
Abstract
Realizing active quantum control of materials near room temperature is one of the ultimate aims for their practical applications. Recent technological breakthroughs demonstrated that optical stimulation may lead to thermally inaccessible hidden states with unique properties. However, most of the reported hidden states were induced around or below liquid nitrogen temperature. Here, we optically manipulated a manganite near its Curie temperature of 300 K, where typically complex phase competitions locate as well as opportunities for new functionality. A room temperature hidden state was formed with threshold behavior evidenced by a femtosecond paramagnetic to ferromagnetic order switching and a structural change distinct from thermal induced lattice expansion in tens of picoseconds accompanying with phonon softening. We propose that such a hidden state originates from the charge transfer between antiferromagnetic chains after strongly correlated spin-charge quantum excitation, which subsequently initiates an orbital polarization rearrangement described as \({\mathrm{Mn}}_{3x^2 - {r}^2/3y^2 - {r}^2}^{3 + }{\mathrm{Mn}}^{4 + } \to {\mathrm{Mn}}^{4 + }{\mathrm{Mn}}_{3z^2 - {r}^2}^{3 + }\) and associated non-thermal lattice change. This study started from room temperature yet near a phase transition point, which suggests a new route to create or manipulate novel phases for practical purpose.
Similar content being viewed by others

Introduction
Exploiting and manipulating novel quantum phases is a central issue in the practical application of materials. A recent proposal is controlling strongly correlated electronic materials actively with femtosecond optical pulses.1,2,3,4,5,6,7,8 The materials, stimulated away from equilibrium by photons, may evolve into states which are energetically almost degenerate but thermally inaccessible, i.e., the so-called hidden states. These states may have attractive magnetic or electrical properties distinct from any other ground states in the equilibrium phase diagram.1 In recent studies, these kinds of hidden states have been successfully observed in strongly correlated materials,2,3,4,5,6,7,8 especially in manganites with charge-orbital order and antiferromagnetic (AFM) arrangements.2,3,6 The photoinduced hidden states were identified typically by an excitation threshold-like behavior, which suggests the cooperative interactions between different orders. However, up to now, most of the reported hidden states were realized based on cryogenic temperature phases of materials. To provide low-cost, compact systems for widespread applications, room temperature hidden states are ultimately pursued.
In this study, we stimulated a paramagnetic (PM) manganite La0.7Ca0.175Sr0.125MnO3 (LCSMO) at room temperature using a femtosecond laser pulse and induced a ferromagnetic (FM) hidden state with a <180 ps lifetime signed by a thermally unprocurable structure change and a phonon softening with distinct threshold behavior. This room temperature hidden state, we suggest, originates from a \({\mathrm{Mn}}_{3x^2 - {r}^2/3y^2 - {r}^2}^{3 + }{\mathrm{Mn}}^{4 + } \to {\mathrm{Mn}}^{4 + }{\mathrm{Mn}}_{3z^2 - {r}^2}^{3 + }\) inter-site charge transfer accompanying with spin, orbital and further lattice change.
Results
Experimental observation of the hidden state
We first designed the mixed-valence manganite LCSMO targeting its FM to PM phase transition at around room temperature, 300 K. Previous neutron scattering measurements demonstrated that, in La0.7A0.3MnO3 (A = Ca, Sr…), as temperature increased, nanoscale CE-type AFM clusters formed gradually and reached a maximum in number around TC.9,10,11,12 Thus, around TC the PM insulating LCSMO is essentially an aggregate of nanoscale CE-type AFM cluster,11,12,13,14 similar to the start points of the previous hidden state reports.2,3,6
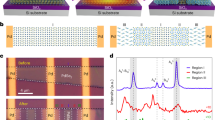
We then manipulated the LCSMO film at room temperature, i.e., close to its Curie temperature, using 40 fs, 800 nm (1.55 eV) pulses with various photon densities. The associate spin, lattice structure, and phonon softening dynamics were tracked experimentally as described in Methods with time-resolved magneto-optical Kerr effect (MOKE), ultrafast X-ray diffraction (XRD), and transient optical reflectivity measurements, respectively. When the excitation was above 2.0 mJ/cm2, we observed a PM to FM photo-switching within 200 fs, structural changes distinct from thermal dynamic lattice expansion in tens of picoseconds and a phonon softening from 40 GHz to 26 GHz up to 180 ps. The experimental evidences of the hidden state formation are presented below in the temporal sequence of their appearance.
The magnetic order change arises right after femtosecond illumination, which is typically monitored by the transient Kerr rotation change (Δθ) in MOKE measurements. Since the LCSMO sample is originally a PM insulator at room temperature, Δθ > 0 means a photoinduced magnetization process as shown in Fig. 1a. At low pump fluences, only a gradual, picosecond magnetization rise is observed. However, once the pump fluence is over 2.0 mJ/cm2, an ultrafast Kerr rotation increase with a formation time of ~200 fs is revealed (Fig. 1d), indicating that a state with magnetization larger than the original PM ground state is built up. This is similar to the recently reported photoinduced magnetic switching at 30 K in a sibling manganite compound, Pr0.7Ca0.3MnO3, from AFM to FM ordering within about 120 fs, which is distinct from the picosecond component without threshold behavior.3 The PM insulating La0.7A0.3MnO3 is actually an aggregate of nanoscale CE-type AFM clusters.11,12,13,14 Therefore, we attribute the femtosecond magnetization process in LCSMO to a PM to FM transition, while this FM state is hidden in the normal equilibrium phase diagram.

Temporal response of a Kerr rotation angle, b [002] transient lattice change, and c 800 nm optical reflectivity of the LCSMO film under different pump fluences at 300 K (F0 = 1 mJ/cm2). d Pump fluence dependence of the Kerr rotation change at 280 fs. e Pump fluence dependence of lattice changes at t = 25 ps (red solid circles) and 170 ps (green open triangles) by time-resolved XRD, and temperature dependence of the thermally induced lattice changes (blue open squares) by static XRD. f Pump fluence dependence of the amplitudes of the 26 and 40 GHz oscillations. All solid smooth curves only serve to guide the eyes. The vertical dashed lines in (d), (e), and (f) highlight the formation threshold of the hidden state. The error bars correspond to the standard error of the mean
The lattice response of this LCSMO film to laser excitation is tracked by the relative out-of-plane lattice change Δd/d along the (002) direction in ultrafast XRD measurements, as presented in Fig. 1b. The lattice constant starts to increase immediately after photoexcitation, peaks at ~25 ps (tmax) and then decays up to 170 ps, the longest experimental time delay. Static temperature dependent XRD measurements without laser illumination demonstrate that as temperature increase from 300 to 460 K, the lattice expands linearly as shown in Fig. 1e with a thermal expansion coefficient of 1.16 × 10−5 K−1 similar to the referential values.15,16 Under laser illumination, if all photons were absorbed and converted to heat finally, the average temperature of the probed area would increase by ΔT = 15 K per 1 mJ/cm2 (see Supplementary Note 1 for the calculation of ΔT). The photoinduced transient lattice change at 170 ps (Fig. 1e) agrees well with the thermal lattice expansion obtained by static XRD, indicating that at 170 ps the illuminated sample reaches a new thermal equilibrium state.
While Δd/d at 25 ps also increases correlatively with pump fluence, a clear onset threshold exists as shown in Fig. 1e, which is distinct from the thermal-like behavior at 170 ps and therefore, we propose that it is due to the formation of a hidden state. Under low pump fluences, the lattice changes at 25 and 170 ps grow with fluences coincidently. However, as the pump fluence increases to 2.0 mJ/cm2, the former starts to accelerate and break away from the latter. As a result, at a pump fluence of 9.4 mJ/cm2 the photoinduced lattice change reaches 2.8 × 10−3 at 25 ps while thermally only 1.6 × 10−3 at 170 ps. The threshold behavior and the evidently larger value of Δd/d indicate that at 25 ps the LCSMO is at a special hidden state which is thermally inaccessible.
To further understand the formation mechanism of the hidden state, we monitored the low temperature lattice dynamics at 80 K (Fig. 2a) where the LCSMO is a FM metal. Similar to the room temperature structure dynamics, under all pump fluences the lattice constant increases immediately after photoexcitation, reaches a maximum, (Δd/d)max at tmax, and then decays. However, this tmax increases from 11 to 36 ps as the pump fluence rises from 1.2 to 8.0 mJ/cm2, while it fixes to 25 ps at room temperature. Furthermore, the (Δd/d)max at 80 K exhibits linear dependence on pump fluence and no threshold behavior is observed. By converting pump fluence to laser induced temperature change ΔT, we found that the lattice expands with a thermal expansion coefficient of 1.24 × 10−5 K−1 (Fig. 2b), which is very close to the value 1.16 × 10−5 K−1 obtained by the static temperature dependent XRD. This indicates that the low temperature lattice dynamic is thermal-like. The different lattice dynamic behaviors at 80 and 300 K suggest that the observed hidden state is a unique state arising from the PM state.

a Temporal response of the [002] transient lattice change of the LCSMO film under different pump fluences at 80 K. b Pump fluence dependence of maximum lattice change (Δd/d)max. The error bars correspond to the standard error of the mean
The appearance of a hidden state is also perceived from the disparate transient reflectivity responses under low and high pump fluences. As shown in Fig. 1c, the transient reflectivity curves from 1.5 to 6.3 mJ/cm2 exhibit oscillations in GHz range, whose amplitude increases with pump fluence. After applying fast Fourier transform (FFT) to the oscillatory part of the reflectivity curves (Supplementary Note 2), we found that under low pump fluences, the reflectivity curve oscillated at 40 GHz. Right above 2.0 mJ/cm2, a new softer 26 GHz oscillation starts to appear (Fig. 1f), which suggests that it is the manifestation of a hidden state, while phonon softening is typically affiliated to structural changes. The interpretation of the phonon softening mechanism is detailed in Supplementary Note 3.
Mechanism analysis of the hidden state
Considering that the photoinduced magnetic order switching, the thermally inaccessible structural changes and phonon softening have the same threshold, 2.0 mJ/cm2, we suggest that although in different timescales and from different order perspectives, they are different manifestations of a same hidden state. Next, we focus on the formation mechanism of this hidden state. In mixed-valence manganites with CE-type AFM order, one-dimensional zig-zag chains with alternating manganesian Mn3+/Mn4+ ions (charge-orbital order) and ferromagnetically aligned spins are antiferromagnetically ordered in the a–b plane (Fig. 3a). In this arrangement, one Mn3+ ion is surrounded by four Mn4+ ions in the same a–b plane and two other Mn3+ ions in the two neighboring a–b planes.2,17,18 The eg orbitals on the Mn3+ site are known to split into a lower 3x2−r2/3y2−r2 level (eg1) and a higher x2−z2/y2−z2 level (eg2) owing to Jahn-Teller (JT) distortion, with the eg electron occupying the lower one, while those eg orbitals on the Mn4+ site are degenerate. After photon illumination in 1–2 eV range, the eg electron on Mn3+ was suggested to be excited priorly.19,20,21 Three transition paths from the lower Mn3+ eg level were proposed as shown schematically in Fig. 3b: (I) to the on-site higher Mn3+ eg level; (II) to the neighboring non-split Mn4+ eg level; (III) to the higher eg level of the neighboring Mn3+. For the employed 1.55 eV photoexcitation, we agree with many other studies on that Transition II is the most preferred path.19,20,21 Transition I is firstly excluded because this intra-site d–d transition is not allowed by the electric dipole selection rule; besides, even if Transition I might happen, ~0.7 eV photoexcitation was enough to overcome the JT splitting energy EJT,22,23 which is obviously much smaller than the 1.55 eV photon energy. Second, Transition III can also be excluded because this transition must overcome EJT (~0.7 eV) as well as an on-site repulsion U coming from the lower level Mn3+ eg electron (1.7 eV),22,23 and their sum, EJT + U, overruns the 1.55 eV photon energy too much. While for Transition II, the 1.55 eV right falls into its excitation energy range EB + 3Vi ± 0.45 eV, where EB ~ 0.8EJT is the breathing distortion energy,24 Vi ~ 0.44 eV (i = 1, 2, 3) is the inter-site Coulomb repulsion from other Mn3+ ions adjacent to the Mn4+ ion marked as 1, 2, and 3 in Fig. 3a,21 and 0.45 eV is the fluctuation of the threshold for the inter-site d–d transition induced by band dispersion.25

a Scheme of the charge-orbital-ordered CE-type AFM cluster. The black dashed lines represent the one-dimensional zig-zag chains. Processes A and B describes the intra- and inter-chain charge transfer induced by 1.55 eV photoexcitation, respectively. b t2g and eg energy levels of Mn ions and possible transition paths of eg electrons of Mn3+ ion. c The ABO3 type lattice structure of LCSMO and the lengths of Mn–O bonds around Mn3+ (left) and Mn4+ (right). d Orbital polarization rearrangement and out-of-plane lattice expansion induced completely by Transition II (B), and Δd/d = 11.4 × 10−3 at 9.4 mJ/cm2
The magnetism state in manganites could be modulated by photons through two routes: a direct electronic transition or lattice–spin interaction. In the first route, the magnetization will be induced by the ultrafast spin-flip/canting driven by the direct electronic transition within 200 fs.3 For Transition II, normally a Mn3+ to Mn4+ charge transfer within the same one-dimensional zig-zag chain (intra-chain) is suggested (marked as A in Fig. 3a). However, because the spins within one zig-zag chain are parallel, femtosecond spin-flip/canting could not be induced by the intra-chain charge transfer.3,18,26 During the second route, a Mn3+ to Mn4+ charge transfer will instantly turn the sample into a JT inactive state within 100–200 fs presented as the first drop in reflectivity (Fig. 1c and S2). Then, the new JT polarons will form with a phonon period of 500–600 fs and thermalize afterward.27,28 Next, they may influence the magnetism state in manganites through lattice–spin interaction, which usually occurs on picosecond timescale limited by the relatively weak lattice–spin coupling.29,30 Thus, Transition II (A) should not be the origin of the femtosecond ferromagnetic hidden state observed by our time-resolved MOKE measurements.
Furthermore, the lattice change induced by Transition II (A) is relatively small compared with the value observed experimentally when the pump fluences above 2 mJ/cm2 at room temperature (Table S1), indicating also that Transition II (A) is not the origin of the hidden state. After photoexcitation, the lattice could change through two paths: orbital rearrangement or lattice thermal expansion. Transition II (A) will induce an orbital rearrangement described as \({\mathrm{Mn}}_{3x^2 - {r}^2/3y^2 - {r}^2}^{3 + }{\mathrm{Mn}}^{4 + } \to {\mathrm{Mn}}^{4 + }{\mathrm{Mn}}_{3x^2 - {r}^2/3y^2 - {r}^2}^{3 + }\) and the overall effect is similar to a position exchange of Mn3+ and Mn4+ (Fig. 4). Thus, the lattice change through orbital rearrangement in Transition II (A) is negligible. While for lattice thermal expansion, assuming that all the absorbed photon energy at 9.4 mJ/cm2 was converted into thermal energy, the induced lattice expansion would be ~1.6 × 10−3, which is much smaller than the observed 2.8 × 10−3 lattice change for the hidden state (Supplementary Note 4).

Schematic illustration of orbital rearrangement induced by Transition II (a) and (b), which subsequently results in the corresponding structure change, during the evolution of the hidden state. The blue and pink backgrounds stand for the status before and after photoexcitation, respectively. The blue arrows describe the AFM order between neighboring chains and the red arrows describe the inter-chain FM coupling after spin-flip/canting induced by electron transition
Recent studies on the magnetic and structural dynamics of manganites proposed that a charge transfer between two neighboring one-dimensional zig-zag chains with antiparallel spins could also occur (marked as B in Fig. 3a), when excited above a certain pump fluence.2,3,18 Compared with Transition II (A), Transition II (B) could result in complicated and significant changes, both in spin and lattice structure. According to a recent report, during this type of transition, a quantum spin fluctuation may be excited, which will result in a femtosecond spin-flip/canting and consequently an inter-chain FM coupling.3 Furthermore, this transition will also induce an orbital polarization rearrangement described as \({\mathrm{Mn}}_{3x^2 - {r}^2/3y^2 - {r}^2}^{3 + }{\mathrm{Mn}}^{4 + } \to {\mathrm{Mn}}^{4 + }{\mathrm{Mn}}_{3z^2 - {r}^2}^{3 + }\),2 which may originate from complex spin–orbital coupling.31 As analyzed below, this transition will result in a distinct out-of-plane lattice expansion. For LCSMO with an ABO3 type structure (Fig. 3c), the interplanar spacing d (002) is nearly the same as the average length of all out-of-plane Mn–O bonds. For the Mn4+ ion, because the eg orbitals are empty, the lattice is undistorted and six Mn–O bonds are co-elongate (1.90 Å) (Fig. 3c).32 However, owing to the occupation of the 3x2−r2/3y2−r2 orbital by one electron, the JT distorted lattice surrounding Mn3+ leads to two long (2.15 Å, in plane) and four short (1.94 Å, two in plane and two out-of-plane) Mn–O bonds (Fig. 3c).32 After illumination through Transition II (B), the orbitals and subsequently the Mn–O bonds are rearranged. The overall effect is that two short out-of-plane Mn–O bonds (1.90 Å) become two longer ones (2.15 Å) (Fig. 3d). Thus, a significant out-of-plane lattice expansion is induced, which we propose is responsible for the experimental observed distinct lattice change. Considering the unique characters of the hidden state, i.e., the magnetic order switching and the significantly larger lattice constant change compared with its thermal dynamic counterpart, we suggest that the observed hidden state originates from Transition II (B).
This deduction is further confirmed by comprehensively analyzing the out-of-plane and in-plane lattice structure dynamics monitored along the [002] and [011] (Fig. S4) directions. As estimated in Supplementary Note 4, if all photons excited Transition II (B), the resulted bond rearrangement at 9.4 mJ/cm2 would generate a lattice expansion of 11.4 × 10−3 in the [002] orientation, which is much larger than the experimentally observed value, 2.8 × 10−3. Considering the threshold behavior of the lattice change, we suggest that at low pump fluences, most, if not all, photons involve in Transition II (A); while once the pump fluence is higher than 2.0 mJ/cm2, they start to participate in Transition II (B). We hence calculated the fractions of pumping photons used for both transitions, and found that at 9.4 mJ/cm2, ~11% photons induce Transition II (B), while the remainders initiate Transition II (A) (detailed in Supplementary Notes 4–6). In a similar way, we calculated the pumping fractions based on the dynamic response of the [011] orientation, and reached the same conclusion, indicating that the hidden state is intrinsically from Transition II (B).
Summary of the physical property evolution during hidden state
In the following, we discuss the physical property evolution of the LCSMO hidden state from Transition II (B). First, as described above, during the initial ~200 fs, a FM hidden state is initiated. The generation of a FM hidden state is suggested to be accompanied with a quasi-instantaneous transient insulator to metal transition by a recent theoretical study18 and verified in a similar system Gd0.55Sr0.45MnO3 experimentally.33 Although we did not track the transient electric transport response of the LCSMO film in this study, we suggest that at ~200 fs the LCSMO is also metallic. Second, although the FM property lasts up to hundreds of picoseconds, the metallic state always decays to an insulating state within 1 ps.33 Thus, at 25 ps, when the lattice structure of the hidden state is established, the illuminated LCSMO is suggested to be a FM insulator. Third, as the lattice starts to decrease from 25 ps, the FM property keeps rising slightly until hundreds of picoseconds. A further analysis of the transient reflectivity oscillations also demonstrated that the hidden state is metastable with a lifetime shorter than 180 ps. We applied FFT to the different segments of the dynamic reflectivity curve obtained at 6.3 mJ/cm2, as presented in Fig. S2c, and found that the 26 GHz oscillation exists only in the first 0–120 ps, decays to 40 GHz during 120–180 ps, and disappears completely in the 180–340 ps segment, where the 40 GHz oscillation arises. This is in accordance with the lattice dynamics, which suggests that at 170 ps the illuminated sample reaches a new thermal equilibrium state, while residual magnetism still exists.
In summary, the development of the hidden state can be described as sketched in Fig. 4: after high-intensity photoexcitation, the inter-chain charge transfer disturbs the original spin ordering, building up a FM metallic hidden state in ~200 fs; this charge transfer also initiates an orbital polarization rearrangement described as \({\mathrm{Mn}}_{3x^2 - {r}^2/3y^2 - {r}^2}^{3 + }{\mathrm{Mn}}^{4 + } \to {\mathrm{Mn}}^{4 + }{\mathrm{Mn}}_{3z^2 - {r}^2}^{3 + }\) and subsequently induces a reorientation of the lattice structure, establishing a new lattice structure within 25 ps as a FM insulator; the hidden state decays to a new thermodynamic insulating equilibrium state through the release of the excessive elastic energy from the newly reoriented sites to the remaining non-reoriented sites in <180 ps but with residual magnetism; by transferring energy to the surrounding environment, the illuminated area finally recovers to the initial PM-insulator status on a nanosecond to microsecond timescale.34,35 However, it should be noted that, after photon illumination, the electron transitions between different AFM chains are excited and induce spin flips on single sites. It is still not clear to us even schematically how those locally site-specific spin flips interact with each other, influence other unexcited sites, assemble together and form a macroscopic observable FM order. Magnetic force microscope measurements with a time resolution better than 100 fs may give us a direct view of this process, which though have not been realized recently limited by the development of such technique.
Discussion
The above analysis suggests that different degrees of freedom have various responses at individual timescales, which results from the complex cooperative interactions among spin, orbital and lattice in strongly correlated materials. We now discuss how those interactions initiate these correlated phenomena. On one hand, theoretical studies3,18 pointed out that charge transfer between AFM chains, which is prohibited in classical spin models but can be well described using quantum spin scenarios, will induce a spin-flip/canting of the migrated electron from Mn3+ to Mn4+ site and subsequently an inter-chain FM coupling. This explains why we observe a FM coupling in LCSMO after intense photoexcitation. On the other hand, the charge transfer disturbs the orbital ordering, resulting in the transient conductivity. Originally, the eg electron is localized to Mn3+, forming a JT polaron due to the orbital and lattice interaction. The photoinduced Mn3+ to Mn4+ charge transfer releases the polaron and the localized eg electron becomes itinerant transiently in the a–b plane.33,36,37 Furthermore, the charge transfer may also initiate a quantum electron tunneling between the a–b planes and open up an inter-plane electron hopping channel, making the LCSMO more conductive.18 Both as intrinsic natures of electrons, the spin and orbital ordering changes are suggested to occur simultaneously after electron transition. This explains the instantaneous conductivity rise accompanying with the spin ordering switch within 200 fs. Next, as shown in Fig. 3a, if an inter-chain charge transfer occurs, the electron migrated to the Mn4+ site should be strongly repulsed by the eg electrons occupying the 3x2−r2 and 3y2−r2 orbitals of the two neighboring Mn3+ sites. Thus, for the excited electron, a 3z2−r2 occupation is preferred. Such orbital occupation results in an out-of-plane lattice stretching,38 i.e., the thermally inaccessible lattice expansion observed by the time-resolved XRD measurement.
Previously a simplified energy transfer scheme was applied to explain the complex interactions among several degrees of freedom.30 Such a scheme may also describe the processes during the development of the observed hidden state as follows: first, the electrons absorb photon energy directly, resulting in the spin and orbital ordering change simultaneously; second, the energy is transferred to the lattice system through either spin–lattice or orbital–lattice interactions (or both), leading to the lattice expansion. However, the actual processes might be much more complex than above. For instance, although several studies suggested that to realize femtosecond magnetization, spin should response to photoexcitation in the first place followed by spin–orbital interaction,3,31 here we propose but could not conclude that ultrafast magnetization takes precedence over lattice changes limited by the lower temporal resolution of our time-resolved XRD compared with that of time-resolved MOKE. Furthermore, there might be interactions between spin and orbital degrees (through angular momentum exchange), which bridge spin and lattice.31,39 Since the above simplified energy transfer scheme does not include the angular momentum exchange between subsystems,40 the interaction between spin and lattice cannot be interpreted directly here. These interesting open questions are critical for a thorough understanding of the evolution of the hidden state and hence for its active control.
In conclusion, we observed a room temperature hidden state formed in 25 ps induced by an 800 nm femtosecond photoexcitation. This hidden state is mainly evidenced by a lattice structure change distinct from the photoinduced thermal expansion as well as a PM to FM ultrafast switching and a 40 to 26 GHz phonon softening above a pump fluence of ~2.0 mJ/cm2. We suggest that the FM hidden state originates from the photoinduced charge transfer between AFM chains in the CE-type AFM clusters, which subsequently initiates an orbital polarization rearrangement described as \({\mathrm{Mn}}_{3x^2 - {r}^2/3y^2 - {r}^2}^{3 + }{\mathrm{Mn}}^{4 + } \to {\mathrm{Mn}}^{4 + }{\mathrm{Mn}}_{3z^2 - {r}^2}^{3 + }\). Similar strategy may be applied to search hidden state in other strongly correlated materials, for example, we may stimulate from a non-superconducting ground state near transition point and seek for superconducting hidden states. This exploration of the hidden state at room temperature, yet near a transition point, provides a new insight to create or manipulate novel phases for practical purpose. Our study also demonstrates that time-resolved XRD is a powerful method for not only identifying hidden states, but also revealing the underlying physical mechanisms.
Methods
Two LCSMO films grown on (001) and (011) perovskite LaAlO3 (LAO) substrates were employed to systematically investigate the in-plane and out-of-plane lattice dynamics. The two LCSMO films with a similar thickness of 200 ± 10 nm were grown by the sol–gel method reported previously.41 The sizes of the films are 10 mm × 10 mm, which is large enough for the measurements of both the irradiated and non-irradiated area simultaneously by time-resolved XRD. The static XRD pattern measured with Cu Kα (Fig. S6a) demonstrates that the LCSMO films are well crystallized with a (002) and (011) diffraction angle of 23.50° and 16.40°, corresponding to an interplanar spacing of 1.934 and 2.732 Å, respectively. The LCSMO films experience a FM-metal to PM-insulator phase transition at ~300 K (TC), as exhibited by the resistivity spectra and the magneto-resistance curves (Fig. S6b, c).
All the time-resolved MOKE, XRD and reflectivity measurements were carried out with a 1 kHz Ti:sapphire laser system delivering 40 fs, 5 mJ pulses at a center wavelength of 800 nm (1.55 eV). In all measurements, 800 nm laser pulses were mainly used as the pump with fluences varying from 0.1 to 9.4 mJ/cm2, well below the damage threshold of LCSMO (~30 mJ/cm2). The absorption coefficient of LCSMO is 9 × 104 cm−1 at 800 nm,42 corresponding to a penetration depth of ~110 nm. In transient MOKE and reflectivity measurements, the probe fluence was fixed at ~ 2 μJ/cm2, which was only 2% of the lowest pump fluence.
In time-resolved XRD measurements with a temporal resolution of ~1 ps, sub-picosecond Cu Kα (1.542 Å) X-ray pulses generated by a home-build table-top X-ray plasma source schemed on Fig. S7 were employed as a transient structural probe. The emitted X-ray was shaped to a divergence of 0.6° by a tungsten slit. Since the (002) diffraction angle of the LCSMO sample is 0.5° smaller than that of the LAO substrate, only the diffraction rocking curve of the LCSMO and its changes within ±0.3° were monitored simultaneously. On the sample, the diameter of the pump beam (~3 mm) was larger than the transverse size of the X-ray spot (~2 mm) to ensure well overlapping in space. A typical diffraction pattern was obtained with a 60-s exposure. Other details for the sample preparation and experimental techniques are described in Supplementary Note 7.
Data Availability
The authors declare that the data supporting the findings of this study are available within the article and its Supplementary Information files.
References
Tokura, Y. Photoinduced phase transition: a tool for generating a hidden state of matter. J. Phys. Soc. Jpn. 75, 011001 (2006).
Ichikawa, H. et al. Transient photoinduced ‘hidden’ phase in a manganite. Nat. Mater. 10, 101–105 (2011).
Li, T. et al. Femtosecond switching of magnetism via strongly correlated spin-charge quantum excitations. Nature 496, 69–73 (2013).
Stojchevska, L. et al. Ultrafast switching to a stable hidden quantum state in an electronic crystal. Science 344, 177–180 (2014).
Fukaya, R. et al. Ultrafast electronic state conversion at room temperature utilizing hidden state in cuprate ladder system. Nat. Commun. 6, 8519 (2015).
Zhang, J. et al. Cooperative photoinduced metastable phase control in strained manganite films. Nat. Mater. 15, 956–960 (2016).
Morrison, V. R. et al. A photoinduced metal-like phase of monoclinic VO2 revealed by ultrafast electron diffraction. Science 346, 445–448 (2018).
Sun, K. et al. Hidden CDW states and insulator-to-metal transition after a pulsed femtosecond laser excitation in layered chalcogenide 1T-TaS2-xSex. Sci. Adv. 4, eaas9660 (2018).
Adams, C. P., Lynn, J. W., Mukovskii, Y. M., Arsenov, A. A. & Shulyatev, D. A. Charge ordering and polaron formation in the magnetoresistive oxide La0.7Ca0.3MnO3. Phys. Rev. Lett. 85, 3954–3957 (2000).
Lynn, J. W. et al. Order and dynamics of intrinsic nanoscale inhomogeneities in manganites. Phys. Rev. B 76, 014437 (2007).
Chen, Y. et al. Polaron formation in the optimally doped ferromagnetic manganites La0.7Sr0.3MnO3 and La0.7Ba0.3MnO3. Phys. Rev. B 78, 212301 (2008).
Maschek, M. et al. Polaronic metal phases in La0.7Sr0.3MnO3 uncovered by inelastic neutron and x-ray scattering. Phys. Rev. B 93, 045112 (2016).
Dai, P. et al. Short-range polaron correlations in the ferromagnetic La1-xCaxMnO3. Phys. Rev. Lett. 85, 2553–2556 (2000).
Adams, C. P. et al. First-order nature of the ferromagnetic phase transition in (La-Ca)MnO3 near optimal doping. Phys. Rev. B 70, 134414 (2004).
Grande, T., Tolchard, J. R. & Selbach, S. M. Anisotropic thermal and chemical expansion in Sr-substituted LaMnO3+δ: implications for chemical strain relaxation. Chem. Mater. 24, 338–345 (2011).
Shayduk, R. et al. Nanoscale heat transport studied by high-resolution time-resolved x-ray diffraction. New J. Phys. 13, 093032 (2011).
Tokura, Y. & Nagaosa, N. Orbital physics in transition-metal oxides. Science 288, 462–468 (2000).
Lingos, P. C. et al. Correlating quasiparticle excitations with quantum femtosecond magnetism in photoexcited nonequilibrium states of insulating antiferromagnetic manganites. Phys. Rev. B 95, 224432 (2017).
Zhao, Y. G. et al. Ultrafast laser induced conductive and resistive transients in La0.7Ca0.3MnO3: charge transfer and relaxation dynamics. Phys. Rev. Lett. 81, 1310–1313 (1998).
Kim, K. H., Lee, S., Noh, T. W. & Cheong, S. W. Charge ordering fluctuation and optical pseudogap in La1-xCaxMnO3. Phys. Rev. Lett. 88, 167204 (2002).
Volja, D., Yin, W.-G. & Ku, W. Charge ordering in half-doped manganites: weak charge disproportion and leading mechanisms. EPL 89, 27008 (2010).
Kovaleva, N. N. et al. Spin-controlled Mott-Hubbard bands in LaMnO3 probed by optical ellipsometry. Phys. Rev. Lett. 93, 147204 (2004).
Yin, W. G., Volja, D. & Ku, W. Orbital ordering in LaMnO3: electron-electron versus electron-lattice interactions. Phys. Rev. Lett. 96, 116405 (2006).
Calderón, M. J., Millis, A. J. & Ahn, K. H. Strain selection of charge and orbital ordering patterns in half-doped manganites. Phys. Rev. B 68, 100401 (2003).
Li, J. et al. Dichotomy in ultrafast atomic dynamics as direct evidence of polaron formation in manganites. npj Quantum Mater. 1, 16026 (2016).
Ehrke, H. et al. Photoinduced melting of antiferromagnetic order in La0.5Sr1.5MnO4 measured using ultrafast resonant soft x-ray diffraction. Phys. Rev. Lett. 106, 217401 (2011).
Beaud, P. et al. Ultrafast structural phase transition driven by photoinduced melting of charge and orbital order. Phys. Rev. Lett. 103, 155702 (2009).
Jang, K.-J. et al. Ultrafast near-infrared spectroscopic study of coherent phonons in the phase-separated manganite La1/4Pr3/8Ca3/8MnO3. Phys. Rev. B 81, 214416 (2010).
Averitt, R. D. et al. Ultrafast conductivity dynamics in colossal magnetoresistance manganites. Phys. Rev. Lett. 87, 017401 (2001).
Muller, G. M. et al. Spin polarization in half-metals probed by femtosecond spin excitation. Nat. Mater. 8, 56–61 (2009).
Kirilyuk, A., Kimel, A. V. & Rasing, T. Ultrafast optical manipulation of magnetic order. Rev. Mod. Phys. 82, 2731–2784 (2010).
Booth, C. H. et al. Lattice effects in La1-xCaxMnO3 (x=0–1): relationships between distortions, charge distribution, and magnetism. Phys. Rev. B 57, 10440–10454 (1998).
Matsubara, M. et al. Ultrafast photoinduced insulator-ferromagnet transition in the perovskite manganite Gd0.55Sr0.45MnO3. Phys. Rev. Lett. 99, 207401 (2007).
Bielecki, J. et al. Two-component heat diffusion observed in LaMnO3 and La0.7Ca0.3MnO3. Phys. Rev. B 81, 064434 (2010).
Ogasawara, T. et al. General features of photoinduced spin dynamics in ferromagnetic and ferrimagnetic compounds. Phys. Rev. Lett. 94, 087202 (2005).
Sheu, Y. M. et al. Polaronic transport induced by competing interfacial magnetic order in a La0.7Ca0.3MnO3/BiFeO3 heterostructure. Phys. Rev. X 4, 021001 (2014).
Zhang, H. et al. Strong influence of polaron-polaron interaction on the magnetoresistance effect in La0.7A0.3MnO3 thin films. Appl. Phys. Lett. 111, 192408 (2017).
Chakhalian, J. et al. Asymmetric orbital-lattice interactions in ultrathin correlated oxide films. Phys. Rev. Lett. 107, 116805 (2011).
Koopmans, B., Kicken, H. H. J. E., van Kampen, M. & de Jonge, W. J. M. Microscopic model for femtosecond magnetization dynamics. J. Magn. Magn. Mater. 286, 271–275 (2005).
Cywiński, Ł. & Sham, L. J. Ultrafast demagnetization in the sp-d model: a theoretical study. Phys. Rev. B 76, 045205 (2007).
Zhang, Y. et al. Large room-temperature magnetoresistance in epitaxial La0.7Ca0.25Sr0.05MnO3 thin films prepared by sol–gel method. J. Sol.-Gel Sci. Technol. 78, 576–581 (2016).
Ren, Y. H., Ebrahim, M., Xu, Z. A. & Lüpke, G. Ultrafast quasi-particle dynamics of charge/orbital ordered and ferromagnetic clusters in La0.7Ca0.3MnO3. New J. Phys. 11, 113013 (2009).
Acknowledgements
The authors thank Dr. Jie Ma for stimulating discussions. This work was supported by the National Natural Science Foundation of China under Grant Nos. 11574196, 11704246, 61222509, and 11721091.
Author information
Authors and Affiliations
Contributions
H.-J.Z. and J.C. designed the research, analyzed the data, and wrote the paper. H.-J.Z. carried out the experiments. Y.-Y.Z., W.-X.D., and X.-D.T. synthesized the samples. R.-Z.L., J.-X.Y., C.-L.C., and K.-D.W. supplied help during the experiments, data analysis, and paper preparation. All authors discussed and made comments on the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhang, H., Zhang, Y., Li, R. et al. Room temperature hidden state in a manganite observed by time-resolved X-ray diffraction. npj Quantum Mater. 4, 31 (2019). https://doi.org/10.1038/s41535-019-0170-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41535-019-0170-3
