
Abstract
Short QT syndrome (SQTS) is a rare but dangerous genetic disease. In this research, we conducted a comprehensive in silico investigation into the arrhythmogenesis in KCNH2 T618I-associated SQTS using a multi-scale human ventricle model. A Markov chain model of IKr was developed firstly to reproduce the experimental observations. It was then incorporated into cell, tissue, and organ models to explore how the mutation provided substrates for ventricular arrhythmias. Using this T618I Markov model, we explicitly revealed the subcellular level functional alterations by T618I mutation, particularly the changes of ion channel states that are difficult to demonstrate in wet experiments. The following tissue and organ models also successfully reproduced the changed dynamics of reentrant spiral waves and impaired rate adaptions in hearts of T618I mutation. In terms of pharmacotherapy, we replicated the different effects of a drug under various conditions using identical mathematical descriptions for drugs. This study not only simulated the actions of an effective drug (quinidine) at various physiological levels, but also elucidated why the IKr inhibitor sotalol failed in SQT1 patients through profoundly analyzing its mutation-dependent actions.
Similar content being viewed by others
Introduction
Short QT syndrome (SQTS) is an inherited and life-threatening cardiac channelopathy characterized by an abnormally short QT interval and increased risks for cardiac arrhythmias and sudden cardiac deaths (SCD). Depending on the mutated genes, the SQTS variants are named SQT1-8 and correlated with various ion channels, including potassium channels (SQT1-3)1,2,3,4,5, calcium channels (SQT4-6)6,7, sodium channels (SQT7)8 and anion exchangers (SQT8)9. Among these variants, SQT1 is the one that presents in the majority of patients, being responsible for up to 15% of all cases10. The SQT1 arises from gain-of-function mutations in the KCNH2 gene, and was first identified in 2004 by Brugada et al. with a substitution mutation N588K1. In 2010, another mutation in the KCNH2 gene was reported in a Chinese family that the threonine at carbon 618 was replaced by an isoleucine (i.e., T618I)11. The ECG screening showed a strong family history of SCD, and characteristics of SQTS including abbreviated QT intervals and peaked T-waves were also observed on the ECG. Additional electrophysiological tests revealed that the IKr was markedly increased in the T618I mutant channel, which was mainly caused by the significantly altered inactivation curve that shifted almost 50 mV towards the depolarized direction11. Despite the above clinical and genetic findings, mechanisms regarding how these molecular changes finally lead to a disastrous malignant arrhythmia remain to be elucidated.
To explore the proarrhythmic effects of the KCNH2 T618I mutation, an in silico investigation was conducted in this study based on a multi-scale virtual heart. First, a biophysically-accurate and validated Markov chain model for IKr was developed to recapitulate the experimental observations at the subcellular level. The developed IKr model was then incorporated into a well-established human ventricle model to determine the functional consequences of the T618I mutation on the action potential (AP) and the transmural heterogeneity of repolarization. Next, the arrhythmogenic substrate in the T618I mutation condition was investigated using tissue models. The sustainability of arrhythmias was also evaluated using realistic 2D ventricular slice and organ models. Finally, in terms of pharmacotherapy, we investigated the potential therapeutic effects of quinidine, a clinically available drug, by incorporating its known interactions with multiple ion channels. We also simulated the actions of IKr inhibitor sotalol and analyzed why it failed in SQT1 patients.
Results
Simulation results of the remodeling effects of KCNH2 T618I mutation
Simulation of I Kr in wild-type and KCNH2 T618I mutation conditions
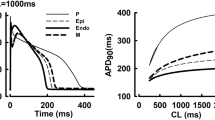
The developed IKr Markov chain model was evaluated by comparing the generated I–V curve with the experimental data from Sun et al.11 (their Fig. 4B). Both the experimental data and the simulated I–V curve were normalized to their corresponding maximum currents (around 10 mV) under the wild-type condition, as shown in Fig. 1A. It can be observed that the model-generated I–V curve fitted well with the experimental data in AP-relevant membrane potential range, and the two main phenotypes of the mutant IKr were successfully recapitulated by the model, namely the right-shifted I–V relationship and the greatly enhanced IKr amplitude. These results suggested that the developed model was able to simulate the effects of the T618I mutation at the ion channel level.

A The fitted current–voltage (I–V) curves in wild-type (black) and T618I (red) conditions using IKr Markov chain models. Experimental data from Sun et al.11 (their Fig. 4B). Error bars represent the standard error of measurement (SEM). B The IKr Markov chain model. C Steady-state (1.25 Hz) action potentials (Ci) and the corresponding IKr (Cii) for EPI cells. Solid and dashed lines represent wild-type and T618I conditions, respectively. D Steady-state action potentials and the corresponding IKr for MIDDLE cells. E Steady-state action potentials and the corresponding IKr for ENDO cells. F The comparison of the peak IKr in wild-type and T618I conditions. G The comparison of transmural APD differences. H The comparison of transmural ERP differences.
Effects of the KCNH2 T618I mutation on action potentials
The developed IKr Markov chain model was then incorporated into the Tusscher-Noble-Noble-Panfilov (TNNP06) model to explore the effects of the KCNH2 T618I mutation at the cellular level. Steady-state APs of different cell types in wild-type and T618I conditions, as well as their corresponding IKr, are presented in Fig. 1. Simulation results suggested that the T618I mutation accelerated the repolarization phase of action potentials through enhancing IKr, which led to abbreviated ventricular APD and ERP.
Though ERP was shortened in all cell types, this effect was not the same in different cell types, with MCELL being the most affected one among three cell types. The differently reduced APD also led to the remodeling of transmural dispersion of repolarization. The membrane potential difference of APD (ΔAPD) between ENDO and MCELL was decreased by almost 60% from 91.7 to 39.7 ms, and the ΔAPD between EPI and MCELL was also reduced from 102.7 to 60.3 ms. Likewise, transmural differences of ERP (ΔERP) were decreased accordingly (Fig. 1G, H).
Influences of the KCNH2 T618I mutation on APD and CV restitution properties and the rate adaption of QT interval
Clinical reports have demonstrated a reduced rate adaption of QT in SQTS patients during exercise tests12. To investigate the mechanism underlying the reduced rate adaption of QT interval, we conducted a series of simulations regarding the restitution properties using standard S1–S2 and dynamic protocols. Depending on the level at which the experiments were performed, these experiments can be organized as APD restitution curve (cellular level), conduction velocity restitution curve (tissue level), and rate adaption curve of QT interval (organ level). Simulation results are illustrated in Fig. 2.

A APD restitution curves obtained from the S1–S2 protocol (Ai) and the dynamic protocol (Aii), where APDs were plotted against PCL and DI, respectively. B CV restitution curves obtained from the S1–S2 protocol (Bi) and the dynamic protocol (Bii). C Rate adaption curve of QT interval.
First, for the cellular level APD restitution properties, it can be observed in Fig. 2Ai, Aii that the APD restitution curves under mutation conditions obtained from either S1–S2 protocol or dynamic protocol showed shallower slopes compared with wild-type conditions, indicating a reduced rate adaption of APD at higher heart rates. Next, for the CV restitution curve at the tissue level, the two protocols showed diverse results. Specifically, under the setting of dynamic protocol, restitution curves in the mutation and wild-type conditions diverged when the PCL was below 700 ms. The conduction velocity of excitation waves in the mutant tissue strand was larger than that in the wild-type tissue at smaller PCLs. In contrast, the S1–S2 protocol generated identical curves (Fig. 2Bii) for these two conditions. The two different measuring results suggested that the change of CV depended on the diastolic interval, and the mutation increased the CV indirectly via the abbreviation of APD. Finally, at the organ level, simulation results demonstrated a shortened QT interval irrespective of heart rate (Fig. 2C). However, the QT discrepancy between the two conditions became smaller at faster heart rates (i.e., shorter PCLs), which was caused by the impaired rate adaption of QT interval in the mutant tissue. In particular, the mutant tissue demonstrated a shallower slope compared with WT, and this phenomenon was in line with the cellular observation that the APD of mutant myocyte also showed a reduced rate adaption.
Effects of the KCNH2 T618I mutation on the temporal vulnerability to the unidirectional conduction block
To investigate the influences of the T618I mutation on tissue’s susceptibility to the unidirectional conduction block, the vulnerable window (VW) was measured separately in wild-type and mutant transmural strands. Figure 3 presents the simulated unidirectional conduction block phenomenon in wild-type and mutation conditions at a position of 7.5 mm away from the endocardial end. It can be observed that the evoked excitation wave propagated unidirectionally towards the epicardial end due to a shorter refractory period of the epicardium. The measured VW width at the specific position was 1.7 ms (from 348.7 to 350.4 ms) in wild-type, and it was enlarged to 2.4 ms (from 267.1 to 269.5 ms) in the T618I condition, indicating a higher temporal vulnerability in the mutant tissue at this position. However, further measurements of other locations on the strand suggested that the overall temporal vulnerability was actually decreased in the mutation condition, and the increased VW was only observed in locations within a local 2-mm segment (approximately from 7.5 to 9.5 mm). The overall distribution of VWs on the strand, and the average VW width in wild-type and T618I conditions, are plotted in Fig. 3C, D.

A The evoked unidirectional conduction block in the wild-type condition. B The unidirectional conduction block in the T618I mutation condition. C Distributions of VWs in the two conditions. D The comparison of the average VW width in the two conditions.
Effects of the KCNH2 T618I mutation on the spatial vulnerability to reentry arrhythmias
The influences of the KCNH2 T618I mutation on the spatial vulnerability to reentry arrhythmias were evaluated by measuring the critical length (CL) in an idealized 2D sheet. The critical length provided a quantitative index for the inducibility of reentry arrhythmia in terms of spatial perspective, and a shorter CL indicated that the tissue was more prone to reentry arrhythmias. The CL was related to the wavelength (the product of ERP and CV) of excitation. In the simulation, the measured CL was up to 47.1 mm in the wild-type condition (Fig. 4B) due to a comparatively long ERP of the wild-type myocytes. In contrast, the CL was 37.1 mm in the mutation condition (Fig. 4C), decreased by almost a quarter comparing to the wild-type tissue. This was due to the accelerated repolarization and abbreviated ERP resulted from the enhanced IKr in the T618I condition.

A The schematic diagram of the idealized 2D sheet. B The critical length of S2 for initiating spiral waves in the wild-type tissue. C The critical length in the T618I mutant tissue. D The comparison of critical length in the two situations.
Effects of the KCNH2 T618I mutation on the dynamic behavior of reentrant spiral waves
To investigate the influences of the T618I mutation on the dynamic behavior of reentrant spiral waves such as the rotation frequency and sustainability, simulation experiments were conducted using 2D and 3D models. First, we initiated spiral waves using S1–S2 protocols in an idealized tissue sheet (75 × 75 mm2) to investigate the influences of the mutation on rotation behavior of spiral waves. Simulation results demonstrated that both conditions produced stable spiral waves and tip trajectories (see Supplementary Fig. 4). However, a significant decrease in wavelength was observed under the mutation condition, and the rotation frequency was increased from 4.08 to 4.55 Hz.
Next, we investigated the influences of T618I mutation on the sustainability of reentry arrhythmias. Simulation experiments were performed on realistic models, i.e., the 2D ventricular slice and 3D bi-ventricle. The S1–S2 protocol was used to initiate the reentrant excitation waves. The simulated reentrant spiral waves on 2D tissue slices for T618I and wild-type conditions were separately demonstrated in Fig. 5A, B. In the mutation condition, the evoked spiral wave circulated around a functionally determined core and lasted 1740 ms with more than 8 cycles. The functional reentry then transformed into anatomical reentry arrhythmia and persisted since then. In contrast, the spiral wave evoked in the wild-type slice was unsustainable, and it self-terminated with a brief lifespan. This was due to the relatively long wavelength of the wild-type spiral wave that could hardly be accommodated within a limited tissue size.

Spiral waves were evoked by the S1–S2 protocol. The stimulating locations of S2 were indicated by the black arrows. A Spiral waves in the wild-type condition with a lifespan of only 480 ms. Snapshots were recorded at 320, 390, 490, 800 ms. B The spiral wave in the T618I mutation condition. Snapshots were recorded at 260, 410, 1090, 1760, 1940, 2330, 2630, 2930 ms. Spiral waves took the form of functional reentry initially before it transformed into anatomical reentry arrhythmia at about 2000 ms and persisted since then. C The comparison of lifespan of spiral waves in two situations.
The above observations in 2D slices were further verified on the realistic 3D bi-ventricle model. The simulated scroll waves in the 3D bi-ventricle models under wild-type and mutation conditions were illustrated in Fig. 6. It can be observed that, in both wild-type and mutation conditions, a premature stimulus applied on the upper part of the left ventricle during the vulnerable window could successfully evoke a unidirectional conduction block, which then transformed into scroll waves in the 3D ventricle. However, the evoked scroll wave in the wild-type ventricle was not sustainable and disappeared after 800 ms (see Fig. 6B). In contrast, the evoked scroll wave in the T618I mutant ventricle persisted throughout the whole simulation period of 10,000 ms, with its functional core anchoring the side wall of the left ventricle.

The S2 stimulus was applied to the upper part of the LV epicardium to initiate scroll waves. A The scroll wave in the wild-type condition. The lifetime was only 410 ms, and the recorded onset and disappearance time of the scroll wave were separately 270 and 680 ms. B The scroll wave in the T618I mutation condition, which persisted throughout the whole simulation period of 10,000 ms.
Simulation results of the actions of quinidine
Actions of quinidine on action potentials of KCNH2 T618I mutation
To investigate the pharmacological mechanisms of quinidine (i.e., a common antiarrhythmic drug for treating SQTS) on the KCNH2 T618I-associated short QT syndrome, we modeled the drug actions by adding two additional states to the Markov chain states (Fig. 7B) as in Whittaker et al.’s study13. Simulation results at ion channel and cellular levels after the addition of 5 μmol quinidine, along with that under control and mutation conditions, are gathered in Fig. 7.

A The fitted current–voltage (I–V) curves in wild-type (black), T618I (red), and quinidine (blue) conditions using IKr Markov chain models. Experimental data from Sun et al.11 (their Fig. 7B). Error bars represent the standard error of measurement (SEM). B The IKr Markov chain model with two additional drug-related states. C–E Steady-state (1.25 Hz) action potentials and the corresponding IKr for EPI, MID, and ENDO cells. F The comparison of the peak IKr. G The comparison of transmural APD differences. H The comparison of transmural ERP differences.
First, it can be observed in Fig. 7A that the I–V curves generated by the Markov model fitted well with the experimental data from Sun et al.11 (their Fig. 7B). Both the experimental data and the simulated I–V curve were normalized to their corresponding maximum currents under the WT condition. The well-fitted I–V curves under various conditions illustrated the rationality of the developed Markov model. In terms of APD, quinidine reversed the abbreviated APD in all cell types (Fig. 7Ci, Di, Ei), and such effect was attributed to the suppressed IKr. As plotted in Fig. 7Cii, Dii, Eii, F, quinidine reduced the IKr almost to its control level. Therefore, the increased repolarizing forces in mutation conditions were attenuated and thereby the APD was restored.
In line with APD, the ERP was also restored after the application of quinidine. In detail, the ERP of EPI and ENDO cells increased by 38.6 and 47.5 ms, respectively, while for MID cells the ERP increased significantly by 83.76 ms. The reversed refractory periods of three cell types in the drug group were comparable to their control levels. In terms of the transmural dispersion of repolarization, ΔAPD and ΔERP were also restored to their control levels (Fig. 7G, H).
To give further insights into the pharmacological actions of quinidine at the subcellular level, we plot the proportion of states under different conditions, as shown in Fig. 8.

A Action potential traces. B The corresponding state proportions under different conditions. The Markov model demonstrated that the enhanced IKr in T618I was due to the loss of inactivation, as evidenced by the significantly dropped proportion of the Inactivation (“I”) state in panels Bi and Bii. Simulation also showed that quinidine exerted its effects by directly deprecating the Open (“O”) state rather than restoring the “I” state (see panels Bii and Biii).
It can be observed in Fig. 8Bi, Bii that there was not much difference in the peak proportion of the Open state between WT and T618I conditions. However, the Open state started earlier in T618I and therefore corresponded to an overall larger current comparing to the WT condition. By further comparing the inactivation state of the two conditions, it can be found that the Inactivation state in the mutation condition was significantly lower than that in WT. Therefore, the enhanced IKr was due to the early opening of the IKr channel, which in turn arose from the loss of inactivation by the T618I mutation. In addition, the simulation results of quinidine (Fig. 8Biii) suggest that the drug did not exert its effects by restoring the lost inactivation in T618I cells, as evidenced by the low level of the Inactivation state. Instead, quinidine suppressed the Open state directly by transferring it into drug-bound states (i.e., OD and ID).
Actions of quinidine on restitution properties and the rate adaption of QT interval
We next tested the drug efficacy in reversing the restitution properties and the rate adaption of QT interval that altered by the T618I mutation. In line with the wild-type and the mutation groups, both standard S1–S2 and dynamic protocols were adopted to generate restitution curves of APD, CV, and QT. Simulation results are illustrated in Fig. 9. Restitution curves under wild-type and mutation conditions were also plotted for comparison purposes.

A APD restitution curves obtained from the S1–S2 protocol (Ai) and the dynamic protocol (Aii), where APDs were plotted against PCL and DI, respectively. B CV restitution curves obtained from the S1–S2 protocol (Bi) and the dynamic protocol (Bii). C The rate adaption curves of QT interval.
Simulations demonstrated that quinidine partially restored the AP adaption at higher pacing rates under mutation conditions, producing a re-steepened restitution curve (Fig. 9Ai, Aii). For the CV restitution curve, quinidine led to a significant decrement of CV, and this influence was observed in both CV-PCL and CV-DI curves. The CV reduction effect of quinidine arose from its inhibitory action on sodium channels. More concretely, quinidine decreased INa by 21.3% at a dose of 5 μmol in our model. Finally, as shown in Fig. 9C, quinidine rescued the rate adaption of QT interval impaired by the T618I mutation. It also prolonged the QT interval in a significant manner regardless of the pacing rate.
Actions of quinidine on the spatial vulnerability to reentry arrhythmias
As suggested in previous sections, the spatial vulnerability was increased in the T618I mutation condition because of the shortened critical length for initiating reentrant excitation waves. In this regard, we investigated the effects of quinidine to explore whether it would reverse the increased spatial vulnerability. Simulation results were plotted in Supplementary Fig. 16. It can be observed that, quinidine greatly extended the CL from 37.1 to 56.6 mm and even exceeded that in the wild-type tissue (47.1 mm). The extended CL indicated the role of quinidine in decreasing spatial vulnerability, and this effect was mainly attributed to the ERP prolonging effects of quinidine through the inhibition of repolarization currents (IKr, IKs, IK1) and INa.
Antiarrhythmic actions of quinidine in terms of the dynamic behavior of reentrant spiral waves
The antiarrhythmic effects of quinidine were also investigated by simulating its actions on the dynamic behavior of reentrant spiral waves. The aforementioned simulation results have demonstrated that the T618I mutation could lead to an increase in the rotation frequency (from 4.08 to 4.55 Hz) of induced spiral waves but without obvious change in the meandering size of the spiral wave tip. After the administration of quinidine (see Supplementary Fig. 5), the tip trajectory still did not show any significant change, but the shortened wavelength caused by the mutation was partially restored. Regarding the rotation frequency, quinidine also exerted antiarrhythmic effects by decreasing the frequency to 3.85 Hz.
In addition to the rotation behavior of spiral waves, the antiarrhythmic effects of quinidine were also evaluated in terms of the sustainability of reentrant arrhythmias. Figure 10A presents the simulated reentrant spiral waves on the 2D tissue slice after administration of quinidine. Experiments showed that the lifespan of the induced spiral waves was only 790 ms, which indicated that quinidine could successfully prevent the sustained reentrant arrhythmias in the T618I mutation condition. In line with the 2D simulation results, the evoked scroll waves (Fig. 10B) were not sustainable in the drug group due to the prolonged refractory distance by quinidine. The scroll waves disappeared at around 900 ms.

A Spiral waves on the 2D ventricular slice. Spiral waves were evoked by the S1–S2 protocol, and the black arrow indicates the stimulating location of S2. Quinidine prevented the persistent arrhythmias, which was evidenced by the disappearance of spiral waves at about 1000 ms. B Scroll waves in the 3D ventricle. In line with the 2D simulation results, the evoked scroll waves were not sustainable and disappeared at about 900 ms.
Parallel simulations using two other models
To prove the robustness of the simulation observations reported in this paper and avoid potential model-dependent results, we conducted additional parallel simulations (i.e., identical experiments but with different cell models) using two other models, namely the O’hara-Rudy dynamic model (the ORd model14) and a contemporary one named ToR-ORd15. We followed a strict experimental procedure to ensure that all the parallel experiments based on ORd and ToR-ORd models were identical to that based on the TNNP06 model. Detailed simulation results using these two cell models were summarized in the Supplementary material. In general, the three models produced highly consistent results under both pathological and pharmacological conditions.
Computational analysis of the poor pharmacological effects of sotalol in SQT1
Previous simulation studies have reported several drugs that may act therapeutic effects13,16. However, few literatures focused on those “failed” drugs, which were supposed to but actually failed to act therapeutic effects. For example, sotalol, a class III antiarrhythmic drug, was known to block IKr under normal conditions, but clinical reports have substantially vetoed its therapeutic effects in both T618I and N588K SQT1 patients17,18. To give a comprehensive analysis of the failure reasons of sotalol in different SQT1 variants, we constructed another Markov model that specifically for N588K, and then developed the drug-bound IKr model for sotalol and compared its actions under WT, T618I, and N588K conditions.
Simulation of I Kr in the KCNH2 N588K mutation condition
Simulated I–V curves of the N588K, along with the experimental data from1, are illustrated in Supplementary Fig. 17. It can be observed that, compared with the T618I mutation, the N588K mutation did not exhibit significant inactivation over the physiological voltage range (−80~20 mV). Actually, simulations showed that the inactivation did not develop until the voltage reached approximately 55 mV. In contrast, while the voltage range over which inactivation of the T618I IKr occurred was also above the physiological range, it was shifted to a smaller extent than the N588K mutation11.
Actions of sotalol on I Kr under WT, T618I, and N588K conditions
We then developed the model of sotalol and compared its actions under WT, T618I, and N588K conditions. The simulated I–V curves under multiple doses of sotalol are illustrated in Supplementary Fig. 18.
It can be observed from Supplementary Fig. 18B, C that, at a clamped voltage of +20 mV, 100 μmol sotalol depressed WT IKr extensively by 49.4%, but only by 7.5% under the N588K condition. Even 500 μmol sotalol suppressed N588K IKr only by 29.7%. These simulation results match well with experimental observations (decreased by 48% for WT, 9.0% for N588K+ 100 μmol sotalol, and 27.0% for N588K+ 500 μmol sotalol1). Another phenomenon is that the inhibition ability of sotalol was significantly restored at more positive voltages, and the gap of drug efficacy between N588K and WT was reduced.
Due to the smaller shift of inactivation in T618I than N588K, the effect of sotalol is also different from that in N588K. Specifically, compared with the decrement of only 29.7% in N588K, 500 μmol sotalol could decrease the mutant IKr by 63.8% in the T618I condition, suggesting that the drug efficacy was less diminished in the latter case. This is in accordance with Sun et al.’s reports that 500 μmol sotalol was able to decrease T618I IKr substantially and such inhibitory effect was comparable to that achieved by 5 μmol quinidine.
We did not conduct more simulations in high-dimensional models since the cell model is enough to demonstrate the failure reasons of sotalol. On the other hand, most high-dimensional simulation results of sotalol can be expected. For example, the critical length would barely change even under 100 μmol sotalol.
Discussion
Main findings
Short QT syndrome is associated with high risks for life-threatening events. In this study, we investigated the proarrhythmic effects of the KCNH2 T618I mutation using a multi-scale human virtual heart. The major findings of this study are as follows: (i) The T618I mutation unevenly shortened the ventricular APD and ER and decreased the transmural dispersion of repolarization, leading to a decreased susceptibility to arrhythmias in terms of temporal domain. (ii) The T618I mutation shortened the tissue excitation wavelength through a reduction of the ERP, therefore increased the spatial vulnerability to reentry arrhythmias. (iii) The T618I mutation caused a series of remodeling effects on the restitution properties, including the flattened APD restitution curve and the reduced rate adaption of the QT interval. (iv) The T618I mutation stabilized the reentrant spiral waves, which was evidenced by the increased rotation frequency and the small meandering size of spiral wave tips. Besides, 2D and 3D simulations consistently showed that the T618I mutation favored the sustenance of reentry arrhythmias on realistic tissues of limited size. (v) Quinidine prolonged APD and ERP in all cell types, and restored the QT interval in the mutation condition. (vi) Quinidine reversed the flattened APD restitution curve under the mutation condition and rescued the rate adaption of the QT interval as well. (vii) Quinidine decreased the spatial vulnerability to reentry arrhythmias, reduced the rotation frequency of spiral waves, and also successfully prevented the sustained spiral/scroll waves in realistic slice and organ models of SQTS. (viii) Clinically relevant doses of sotalol failed to inhibit mutant IKr, but the failure reasons were different between N588K and T618I.
Relevance to previous experimental studies
The existing understanding of SQTS is mainly derived from three experimental models, namely drug-induced models, induced pluripotent stem cell (iPSC)-based models, and transgenic animal models. First, drugs like pinacidil and specific IKr agonists can act on outward potassium currents and lead to reduction of APD and QT interval, therefore they were used in early studies to mimic SQTS19,20,21,22. Although drug-induced SQTS models have allowed for a better understanding of the SQTS mechanism, the drug may also activate ion channels not implicated in SQTS, or affect IKr biophysics in a different way from the mutation induced functional remodeling23. Therefore, experimental SQTS models based on induced pluripotent stem cell cardiomyocytes (iPSC-CMs) and transgenic animals have emerged as new tools for investigating SQTS. Guo et al.24 generated iPSC-CMs with KCNH2 T618I from skin fibroblasts and demonstrated that iPSC-CMs could recapitulate the increased current density of IKr and abnormal AP phenotype featured by shortened APD. Similar techniques were also adopted for generating iPSC-CMs of another SQT1 subtype carrying KCNH2 N588K25. El-Battrawy et al.26 established a cellular model of SQTS using hiPSC-CMs, and most of our simulation results were consistent with their observations. Specifically, they showed that most of the AP biomarkers including resting potential (RP), action potential amplitude (APA), maximal upstroke velocity (Vmax) did not exhibit statistically significant differences between WT and SQTS groups. APD50 and APD90 were the only two parameters that showed differences. In our simulations, take EPI cells as examples, the RP changed from −85.47 mV (WT) to −85.62 mV (mutation), the APA changed from 127.01 mV (WT) to 127.15 mV (mutation), and the Vmax changed from 387.23 mV (WT) to 390.81 mV (mutation). The insensible changes of the above three parameters are consistent with El-Battrawy et al.’s observations. On the other hand, our simulations reproduced the significantly decreased APD50 and APD90 in experiments. Specifically, APD90 dropped from 308.94 ms (WT) to 235.16 ms (mutation), and APD50 decreased from 276.16 ms (WT) to 203.12 ms (mutation). Therefore, the constructed model in this study is able to recapitulate the phenotype of SQT1 at the cellular level. The simulation results support El-Battrawy et al.’s observations.
El-Battrawy et al. reported an interesting observation that the intracellular Ca2+ level was increased in the SQTS group. Arrhythmia events (manifested as early or delayed afterdepolarization-like Ca2+ transients) were also frequently observed. To explore the underlying mechanisms, we conducted additional experiments and recorded the changes in Ca2+ transients under mutation conditions. However, our simulation showed that the intracellular Ca2+ concentration was decreased and no EAD-like Ca2+ activities were observed (see Supplementary Fig. 1). Identical observations were observed in another two cell models (i.e., ORd and ToR-ORd) so that the factor of model-dependency can be excluded. In our simulations, the decreased Ca2+ concentration was attributed to the action potential abbreviation-induced ICaL decreasing, which in turn affected the intracellular Ca2+ released from the sarcoplasmic reticulum. Noted that the decreased Ca2+ was also observed in another simulation study27. Therefore, the underlying mechanisms for the increased Ca2+ in SQTS were currently unknown. Although the increased Ca2+ was not reproduced in simulations, the observed afterdepolarization activities in26 are of great significance for understanding arrhythmogenesis in SQTS patients. Current studies have revealed the increased vulnerability to arrhythmias (substrates for arrhythmias) in hearts with SQTS mutations, while few is known about the actual “triggers”. Therefore, the EAD-like Ca2+ activities warrant further investigations by means of wet experiments.
In terms of the pharmacotherapy, Zhao et al.28 tested the effects of six clinical drugs on hiPSC-CMs. Among six tested drugs, three of them, namely ivabradine, ajmaline, and mexiletine, were observed to prolong APD and reduce arrhythmic events. In ref. 27, Jæger et al. computed the optimal drug combinations for SQT1 based on a suite of computational models including hiPSC-CMs, rabbit ventricular myocytes, and adult human ventricular myocytes. They proposed a reliable in silico approach for assessing the combined efficacy of multiple drugs, and they suggested that simultaneous induction of INaL and blockade of IKr could be a promising strategy for SQT1.
Despite the great achievements of hiPSC-CMs, cell models cannot reflect those dynamic behaviors at the tissue level. In view of this, Shinnawi et al.29 established a iPSC-based 2D model named “hiPSC-CCSs” (i.e., human iPSC-derived cardiac cell sheets) to study the tissue-level SQTS phenotype. Finally, transgenic SQTS animals are the third experimental measure to investigate the mechanisms underlying SQTS. Compared with iPSC-based myocytes and tissue models, transgenic SQTS animals can mimic the human disease phenotype on all levels (from ion current to whole-heart and in vivo levels). For example, Odening et al.23 raised transgenic SQT1 rabbits carrying KCNH2 N588K by which the whole-heart and in vivo phenotypes of SQT1 such as the increased QT-dispersion in 12-lead ECG and the lack of QT interval adaption to heart rates were observed.
The virtual heart model in this study provided another reliable tool for investigating the mechanisms of SQTS and evaluating the pharmacotherapeutic effects of antiarrhythmic drugs. Different from some current data-driven models30, the presented model recapitulated many clinically or experimentally observed important phenotypes of SQT1 as well as the drug effects at multiple levels, thus facilitating our understanding of the mechanism underlying SQTS. Specifically, at the cellular level, the cell model reproduced the abbreviated APD and ERP widely reported in previous experimental research22,31. In particular, the APD restitution-plots (Fig. 2A) recapitulated the reduced rate adaption of APD, which was a critical cellular level phenotype of SQTS23,29. For the phenotype at the tissue level, Shinnawi et al. generated iPSC-based tissue of SQTS and demonstrated the marked shortening of wavelength without affecting CV, the increased susceptibility for reentry arrhythmias, and the increased rotor frequency29. In line with these observations, our tissue model also produced an unaffected CV (at normal heart rates) and shortened wavelength. Increased spatial vulnerability and rotor frequency were also observed in our simulation. Third, the model successfully recapitulated the impaired QT interval adaption to heart rates. The reduced rate adaption of QT particularly at fast heart rates is an important ECG phenotype of SQTS23,32 and has been utilized as a diagnostic tool in clinical practice12,33. Finally, the drug quinidine showed a strong ability to rescue abnormal SQTS phenotypes at different levels. Available experimental and clinical studies have reached a consensus on the efficacy of ranolazine in saving SQTS hearts from arrhythmias33,34,35. Our simulation results regarding the effects of quinidine were consistent with these studies. As summarized in the Main Finding section, simulations demonstrated that quinidine prolonged APD and restored QT interval at cellular and organ levels, respectively. The decreased inducibility of arrhythmias and reduced rotor frequency predicted by the model after the “administration” of quinidine were also observed in the iPSC-based tissue model29.
This study provides theoretical insights into the difference of drug efficacy between quinidine and sotalol. Based on evaluation results of five drugs, McPate et al. summarized that SQT1 might be more responsive to those hERG blockers that do not depend strongly on inactivation for their potency36. Particularly, they suggested that quinidine’s ability to block N588K-hERG at therapeutic concentrations might derive from its comparative insensitivity to attenuation of hERG inactivation. In accordance with McPate et al.’s findings, our simulation results revealed that quinidine’s ability to block hERG depended more on the Open state and was less sensitive to the change of the Inactivation state compared with sotalol. This can be reflected in a quantitative way by introducing the ratio kA/kI, where kA and kI are transition rates from Open (O) to Drug-bound Open (OD), and from Inactivation (I) to Drug-bound Inactivation (ID), respectively. The ratio is able to demonstrate the extent to which the blocking capacity of a drug depends on the Open state (alternatively, not depend on the Inactivation state). The kA/kI are separately 23.8 and 1.50 for quinidine and sotalol (see Eqs. (66), (68), (85), (87) in Methods), which clearly shows the discrepancy of sensitivity to inactivation between these two drugs. The higher kA/kI value of quinidine suggests that it is less sensitive to inactivation than sotalol, which is consistent with McPate’s inference.
Of note, there are some simulation results that do not agree with previously reported experimental findings. Specifically, experiments based on iPSC-derived cell sheets demonstrated that there was no obvious difference between the CVs measured under conditions of mutation and quinidine administration29. In contrast, our simulations showed that the CVs diverge significantly under these two conditions (Fig. 9B). In our model, the drug effects of quinidine were simulated by incorporating its dose-dependent influences on multiple ion channels, among which the suppression of INa would cause a direct impairment on the conduction of excitations. Quinidine is a widely acknowledged class Ia antiarrhythmic agent, and the unchanged CV in experiments might be due to the considerably slow velocity of only around 2.5 cm/s in the iPSC-derived cell sheet (for comparison, the CV is about 70 cm/s in physiological conditions and in our simulations). Another inconsistence that may arise from the slow conduction velocity was the meandering activity of rotor tips. Shinnawi et al.29 showed that the induced spiral wave in the wild-type cell sheet meandered greatly, while the spiral wave in the mutation condition became more stable with decreased meandering distance of the rotor tip. Although the model recapitulated the stable spiral wave and increased rotor frequency, it failed to reproduce the meandering behavior of the spiral wave tip under the wild-type condition.
As far as we are concerned, this study is the first one to simulate failed drugs using identical mathematical equations. For example, Luo et al.16 found that E-4031 and disopyramide failed to exert noticeable effects on APD; however, the interaction of drugs with IKr was modeled based on simple Hill functions and different parameters (e.g., IC50) were used for WT and mutation conditions. In contrast, we modeled the effects of sotalol by adding exactly the same two states (along with identical association/dissociation rates, see Eqs. (85)–(88), (98)–(101) in Methods) to the original 5-state Markov chain model. Another simulation study by Whittaker et al.13 discussed the potential antiarrhythmic effects of disopyramide and quinidine on SQT1. However, the significantly different effects of those failed class III drugs were neither simulated nor analyzed. In our study, the two pharmacological states were able to account for various conditions. It could not only reproduce the significant IKr-blocking effects of class III drugs (i.e., sotalol) under the WT condition, but also accurately replicate the decreased efficacy under T618I and N588K mutations.
Mutation-dependent failure reasons of sotalol in SQT1 patients
Sotalol is a typical class III antiarrhythmic drug known to block IKr, but clinical studies have proved it to be a failed drug therapy for SQT1 patients17,25. In addition, experimental investigations demonstrated that the loss of drug effects were not the same among different SQT1 mutations11. Our simulations showed that, for N588K, the inhibition ability of sotalol on IKr was significantly diminished and even high doses of sotalol (500 μmol) did not exert enough inhibitory effects over the physiological membrane potential range. McPate et al. predicted that for drugs strongly depend on inactivation, the gap between potency of inhibition of N588K and WT IKr might be smaller at more positive voltages. This assumption is also proved using our models. To be more specific, when clamped at 20 mV, 100 μmol sotalol blocked IKr by 49.4% and 7.5% under WT and N588K conditions, respectively, and such gap was significantly restored at more positive voltages (>60 mV, see Supplementary Fig. 18). Despite the restored inhibiting ability, the voltages that sotalol acted inhibitory effects were out of physiological action potential range (about −90 to 20 mV).
Compared with N588K, the drug efficacy was less diminished in the T618I condition. For example, 500 μmol sotalol was still able to inhibit IKr by 63.8% at +20 mV under the T618I condition, which is in accordance with Sun et al.’s experimental observations that 500 μmol sotalol achieved comparable inhibitory effect to 5 μmol quinidine11. However, the required dose (i.e., 500 μmol) is beyond the plasma concentrations (88.4–265.2 μmol) during long-term oral therapy17.
Taken together, this study reveals mutation-dependent failure reasons of sotalol. Although the failure of sotalol can be generally explained as its high sensitivity to the Inactivation state, the detailed processes are not exactly the same for N588K and T618I. Through a review of past literature, there are few studies building Markov models for this drug, and even fewer in silico research focused on the reason why sotalol failed in SQT1 patients. In this regard, Brennan et al.37 proposed a 10-state Markov model of sotalol where two parallel state groups (i.e., each group had five states: C1, C2, C3, I, O) were included. The model was validated on the inhibition time course of sotalol (Fig. 3 in ref. 38), and again, the significantly different effects of sotalol under control and T618I conditions were not simulated or discussed in their study. Besides, the model proposed by Brennan et al. was based on “guarded-receptor theory”38, i.e., the drug molecule binds with only the open state. However, previous studies have shown that the binding of drugs with the inactivation state is also necessary for modeling drug actions13,39. Our study also supports the necessity of adding the drug-bound inactivation (“ID”) state, as the model could replicate the altered drug efficacy of sotalol only when both OD and ID were incorporated.
Limitations
Previous studies have suggested that IKr is not the only changed ion current in SQT1 patients. Specifically, Guo et al.24 generated iPSC-CMs from a SQT patient carrying KCNH2 T618I mutation, where the mRNA-level expression of SCN5A, KCNQ1, and CACNA1C were all significantly elevated. In line with these increased mRNA-level expressions, the corresponding currents including INa, IKs, and ICaL were all significantly increased. Such remodeling effects were observed even after the correction of T618I mutation in SQTS iPSC-CMs. In addition to T618I, electrical remodeling was also observed in another common variant of SQT1, KCNH2 N588K. Cellular experiments based on transgenic rabbits that carried KCNH2 N588K reported that IK1 was decreased while IKs was increased in SQT1 myocytes compared with WT23. The above observations together indicate that the consequences of mutations to a specific gene are not limited to its corresponding ionic current, but may also result in remodeling of multiple ion channels. Such remodeling effects are not considered in the present study but warrant further investigations in the future.
The monodomain model, rather than the bidomain model, was adopted in this study. According to previous research, the monodomain model is significantly superior to the bidomain model in terms of computational cost, and in some cases the monodomain model can be more than 10 times faster for same problems40. On the other hand, previous literatures that discussed the difference in accuracy between bidomain and monodomain models reported that these two models produced almost identical results except in some cases where stimulus were injected into the extracellular space (e.g., defibrillation). Specifically, Potse et al.41 investigated the impact of the monodomain assumption on simulated propagation by comparing its results with a bidomain model. They concluded that differences between the two models were extremely small, and repeated experiments with simulated ischemia or sodium conductivity reduced by 90% arrived at the same conclusion. Bourgault et al.42 compared monodomain and bidomain models in terms of the activation time, and they performed simulations on both idealized geometry and realistic 2D slice. They concluded that the discrepancy between the two models was of order 1% or even below, and it was smaller than the discretization error resulting from commonly used mesh size. A good review by Clayton et al.43 summarized that if there is no injection of current into the extracellular space, AP propagation provided by mono- and bidomain models are close to each other even under the condition of unequal anisotropy ratio. In our case, there was no stimulus applied to the extracellular space, and therefore we used the monodomain model for simulations.
It should also be noted that the mechanical contraction was not considered in this study. All the ventricular models including the idealized models and the realistic 2D slice and the 3D ventricle were assumed to be stationary. However, the ventricle was known to contract during its repolarization phase, and the geometry would be altered. The altered geometry, along with the mechano-electric feedback, may further influence the T-wave configuration and the electrophysiological behavior44,45. Therefore, the potential roles of mechanical contraction and mechano-electric feedback warrant further investigations.
Conclusion
This study concluded that the arrhythmogenesis of the KCNH2 T618I mutation arises from a series of electrophysiological alterations caused by the enhanced IKr. The T618I mutation shortened the critical length for initiating reentrant excitation waves by abbreviating the ventricular ERP, therefore facilitated the genesis and maintenance of reentry arrhythmias. The antiarrhythmic drug quinidine was suggested to be an effective treatment as it prolonged the cellular ERP, restored the abbreviated QT interval, and also removed substrates for reentry arrhythmias in SQTS patients by not only prolonging the critical length for initiating spiral waves but also preventing the persistence of those evoked ones. These observations were highly consistent when simulations were performed using three apparently different cell models, suggesting the robustness of the developed Markov model and the model-independence of the reported findings. In summary, the study establishes a causal linkage between the genetic mutation and the organ-level ventricular arrhythmias and clinical ECG observations, and the developed Markov IKr model and all the upper level models ranging from cells to organs together constitute a reliable platform for not only the investigation of the underlying mechanisms of SQT1, but also the screening of effective antiarrhythmic drugs for SQT1 patients.
Methods
Development of Markov chain models for I Kr in wild-type and KCNH2 T618I mutation conditions
The Hodgkin–Huxley model (HH model) assumes that the channel gates are independent; however, experiments have shown that activation and inactivation processes are typically dependent on each other. As a result, explicit representations of ion channel states are necessary, as in the case of Markov chain models. Jæger et al. mentioned that the Markov model is able to give a more realistic representation of both the effect of mutations and the effect of drugs46, and the superiority of the Markov model was also explicitly demonstrated in other simulation studies47. Therefore, the Markov chain model of IKr developed in this study is more accurate than the HH-based IKr formulations. A general introduction to the use of Markov models can be found in ref. 48.
The Markov chain model for IKr in this study was based on a previous IKr model by Clancy and Rudy49, as illustrated in Fig. 1B. The model contained five states, including an open state (O), an inactivated state (I), and three closed states (C1, C2, C3). The model parameters were optimized by minimizing the least-squared difference between the model-generated current–voltage (I–V) curve and the experimentally recorded I–V data. The optimizing process was performed using the LMFIT package that implemented in Python. Model equations and the fitted transition rates are listed in Eqs. (1)–(25).
I Kr Markov chain model formulations
Transition rates for the wild-type channel (ms−1)
Transition rates for the T618I mutant channel (ms−1)
Transition rates for the N588K mutant channel (ms−1)
Modeling multi-channel effects of quinidine
Quinidine was known to affect multiple ionic currents including IKr11,50, IKs51, IK152, Ito52, ICaL53, INa54,55, and INaL56. In this study, we incorporated all these reported ionic currents into their corresponding ion channel models. Specifically, for the IKr, two additional states related to drugs were added to the Markov chain model as in13 (Fig. 7B). Model equations and the fitted transition rates are listed in Eqs. (35)–(69).
IKr Markov chain model formulations
Transition rates for the T618I mutant channel after the application of quinidine (ms−1)
Transition rates for the N588K mutant channel after the application of quinidine (ms−1)
The effects of quinidine on other channel currents
For the other currents, the dose-dependent drug effects were modeled based on the “pore block” theory57. The fitted equations are as Eqs. (70)–(75).
“kcurrent” in above equations is the blocking factor that represents the reduction ratio of the maximum conductance of the targeted ion channel. “[QUIN]” represents the concentration of quinidine in μmol. The fitting results are illustrated in Supplementary Fig. 19. Experimental data sources11,50,51,52,53,54,55,56 are indicated in the figure caption.
Modeling the effect of sotalol on I Kr
The Markov model of sotalol on IKr was identical to that of quinidine (Fig. 7B and Eqs. (35)–(43)), except that the transition rates were adjusted to recapitulate experimental observations. The transition rates under T618I and N588K conditions are listed in subsections “Transition rates for the T618I mutant channel after the application of sotalol (ms−1)” and “Transition rates for the N588K mutant channel after the application of sotalol (ms−1)”, respectively.
Transition rates for the T618I mutant channel after the application of sotalol (ms−1)
Transition rates for the N588K mutant channel after the application of sotalol (ms−1)
Single cell simulation
The human ventricular myocyte model developed by Ten Tusscher et al. (TNNP06 model)58 was used in this study. The electrophysiological behavior at the cellular level was described as:
where Vm is the membrane potential, Iion and Istim are separately the total ionic current and the stimulating current, Cm is the membrane capacitance.
Next, we replaced the original IKr in the TNNP06 model by the aforementioned Markov chain model. Besides, the heterogeneity of IKr in different cell types was considered in this study, and the ratio of the maximum conductance of IKr in EPI:MCELL:ENDO was set to 1.6:1.0:1.0 based on previous experimental measurements59. The modified TNNP06 model was paced to achieve its steady state by fifty supra-threshold stimuli (52 pA/pF, 1 ms) in a frequency of 1.25 Hz, which corresponds to a normal heart rate of 75 beats/min. The last AP was recorded for measuring its parameters, e.g., APD, overshoot, etc.
The effective refractory period (ERP) was measured by a standard S1–S2 protocol. Specifically, fifty S1 stimuli were applied at 1.25 Hz before a premature stimulus (S2) was applied. The S2 had the same amplitude and duration as S1, and it would not be able to evoke a new action potential if being applied within the refractory period after the last S1. The protocol was conducted iteratively by gradually reducing the S1–S2 interval, and the ERP was measured as the smallest diastolic interval when the S2 was able to evoke an AP that reached 80% of the overshoot of the preceding AP60.
Multi-scale virtual human ventricle
The propagation of excitation waves was described using the monodomain equation43,
where D is the diffusion coefficient tensor for describing the intercellular electrical coupling via gap junctions.
For one-dimensional (1D) simulations, a 15 mm long transmural 1D strand of 100 nodes with spacing 0.15 mm was constructed. The strand length was consistent with the normal range of human transmural ventricle width (4–14 mm) in previous studies61,62. The ratio of ENDO:MCELL:EPI was set to 37:26:37 as in previous study14. The diffusion coefficient was set to 0.154 mm2/ms to get a conduction velocity of 71.9 cm/s, which was very close to the 70 cm/s that recorded in the human myocardium63.
The 1D strand was expanded in the y direction to form a 15 × 60 mm2 two-dimensional (2D) tissue sheet. Isotropic cell-to-cell coupling was assumed in the idealized model, and the isotropic coefficient was kept the same as in the 1D strand. For the realistic 2D ventricle tissue slice, the D was anisotropic and the coefficients along and perpendicular to the fiber orientation were set to 0.154 and 0.0385 mm2/ms, respectively. For the realistic three-dimensional (3D) bi-ventricle geometry, the anisotropic coefficients were the same as in the 2D realistic tissue slice. The anatomical geometries of realistic 2D ventricular slice and 3D ventricle were reconstructed from DT-MRI64,65.
Generation of pseudo-ECG
The pseudo-ECG was generated using Eqs. (104) and (105)66:
where \(\Phi\) is a unipolar potential generated by the tissue, r is the distance between a source point and the virtual electrode, \(\sigma _{\it{i}}\) and \(\sigma _{\it{e}}\) stand for intracellular and extracellular conductivities, respectively, and \({\int} {}\) is the domain of integration.
The corrected QT interval (QTc) was obtained from the simulated ECG. It was measured as the interval from the earliest onset of the depolarization wave to the end of the T-wave. The measured QT was corrected for heart rate using the Bazett equation:
where RR is the interval between two consecutive R waves.
APD and CV restitution curves and rate adaption curve of QT interval
Two protocols namely the standard S1–S2 protocol and the dynamic protocol were adopted to determine the APD and CV restitution curves. More concretely, for the APD restitution curve (APDR), a series of stimuli (S1) at a frequency of 1.25 Hz were applied to the single cell model until it reached its steady states. A premature stimulus (S2) was then delivered at some diastolic interval (DI) after the last AP of S1. The APDR could be obtained by plotting the APD of the S2-induced AP against DI. In addition to the APD-DI restitution curve generated by using the S1–S2 protocol mentioned above, the APD-PCL was also determined by the dynamic protocol. The cell model was paced 50 times with an initial cycle length of 1000 ms, and the APD of the last AP was recorded, after which the cycle length was decreased to measure the new APD. The APDR was determined by plotting APD against PCL (pacing cycle length).
For the CV restitution curve, S1 stimuli of 1.25 Hz were applied to a 15-mm long tissue strand. Then S2 was applied at a DI after the last S1 excitation wave. The CV of the S2-induced excitation wave was measured and plotted against DI. In addition, similar to the APD-PCL curve, CV-PCL restitution curve was obtained using the dynamic protocol.
For the rate adaption curve of QT interval, similar dynamic protocol was used. The 1D tissue strand was paced 10 times under each PCL to generate a “steady-state” pseudo-ECG where the QT was measured using the aforementioned method.
Measurement of the vulnerable window to the unidirectional conduction block
The vulnerable window (VW) is a certain period when a premature stimulus applied to the refractory tail of the previous excitation wave will result in a unidirectional conduction block. The VW was measured using the S1–S2 protocol in the 1D strand model. Specifically, the S1 stimulus was applied to the endocardial end of the strand to initiate a conditioning excitation wave, and S2 was then applied to a local segment of 0.45 mm on the strand. The exact time window when the unidirectional conduction block occurred was recorded as the VW. Above process was repeated for all locations on the strand to obtain the overall distribution of VWs.
Measurement of the critical length to the initiation of reentrant spiral waves
The critical length (CL) is the minimal length of the stimulating area for accommodating the evoked spiral wave in a limited tissue size. The CL was measured using the S1–S2 protocol on an idealized 2D sheet model. Similar to the S1–S2 used in 1D simulations, the S1 stimulus was applied to the endocardial side of the tissue sheet to initiate a conditioning wave, and a followed S2 was applied to a local epicardial region during the measured VW. The S2 was always able to evoke a unidirectionally propagated excitation wave, but the wave could evolve into a reentrant spiral wave only when the length of the stimulating region was sufficiently long. The minimum length for a successful initiation of reentrant spiral wave was recorded as the CL.
Initiation of reentry arrhythmias in the anatomical 2D ventricular slice and 3D ventricle
Reentry arrhythmias in anatomical geometries were initiated using the S1–S2 protocol as well. In the ventricular 2D slice and 3D ventricle, a series of S1 stimuli were applied sequentially to multiple sites on the endocardium as in67 to reproduce the activation timing sequence of the Purkinje system68. The reentry arrhythmia that took the form of spiral waves in 2D tissue was initiated by a premature stimulus applied to a local slender region on the epicardium during the refractory tail of the previous conditioning wave (i.e., the vulnerable window). Similarly, the reentry arrhythmia (took the form of scroll waves) in the 3D ventricle was also initiated by a premature stimulus, which was applied to the upper part of the left ventricle during the VW.
Numerical details
The differential equations for the membrane potential and the gating variables were solved by the forward Euler method with a time step of 0.02 ms. For computing state probabilities in the Markov model, the backward Euler method was implemented to avoid using small time step. The spatial resolution was 0.15 mm in 1D strands and idealized 2D tissue sheets. In the realistic 2D ventricular slice and 3D ventricle models, the spatial resolution was set to 0.2 mm to be in line with the anatomical geometry reconstructed from DT-MRI64,65. Neumann (non-flux) boundary conditions were adopted to deal with the geometry boundaries69.
For solving the differential equations in 1D strand and idealized 2D sheet models, OpenMP parallelization scheme was utilized70. For the realistic 2D and 3D models, the optimized GPU algorithm in71 was implemented on a Quadro P4000 “Pascal” GPU with 1792 CUDA cores. The host system for the Quadro GPU was a Lenovo ThinkStation P330 with 8 Intel Core i7-9700K CPU cores at 3.6 GHz.
Data availability
All the experimental data used in this study have been referred to in the relevant texts and figure captions. The generated data supporting the conclusion of this article are available within the article and its additional files.
Code availability
The computer simulation codes used to generate the results in this study can be found at https://github.com/ShugangZhang/T618I.
References
Brugada, R. et al. Sudden death associated with short-QT syndrome linked to mutations in HERG. Circulation 109, 30–35 (2004).
Hong, K., Bjerregaard, P., Gussak, I. & Brugada, R. Short QT syndrome and atrial fibrillation caused by mutation in KCNH2. J. Cardiovasc Electrophysiol. 16, 394–396 (2005).
Bellocq, C. et al. Mutation in the KCNQ1 gene leading to the short QT-interval syndrome. Circulation 109, 2394–2397 (2004).
Hong, K. et al. De novo KCNQ1 mutation responsible for atrial fibrillation and short QT syndrome in utero. Cardiovasc. Res. 68, 433–440 (2005).
Priori, S. G. et al. A novel form of short QT syndrome (SQT3) is caused by a mutation in the KCNJ2 gene. Circ. Res. 96, 800–807 (2005).
Antzelevitch, C. et al. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death. Circulation 115, 442–449 (2007).
Templin, C. et al. Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6). Eur. Heart J. 32, 1077–1088 (2011).
Hong, K., Hu, J., Yu, J. & Brugada, R. Concomitant Brugada-like and short QT electrocardiogram linked to SCN5A mutation. Eur. J. Hum. Genet 20, 1189–1192 (2012).
Thorsen, K. et al. Loss-of-activity-mutation in the cardiac chloride-bicarbonate exchanger AE3 causes short QT syndrome. Nat. Commun. 8, 1–10 (2017).
Campuzano, O. et al. Recent advances in short QT syndrome. Front. Cardiovasc. Med. 5, 149 (2018).
Sun, Y. et al. A novel mutation in the KCNH2 gene associated with short QT syndrome. J. Mol. Cell Cardiol. 50, 433–441 (2011).
Giustetto, C. et al. Usefulness of exercise test in the diagnosis of short QT syndrome. Europace 17, 628–634 (2015).
Whittaker, D. G., Ni, H., Benson, A. P., Hancox, J. C. & Zhang, H. Computational analysis of the mode of action of disopyramide and quinidine on heRG-linked short QT syndrome in human ventricles. Front. Physiol. 8, 759 (2017).
O’Hara, T., Virág, L., Varró, A. & Rudy, Y. Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol. 7, e1002061 (2011).
Tomek, J. et al. Development, calibration, and validation of a novel human ventricular myocyte model in health, disease, and drug block. Elife 8, e48890 (2019).
Luo, C., Wang, K. & Zhang, H. In silico assessment of the effects of quinidine, disopyramide and E-4031 on short QT syndrome variant 1 in the human ventricles. PLoS One 12, e0179515 (2017).
Giustetto, C. et al. The lack of effect of sotalol in short QT syndrome patients carrying the T618I mutation in the KCNH2 gene. Hear Case Rep. 1, 373–378 (2015).
El-Battrawy, I. et al. Impact of antiarrhythmic drugs on the outcome of short QT syndrome. Front. Pharmacol. 10, 771 (2019).
Milberg, P. et al. Reduction of dispersion of repolarization and prolongation of postrepolarization refractoriness explain the antiarrhythmic effects of quinidine in a model of short QT syndrome. J. Cardiovasc. Electrophysiol. 18, 658–664 (2007).
Extramiana, F. & Antzelevitch, C. Amplified transmural dispersion of repolarization as the basis for arrhythmogenesis in a canine ventricular-wedge model of short-QT syndrome. Circulation 110, 3661–3666 (2004).
Nof, E., Burashnikov, A. & Antzelevitch, C. Cellular basis for atrial fibrillation in an experimental model of short QT1: Implications for a pharmacological approach to therapy. Hear Rhythm 7, 251–257 (2010).
Patel, C. & Antzelevitch, C. Cellular basis for arrhythmogenesis in an experimental model of the SQT1 form of the short QT syndrome. Hear Rhythm 5, 585–590 (2008).
Odening, K. E. et al. Transgenic short-QT syndrome 1 rabbits mimic the human disease phenotype with QT/action potential duration shortening in the atria and ventricles and increased ventricular tachycardia/ventricular fibrillation inducibility. Eur. Heart J. 40, 842–853 (2019).
Guo, F. et al. Patient-specific and gene-corrected induced pluripotent stem cell-derived cardiomyocytes elucidate single-cell phenotype of short QT syndrome. Circ. Res. 124, 66–78 (2019).
Huang, M. et al. Effects of antiarrhythmic drugs on hERG gating in human-induced pluripotent stem cell-derived cardiomyocytes from a patient with short QT syndrome type 1. Front. Pharmacol. 12, 675003 (2021).
El-Battrawy, I. et al. Modeling Short QT syndrome using human-induced pluripotent stem cell-derived cardiomyocytes. J. Am. Heart Assoc. 7, e007394 (2018).
Jæger, K. H., Edwards, A. G., Giles, W. R. & Tveito, A. A computational method for identifying an optimal combination of existing drugs to repair the action potentials of SQT1 ventricular myocytes. PLoS Comput. Biol. 17, e1009233 (2021).
Zhao, Z. et al. Drug testing in human-induced pluripotent stem cell–derived cardiomyocytes from a patient with short QT syndrome type 1. Clin. Pharm. Ther. 106, 642–651 (2019).
Shinnawi, R. et al. Modeling reentry in the short QT syndrome with human-induced pluripotent stem cell–derived cardiac cell sheets. J. Am. Coll. Cardiol. 73, 2310–2324 (2019).
Li, Z., Jiang, M., Wang, S. & Zhang, S. Deep learning methods for molecular representation and property prediction. Drug Discov. Today 27, 103373 (2022).
Cordeiro, J. M., Brugada, R., Wu, Y. S., Hong, K. & Dumaine, R. Modulation of IKr inactivation by mutation N588K in KCNH2: A link to arrhythmogenesis in short QT syndrome. Cardiovasc. Res. 67, 498–509 (2005).
Du, C., Zhang, H., Harmer, S. C. & Hancox, J. C. Identification through action potential clamp of proarrhythmic consequences of the short QT syndrome T618I hERG ‘hotspot’ mutation. Biochem. Biophys. Res. Commun. 596, 49–55 (2022).
Hancox, J. C., Whittaker, D. G., Du, C., Stuart, A. G. & Zhang, H. Emerging therapeutic targets in the short QT syndrome. Expert Opin. Ther. Targets 22, 439–451 (2018).
Bjerregaard, P. Diagnosis and management of short QT syndrome. Hear Rhythm 15, 1261–1267 (2018).
Dewi, I. P. & Dharmadjati, B. B. Short QT syndrome: the current evidences of diagnosis and management. J. Arrhythmia 36, 962–966 (2020).
McPate, M. J., Duncan, R. S., Hancox, J. C. & Witchel, H. J. Pharmacology of the short QT syndrome N588K-hERG K + channel mutation: differential impact on selected class I and class III antiarrhythmic drugs. Br. J. Pharm. 155, 957–966 (2008).
Brennan, T. P., Fink, M., Rodriguez, B. & Tarassenko, L. T. Modelling effects of sotalol on action potential morphology using a novel Markov model of the HERG channel. In IFMBE Proceedings (eds. Jarm, T., Kramar, P. & Zupanic, A.) 50–53 (Springer, Berlin, Heidelberg 2007).
Brennan, T., Fink, M. & Rodriguez, B. Multiscale modelling of drug-induced effects on cardiac electrophysiological activity. Eur. J. Pharm. Sci. 36, 62–77 (2009).
Perrin, M. J., Kuchel, P. W., Campbell, T. J. & Vandenberg, J. I. Drug binding to the inactivated state is necessary but not sufficient for high-affinity binding to human ether-à-go-go-related gene channels. Mol. Pharm. 74, 1443–1452 (2008).
Clayton, R. H. & Panfilov, A. V. A guide to modelling cardiac electrical activity in anatomically detailed ventricles. Prog. Biophysics Mol. Biol. 96, 19–43 (2008).
Potse, M., Dube, B., Vinet, A. & Cardinal, R. A comparison of monodomain and bidomain propagation models for the human heart. Conf. Proc. IEEE Eng. Med. Biol. Soc. 2006, 3895–3898 (2006).
Bourgault, Y. & Pierre C. Comparing the bidomain and monodomain models in electro-cardiology through convergence analysis. HAL Arch. 1–19 (2010).
Clayton, R. H. et al. Models of cardiac tissue electrophysiology: progress, challenges and open questions. Prog. Biophys. Mol. Biol. 104, 22–48 (2011).
Adeniran, I., Hancox, J. & Zhang, H. In silico investigation of the short QT syndrome, using human ventricle models incorporating electromechanical coupling. Front. Physiol. 4, 166 (2013).
Zhang, H., Kharche, S., Holden, A. V. & Hancox, J. C. Repolarisation and vulnerability to re-entry in the human heart with short QT syndrome arising from KCNQ1 mutation—a simulation study. Prog. Biophys. Mol. Biol. 96, 112–131 (2008).
Jæger, K. H., Wall, S. & Tveito, A. Computational prediction of drug response in short QT syndrome type 1 based on measurements of compound effect in stem cell-derived cardiomyocytes. PLoS Comput. Biol. 17, e1008089 (2021).
Adeniran, I., McPate, M. J., Witchel, H. J., Hancox, J. C. & Zhang, H. Increased vulnerability of human ventricle to re-entrant excitation in hERG-linked variant 1 short QT syndrome. PLoS Comput. Biol. 7, e1002313 (2011).
Tveito, A. & Lines, G. T. Computing characterizations of drugs for ion channels and receptors using Markov models. Springer Nature 1, 1–261 (2016).
Clancy, C. E. & Rudy, Y. Cellular consequences of HERG mutations in the long QT syndrome: precursors to sudden cardiac death. Cardiovasc. Res. 50, 301–313 (2001).
El Harchi, A., Melgari, D., Zhang, Y. H., Zhang, H. & Hancox, J. C. Action potential clamp and pharmacology of the variant 1 short QT syndrome T618I hERG K+ Channel. PLoS One 7, e52451 (2012).
Kang, J., Chen, X. L., Wang, L. & Rampe, D. Interactions of the antimalarial drug mefloquine with the human cardiac potassium channels KvLQT1/minK and HERG. J. Pharm. Exp. Ther. 299, 290–296 (2001).
Nenov, N. I., Crumb, W. J., Pigott, J. D., Harrison, L. H. & Clarkson, C. W. Quinidine interactions with human atrial potassium channels developmental aspects. Circ. Res 83, 1224–1231 (1998).
Zhang, Y. H. & Hancox, J. C. Mode-dependent inhibition by quinidine of NA+-CA2+ exchanger current from guinea-pig isolated ventricular myocytes. Clin. Exp. Pharm. Physiol. 29, 777–781 (2002).
Koumi, S. I. et al. Sodium channel states control binding and unbinding behaviour of antiarrhythmic drugs in cardiac myocytes from the Guinea pig. Cardiovasc. Res. 26, 1199–1205 (1992).
Kramer, J. et al. MICE models: superior to the HERG model in predicting torsade de pointes. Sci. Rep. 3, 2100 (2013).
Wu, L. et al. Role of late sodium current in modulating the proarrhythmic and antiarrhythmic effects of quinidine. Hear Rhythm 5, 1726–1734 (2008).
Yuan, Y., Bai, X., Luo, C., Wang, K. & Zhang, H. The virtual heart as a platform for screening drug cardiotoxicity. Br. J. Pharmacol. 172, 5531–5547 (2015).
Ten Tusscher, K. H. & Panfilov, A. V. Alternans and spiral breakup in a human ventricular tissue model. Am. J. Physiol. Circ. Physiol. 291, H1088–H1100 (2006).
Szabó, G. et al. Asymmetrical distribution of ion channels in canine and human left-ventricular wall: epicardium versus midmyocardium. Pflügers Arch. 450, 307–316 (2005).
Zhang, H., Tao, T., Kharche, S. & Harrison, S. M. Modelling changes in transmural propagation and susceptibility to arrhythmia induced by volatile anaesthetics in ventricular tissue. J. Theor. Biol. 257, 279–291 (2009).
Drouin, E., Charpentier, F., Gauthier, C., Laurent, K. & Le Marec, H. Electrophysiologic characteristics of cells spanning the left ventricular wall of human heart: evidence for presence of M cells. J. Am. Coll. Cardiol. 26, 185–192 (1995).
Yan, G. X., Shimizu, W. & Antzelevitch, C. Characteristics and distribution of M cells in arterially perfused canine left ventricular wedge preparations. Circulation 98, 1921–1927 (1998).
Taggart, P. et al. Inhomogeneous transmural conduction during early ischaemia in patients with coronary artery disease. J. Mol. Cell Cardiol. 32, 621–630 (2000).
Seemann, G., Keller, D. U. J., Weiss, D. L. & Dossel O. Modeling human ventricular geometry and fiber orientation based on diffusion tensor MRI. Comp. Cardiol. 33, 801–804 (2006).
Weiss, D. L., Keller, D. U. J., Seemann, G. & Dössel, O. The influence of fibre orientation, extracted from different segments of the human left ventricle, on the activation and repolarization sequence: a simulation study. Europace 9(suppl_6), vi96–vi104 (2007).
Lu, W., Wang, K., Zhang, H. & Zuo, W. Simulation of ECG under ischemic condition in human ventricular tissue. Comp. Cardiol. 185–188 https://ieeexplore.ieee.org/abstract/document/5737940 (2010).
Adeniran, I., El Harchi, A., Hancox, J. C. & Zhang, H. Proarrhythmia in KCNJ2-linked short QT syndrome: insights from modelling. Cardiovasc. Res. 94, 66–76 (2012).
Weiss, D. L. et al. Modeling of heterogeneous electrophysiology in the human heart with respect to ECG genesis. Comp. Cardiol. 49–52 https://ieeexplore.ieee.org/document/4745418 (2007).
Colman, M. A., Holmes, M., Whittaker, D. G., Jayasinghe, I. & Benson, A. P. Multi-scale approaches for the simulation of cardiac electrophysiology: I – Sub-cellular and stochastic calcium dynamics from cell to organ. Methods 185, 49–59 (2021).
Dagum, L. & Menon, R. OpenMP: an industry standard API for shared-memory programming. IEEE Comput Sci. Eng. 5, 46–55 (1998).
Xia, Y., Wang, K. & Zhang, H. Parallel optimization of 3D cardiac electrophysiological model using GPU. Comput. Math. Methods Med. 2015, 1–10 (2015).
Acknowledgements
This work was supported by the Shandong Provincial Postdoctoral Program for Innovative Talents (grantee S.Z.), the Natural Science Foundation of Shandong Province (ZR2021MF011, ZR2021QF023, ZR2022QF111), the Shandong Key Science and Technology Innovation Project (2021CXGC011003), the Fundamental Research Funds for the Central Universities (21CX06018A), and the National Natural Science Foundation of China (62202498). The authors would like to thank Pei-Chi Yang and Colleen E. Clancy of the University of California Davis, and Shanzhuo Zhang of Baidu, Inc., for their helpful discussions and valuable suggestions.
Author information
Authors and Affiliations
Contributions
Conceptualization: W.L. Methodology: S.Z., S.W. Software: S.Z., W.L., M.J. Validation: S.Z., W.L., Z.L. Formal analysis: S.Z. Investigation: S.Z. Resources: W.L., F.Y., Z.L. Data curation: S.Z., W.L., X.W. Writing—original draft preparation: S.Z. Writing—review and editing: W.L., F.Y., Z.L., S.W., M.J., X.W., Z.W. Visualization: S.Z., F.Y. Supervision: W.L.., Z.W. Project administration: W.L., Z.W. Funding acquisition: S.Z., W.L., Z.W.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, S., Lu, W., Yang, F. et al. Computational analysis of arrhythmogenesis in KCNH2 T618I mutation-associated short QT syndrome and the pharmacological effects of quinidine and sotalol. npj Syst Biol Appl 8, 43 (2022). https://doi.org/10.1038/s41540-022-00254-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41540-022-00254-5
