (BTK) inhibitors from 2012 to 2017: a mini-review. Eur. J. Med. Chem. 151, 315–326 (2018).
Pal Singh, S., Dammeijer, F. & Hendriks, R. W. Role of Bruton’s tyrosine kinase in B cells and malignancies. Mol. Cancer 17, 57 (2018).
Di Paolo, J. A. et al. Specific BTK inhibition suppresses B cell- and myeloid cell-mediated arthritis. Nat. Chem. Biol. 7, 41–50 (2011).
Kiefer, F. et al. The Syk protein tyrosine kinase is essential for Fcgamma receptor signaling in macrophages and neutrophils. Mol. Cell Biol. 18, 4209–4220 (1998).
Horwood, N. J. et al. Bruton’s tyrosine kinase is required for TLR2 and TLR4-induced TNF, but not IL-6, production. J. Immunol. 176, 3635–3641 (2006).
Doyle, S. L., Jefferies, C. A., Feighery, C. & O’Neill, L. A. Signaling by Toll-like receptors 8 and 9 requires Bruton’s tyrosine kinase. J. Biol. Chem. 282, 36953–36960 (2007).
Liu, X. et al. Intracellular MHC class II molecules promote TLR-triggered innate immune responses by maintaining activation of the kinase BTK. Nat. Immunol. 12, 416–424 (2011).
Rijvers, L. et al. Human T-bet + B cell development is associated with BTK activity and suppressed by evobrutinib. JCI Insight https://doi.org/10.1172/jci.insight.160909 (2022).
Gruber, R. et al. Central effects of BTK inhibition in neuroinflammation. Neurology 94, 808 (2020).
Gruber, R. et al. Evaluating the effect of BTK inhibitor tolebrutinib in human microglia. Mult. Scler. 27, P391 (2021).
Cui, L. Y., Chu, S. F. & Chen, N. H. The role of chemokines and chemokine receptors in multiple sclerosis. Int. Immunopharmacol. 83, 106314 (2020).
Kowarik, M. C. et al. CXCL13 is the major determinant for B cell recruitment to the CSF during neuroinflammation. J. Neuroinflammation 9, 93 (2012).
Ransohoff, R. M. & Perry, V. H. Microglial physiology: unique stimuli, specialized responses. Annu. Rev. Immunol. 27, 119–145 (2009).
Luo, C. et al. The role of microglia in multiple sclerosis. Neuropsychiatr. Dis. Treat. 13, 1661–1667 (2017).
Dong, Y. & Yong, V. W. When encephalitogenic T cells collaborate with microglia in multiple sclerosis. Nat. Rev. Neurol. 15, 704–717 (2019).
Kamma, E., Lasisi, W., Libner, C., Ng, H. S. & Plemel, J. R. Central nervous system macrophages in progressive multiple sclerosis: relationship to neurodegeneration and therapeutics. J. Neuroinflammation 19, 45 (2022).
Puthenparampil, M. et al. BAFF index and CXCL13 levels in the cerebrospinal fluid associate respectively with intrathecal IgG synthesis and cortical atrophy in multiple sclerosis at clinical onset. J. Neuroinflammation 14, 11 (2017).
Jain, R. W. & Yong, V. W. B cells in central nervous system disease: diversity, locations and pathophysiology. Nat. Rev. Immunol. 22, 513–524 (2021).
Arneth, B. M. Impact of B cells to the pathophysiology of multiple sclerosis. J. Neuroinflammation 16, 128 (2019).
Flach, A. C. et al. Autoantibody-boosted T-cell reactivation in the target organ triggers manifestation of autoimmune CNS disease. Proc. Natl Acad. Sci. USA 113, 3323–3328 (2016).
Owens, T. D. et al. Phase 1 clinical trial evaluating safety, exposure and pharmacodynamics of BTK inhibitor tolebrutinib (PRN2246, SAR442168). Clin. Transl. Sci. 15, 442–450 (2022).
Caldwell, R. D. et al. Discovery of evobrutinib: an oral, potent, and highly selective, covalent Bruton’s tyrosine kinase (BTK) inhibitor for the treatment of immunological diseases. J. Med. Chem. 62, 7643–7655 (2019).
Comi, G. et al. Role of B cells in multiple sclerosis and related disorders. Ann. Neurol. 89, 13–23 (2021).
Cencioni, M. T., Mattoscio, M., Magliozzi, R., Bar-Or, A. & Muraro, P. A. B cells in multiple sclerosis — from targeted depletion to immune reconstitution therapies. Nat. Rev. Neurol. 17, 399–414 (2021).
Nyhoff, L. E. et al. Bruton’s tyrosine kinase is not essential for B cell survival beyond early developmental stages. J. Immunol. 200, 2352–2361 (2018).
Bame, E. et al. Next-generation Bruton’s tyrosine kinase inhibitor BIIB091 selectively and potently inhibits B cell and Fc receptor signaling and downstream functions in B cells and myeloid cells. Clin. Transl. Immunol. 10, e1295 (2021).
de Rooij, M. F. et al. The clinically active BTK inhibitor PCI-32765 targets B-cell receptor- and chemokine-controlled adhesion and migration in chronic lymphocytic leukemia. Blood 119, 2590–2594 (2012).
Herman, S. E. et al. Ibrutinib-induced lymphocytosis in patients with chronic lymphocytic leukemia: correlative analyses from a phase II study. Leukemia 28, 2188–2196 (2014).
Francesco, M. et al. PRN2246, a potent and selective blood–brain barrier penetrating BTK inhibitor, exhibits efficacy in central nervous system immunity. Mult. Scler. 23, P989 (2017).
Bassani, C. et al. The role of human and mouse BTK in myeloid cells (P3-4.006). Neurology 98, 2707 (2022).
Boschert, U. et al. T cell mediated experimental CNS autoimmunity induced by PLP in SJL mice is modulated by evobrutinib (M2951): a novel Bruton’s tyrosine kinase inhibitor. Mult. Scler. 23, P678 (2017).
Bhargava, P. et al. Imaging meningeal inflammation in CNS autoimmunity identifies a therapeutic role for BTK inhibition. Brain 144, 1396–1408 (2021).
Li, R. et al. BTK inhibition limits B-cell–T-cell interaction through modulation of B-cell metabolism: implications for multiple sclerosis therapy. Acta Neuropathol. 143, 505–521 (2022).
Wang, J. et al. Targeting microglia and macrophages: a potential treatment strategy for multiple sclerosis. Front. Pharmacol. 10, 286 (2019).
Mangla, A. et al. Pleiotropic consequences of Bruton tyrosine kinase deficiency in myeloid lineages lead to poor inflammatory responses. Blood 104, 1191–1197 (2004).
Alankus, Y., Grenningloh, R., Haselmayer, P., Bender, A. & Bruttger, J. BTK inhibition prevents inflammatory macrophage differentiation: a potential role in MS. Mult. Scler. 24, P557 (2018).
Pellerin, K. et al. MOG autoantibodies trigger a tightly-controlled FcR and BTK-driven microglia proliferative response. Brain 144, 2361–2374 (2021).
Weber, M. et al. Fenebrutinib reduces disease activity in a mouse model of infammatory multiple sclerosis, which is associated with reduced microglial activation. Mult. Scler. 27, P680 (2021).
Gruber, R. et al. Decoding Bruton’s tyrosine kinase signaling in neuroinflammation. Mult. Scler. 26, P0311 (2020).
Gruber, R. et al. Evaluating the effect of a Bruton’s tyrosine kinase inhibitor in a murine experimental autoimmune encephalomyelitis model of multiple sclerosis. Mult. Scler. 28, P174 (2022).
Geladaris, A. et al. Targeting BTK in chronic CNS autoimmunity inhibits activation of microglia. Mult. Scler. 27, P971 (2021).
Barr, H. et al. Microglial BTK signaling regulates immune-mediated cortical demyelination. Mult. Scler. 27, P196 (2021).
Brullo, C., Villa, C., Tasso, B., Russo, E. & Spallarossa, A. BTK inhibitors: a medicinal chemistry and drug delivery perspective. Int. J. Mol. Sci. 22, 7641 (2021).
Zain, R. & Vihinen, M. Structure–function relationships of covalent and non-covalent BTK inhibitors. Front. Immunol. 12, 694853 (2021).
Baillie, T. A. Targeted covalent inhibitors for drug design. Angew. Chem. Int. Ed. Engl. 55, 13408–13421 (2016).
Crawford, J. J. et al. Discovery of GDC-0853: a potent, selective, and noncovalent Bruton’s tyrosine kinase inhibitor in early clinical development. J. Med. Chem. 61, 2227–2245 (2018).
Weber, M. et al. Fenebrutinib demonstrates the highest potency of Bruton tyrosine kinase inhibitors (BTKis) in phase 3 clinical development for multiple sclerosis (MS). Neurology 96, 4437 (2021).
Turner, T., Brun, P., Ofengheim, D. & Gruber, R. Comparative CNS pharmacology of tolebrutinib versus other BTK inhibitor candidates for treating MS. Americas Committee for Treatment and Research in Multiple Sclerosis (ACTRIMS) Forum (West Palm Beach, FL, USA) 162 (ACTRIMS, 2022).
Piasecka-Stryczynska, K. et al. Concentration of evobrutinib, a BTK inhibitor, in cerebrospinal fluid during treatment of patients with relapsing multiple sclerosis in a phase 2 study. Mult. Scler. Relat. Disord. 51, 103001 (2021).
Smith, P. et al. Phase 1 clinical trial of PRN2246 (SAR441268), a covalent BTK inhibitor demonstrates safety, CNS exposure and therapeutic levels of BTK occupancy. Mult. Scler. 25, P072 (2019).
Lipsky, A. & Lamanna, N. Managing toxicities of Bruton tyrosine kinase inhibitors. Hematol. Am. Soc. Hematol. Educ. Program 2020, 336–345 (2020).
Fiorcari, S. et al. BTK inhibition impairs the innate response against fungal infection in patients with chronic lymphocytic leukemia. Front. Immunol. 11, 2158 (2020).
Tam, C. S. et al. A randomized phase 3 trial of zanubrutinib vs ibrutinib in symptomatic Waldenstrom macroglobulinemia: the ASPEN study. Blood 136, 2038–2050 (2020).
Rotstein, D. L. All Bruton’s tyrosine kinase inhibitors have similar efficacy and risks: no. Mult. Scler. 28, 1500–1502 (2022).
Montalban, X. et al. Placebo-controlled trial of an oral BTK inhibitor in multiple sclerosis. N. Engl. J. Med. 380, 2406–2417 (2019).
Reich, D. S. et al. Safety and efficacy of tolebrutinib, an oral brain-penetrant BTK inhibitor, in relapsing multiple sclerosis: a phase 2b, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 20, 729–738 (2021).
Vermersch, P. et al. MRI and clinical outcomes of evobrutinib, a Bruton’s tyrosine kinase inhibitor, in relapsing multiple sclerosis over 2.5 years of the open-label extension to a phase 2 trial. Mult. Scler. 28, P731 (2022).
Montalban, X. et al. Safety and efficacy of evobrutinib, a Bruton tyrosine kinase inhibitor, in relapsing multiple sclerosis over 2.5 years of the open-label extension to a phase 2 trial. Int. J. MS Care 21, DMT02 (2022).
Oh, J. et al. MRI, safety, and efficacy outcomes in patients with relapsing MS: 18-month results from the long-term extension study of tolebrutinib. Americas Committee for Treatment and Research in Multiple Sclerosis (ACTRIMS) Forum (West Palm Beach, FL, USA) 102 (ACTRIMS, 2022).
Kuhle, J. et al. Evobrutinib, a Bruton’s tyrosine kinase inhibitor, decreases neurofilament light chain levels over 2.5 years of treatment in patients with relapsing multiple sclerosis. Mult. Scler. 28, EP1021 (2022).
Syed, S. et al. Efficacy and safety of tolebrutinib in patients with highly active relapsing MS: subgroup analysis of the phase 2b study. Neurology 96, 2260 (2021).
Lee, D. S. W., Rojas, O. L. & Gommerman, J. L. B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat. Rev. Drug Discov. 20, 179–199 (2021).
Correale, J. BTK inhibitors as potential therapies for multiple sclerosis. Lancet Neurol. 20, 689–691 (2021).
Häusser-Kinzel, S. & Weber, M. S. The role of B cells and antibodies in multiple sclerosis, neuromyelitis optica, and related disorders. Front. Immunol. 10, 201 (2019).
Jeske, S. S. et al. Adenosine-producing regulatory B cells in head and neck cancer. Cancer Immunol. Immunother. 69, 1205–1216 (2020).
Lünemann, J. D., Ruck, T., Muraro, P. A., Bar-Or, A. & Wiendl, H. Immune reconstitution therapies: concepts for durable remission in multiple sclerosis. Nat. Rev. Neurol. 16, 56–62 (2020).
Ruck, T., Nimmerjahn, F., Wiendl, H. & Lünemann, J. D. Next-generation antibody-based therapies in neurology. Brain 145, 1229–1241 (2022).
Steinmaurer, A., Wimmer, I., Berger, T., Rommer, P. S. & Sellner, J. Bruton’s tyrosine kinase inhibition in the treatment of preclinical models and multiple sclerosis. Curr. Pharm. Des. 28, 437–444 (2022).
Bauer, R. A. Covalent inhibitors in drug discovery: from accidental discoveries to avoided liabilities and designed therapies. Drug Discov. Today 20, 1061–1073 (2015).
Timofeeva, N. & Gandhi, V. Ibrutinib combinations in CLL therapy: scientific rationale and clinical results. Blood Cancer J. 11, 79 (2021).
Tsouki, F. & Williams, A. Multifaceted involvement of microglia in gray matter pathology in multiple sclerosis. Stem Cell 39, 993–1007 (2021).
Sucksdorff, M. et al. Brain TSPO-PET predicts later disease progression independent of relapses in multiple sclerosis. Brain 143, 3318–3330 (2020).
Becker, A. et al. Safety, tolerability, pharmacokinetics, target occupancy, and concentration-QT analysis of the novel BTK inhibitor evobrutinib in healthy volunteers. Clin. Transl. Sci. 13, 325–336 (2020).
Haselmayer, P. et al. Efficacy and pharmacodynamic modeling of the BTK inhibitor evobrutinib in autoimmune disease models. J. Immunol. 202, 2888–2906 (2019).
Herman, A. E. et al. Safety, pharmacokinetics, and pharmacodynamics in healthy volunteers treated with GDC-0853, a selective reversible Bruton’s tyrosine kinase inhibitor. Clin. Pharmacol. Ther. 103, 1020–1028 (2018).
Angst, D. et al. Discovery of LOU064 (remibrutinib), a potent and highly selective covalent inhibitor of Bruton’s tyrosine kinase. J. Med. Chem. 63, 5102–5118 (2020).
Gabizon, R. & London, N. A fast and clean BTK inhibitor. J. Med. Chem. 63, 5100–5101 (2020).
Kaul, M. et al. Remibrutinib (LOU064): a selective potent oral BTK inhibitor with promising clinical safety and pharmacodynamics in a randomized phase I trial. Clin. Transl. Sci. 14, 1756–1768 (2021).
Zhang, B. et al. Orelabrutinib, a potent and selective Bruton’s tyrosine kinase inhibitor with superior safety profile and excellent PK/PD properties. Cancer Res. 80, CT132 (2020).
Nimmerjahn, F. & Ravetch, J. Fcγ receptors as regulators of immune responses. Nat. Rev. Immunol. 8, 34–47 (2008).
Bonilla, F. A. & Oettgen, H. C. Adaptive immunity. J. Allergy Clin. Immunol. 125, S33–S40 (2010).
Netea, M. G. et al. Defining trained immunity and its role in health and disease. Nat. Rev. Immunol. 20, 375–388 (2020).
Medzhitov, R. & Janeway, C. Jr. Innate immune recognition: mechanisms and pathways. Immunol. Rev. 173, 89–97 (2000).
Li, Q. & Barres, B. A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 18, 225–242 (2018).
Mrdjen, D. et al. High-dimensional single-cell mapping of central nervous system immune cells reveals distinct myeloid subsets in health, aging, and disease. Immunity 48, 599 (2018).
Lloyd, A. F. & Miron, V. E. The pro-remyelination properties of microglia in the central nervous system. Nat. Rev. Neurol. 15, 447–458 (2019).
Baaklini, C. S., Rawji, K. S., Duncan, G. J., Ho, M. F. S. & Plemel, J. R. Central nervous system remyelination: roles of glia and innate immune cells. Front. Mol. Neurosci. 12, 225 (2019).
Acknowledgements
The research of authors is supported by the German Research Council (DFG) grants SFB-CRC TR128 A09 and SFB-CRC TR128 Z02 and funding from Deutsche Myasthenie Gesellschaft, all to H.W. Editorial and graphics assistance was provided by Elevate Scientific Solutions, a division of Envision Pharma Group, and funded by Sanofi. Writing assistance (assistance with drafting of the manuscript text and tables as directed by the authors, data checking and incorporation of comments from reviewers) was provided by R. J. Hogan and C. Underwood of Elevate Scientific Solutions.
Author information
Authors and Affiliations
Contributions
J.K. and H.W. researched data for the article and J.K. wrote the first draft. All authors contributed substantially to discussions of the manuscript content, critically reviewed the drafts and approved the final version for submission.
Corresponding author
Ethics declarations
Competing interests
J.K. declares that she has received honoraria for lecturing from Biogen, Merck, Mylan, Novartis, Roche, Sanofi and Teva and has received financial research support from Amicus Therapeutics and Sanofi. A.B.-O. declares that he has received grant support to the University of Pennsylvania from Biogen Idec, EMD Serono, Novartis and Roche Genentech. He has participated as a speaker in meetings sponsored by and received consulting fees from Accure, Atara Biotherapeutics, Biogen, Bristol-Myers Squibb, GlaxoSmithKline, Gossamer, Janssen, Medimmune, EMD Serono, Novartis, Roche Genentech and Sanofi. T.J.T. is an employee of and may have ownership interests in Sanofi. H.W. declares that he has acted as a member of the Scientific Advisory Boards of Abbvie, Alexion, Argenx, Bristol Myers Squibb, Janssen, Merck and Novartis. He also declares that he has received speaker’s honoraria and travel support from Alexion, Biogen, Bristol-Myers Squibb, F. Hoffmann-La Roche, Genzyme, Merck, Neurodiem, Novartis, Roche, Teva and WebMD Global and acts as a paid consultant for Abbvie, Actelion, Argenx, Biogen, Bristol-Myers Squibb, EMD Serono, Fondazione Cariplo, Gossamer Bio, Idorsia, Immunic, Immunovant, Janssen, Lundbeck, Merck, NexGen, Novartis, PSI Contract Research Organization, Roche, Sanofi, UCB and Worldwide Clinical Trials. His research is funded by Alexion, Amicus Therapeutics, Argenx, Biogen, CSL Behring, F. Hoffmann-La Roche, Genzyme, Merck, Novartis, Roche and UCB.
Peer review
Peer review information
Nature Reviews Neurology thanks Marco Salvetti, who co-reviewed with Gianmarco Bellucci; Roland Liblau; and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Review criteria PubMed was searched between 10 November 2021 and 5 May 2022 using terms related to BTK (Bruton’s tyrosine kinase, BTKi*, BTK inhibit*, Bruton’s tyrosine kinase inhibit*) in combination with the following terms: MS OR multiple sclerosis, immune cell*, cancer OR tumour OR tumour OR malignanc*, immune deficienc* OR immune disease* OR immunodeficienc*, mechanism of action OR MOA, B cell OR microglia OR macrophage, pharmacodynamic* OR pharmacokinetic* OR pharmacolog*, safe* OR adverse effect* OR adverse event*. Searches did not use date, language or other filters. All article types were included if the topic was potentially relevant. Results were initially selected according to the article title and then were screened on the basis of the abstract. The full text was retrieved for all potentially relevant articles.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Krämer, J., Bar-Or, A., Turner, T.J. et al. Bruton tyrosine kinase inhibitors for multiple sclerosis. Nat Rev Neurol 19, 289–304 (2023). https://doi.org/10.1038/s41582-023-00800-7
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41582-023-00800-7
This article is cited by
-
BTK inhibitor falters in multiple sclerosis trials
Nature Reviews Neurology (2024)
-
Drugs Targeting CD20 in Multiple Sclerosis: Pharmacology, Efficacy, Safety, and Tolerability
Drugs (2024)
-
Emerging imaging markers in radiologically isolated syndrome: implications for earlier treatment initiation
Neurological Sciences (2024)
-
Targeting epigenetic and posttranslational modifications regulating ferroptosis for the treatment of diseases
Signal Transduction and Targeted Therapy (2023)
-
MRI as a biomarker of the smouldering component of multiple sclerosis: time to wake up
European Radiology (2023)
