
Abstract
The broad employment of clean hydrogen through water electrolysis is restricted by large voltage requirement and energy consumption because of the sluggish anodic oxygen evolution reaction. Here we demonstrate a novel alternative oxidation reaction of green electrosynthesis of valuable 3,3’-diamino-4,4’-azofurazan energetic materials and coupled with hydrogen production. Such a strategy could greatly decrease the hazard from the traditional synthetic condition of 3,3’-diamino-4,4’-azofurazan and achieve low-cell-voltage hydrogen production on WS2/Pt single-atom/nanoparticle catalyst. The assembled two-electrode electrolyzer could reach 10 and 100 mA cm–2 with ultralow cell voltages of 1.26 and 1.55 V and electricity consumption of only 3.01 and 3.70 kWh per m3 of H2 in contrast of the conventional water electrolysis (~5 kWh per m3). Density functional theory calculations combine with experimental design decipher the synergistic effect in WS2/Pt for promoting Volmer–Tafel kinetic rate during alkaline hydrogen evolution reaction, while the oxidative-coupling of starting materials driven by free radical could be the underlying mechanism during the synthesis of 3,3’-diamino-4,4’-azofurazan. This work provides a promising avenue for the concurrent electrosynthesis of energetic materials and low-energy-consumption hydrogen production.
Similar content being viewed by others


Introduction
Excessive consumption of fossil fuels leads to the aggravating environmental pollution, and greenhouse effect. Hydrogen (H2) is considered as one of the promising alternative energy carrier benefiting from its sustainable, zero-carbon emission, regenerative, and high intrinsic energy density (≈282 kJ mol−1) nature1,2. Electrochemical overall water splitting (OWS) powered by abundant solar or wind energy sources etc. represents a sustainable approach to produce H23. Yet, its implementation faces challenges of heavy electricity consumption (>4.5 kWh m−3 H2) due to the low-energy conversion efficiency and high operating voltage (>1.8 V), majorly originated from the sluggish four-electron transfer kinetics of oxygen evolution reaction (OER) in the anode2,4,5. In extreme cases, water electrolysis produces H2/O2 mixtures crossover through the membrane may give rise to gas explosive, causing considerable security risks6. In addition, the O2 obtained from water electrolysis is relatively less valuable because it can be expediently obtained from air7,8. In parallel to the design of advanced electrocatalysts for OER, considerable research efforts are required to explore alternative technological concepts to surpass the conventional OWS technology and tackle the above drawbacks brought by OER9.
Given the challenges in conventional OWS, efficient alternative strategies have been recently employed. Among them, the co-electrolysis system, appending different electrolytes in the anode and coupled with HER to integrate hybrid electrolysis, have witnessed increasing attention10. For instance, the HER paired with some thermodynamically more favorable electrochemical oxidation reactions such as hydrazine11,12,13, urea14,15, alcohols1,16, aldehyde17,18 etc., significantly lowering the required cell voltage of OWS for low-energy-consumption hydrogen production at the cathode. Meanwhile, the approach simultaneously provides either value-added fine chemicals or safe gas from pollutant degradation (e.g., N2) at the counter side. Moreover, this co-electrolysis strategy could avoid O2 generation and eliminate the risk of explosive H2/O2 mixtures, making highly-efficient membrane-less OWS reactors possible19. In view of this co-electrolysis strategy is still in the initial stage, intensive research should be conducted to explore more promising alternative electrooxidation reactions in the configuration of hybrid OWS system, particularly for the green and sustainable pathway to synthesize value-added chemicals, so that to expand the versatility and enhance the industrialization possibility of this strategy8.
Furazan chemistry has proceed over 140 years, many derivatives have been synthesized and used for diverse applications, ranging from biological medicine to explosive and propellant ingredients20. Among them, the azofurazan structural motif exists in the form of crown ether analogs and considered as biologically active molecules suppressant of the soluble guanylate cyclase20. The azofurazan framework characterized with high-nitrogen and high heats of formation features have been most extensively researched as energetic materials (EMs) due to the large number of N–N and C–N bonds and the thermodynamically favored formation of N221. However, the traditional pathway for the synthesis of azofurazan EMs is an oxidation reaction from aminofurazans that often involves high-temperature synthetic condition and hazardous reagents such as KMnO4/H+, CrO3/AcOH, (NH4)2S2O8 or various of organic reagents, which are a potential danger for the safety hazard and environmental pollution. In addition, the separation of product from excess of an oxidant and other by-products is difficult20,21. These drawbacks have compelled in a host of ecological concerns, the green synthetic techniques have been deserved great attention in the modern synthetic chemist. One such green technique, electrosynthesis has gone through a renaissance of sorts and considered an environmentally benign methodology in recent years22,23,24. The electron in electrosynthesis represents a clean and environmentally friendly reagent for direct oxidation, as it produces no waste instead of hazardous oxidizing agents. The electron offers greater economy in contrast to chemical redox reagents in view of the reagent cost per mole of product produced25. Moreover, mild synthetic condition is feasible because the energy supplied to the electrode could be controlled by the applied cell voltage and current density instead of the heat or high pressure. In terms of the advantages that the synthetically organic electrochemistry offers, many pioneer literatures have been reported detailing the utility of electro-synthetic organic chemicals through the coupling of C–H/X–H (X = C, O, S, N etc.) to construct C–C, C–O, C–S bonds etc23,24. Unfortunately, there has been a notable lack of researches on the electrosynthesis of organic EMs, especially for electrochemical N=N bond formations to generate azofurazan EMs20,26. More importantly, the formation of N=N bond was found thermodynamically more favorable than OER during the electrosynthesis of some azo EMs27,28. Therefore, it is speculated that the sluggish OER is replaced by electrosynthesis of azofurazan EMs and coupled with OWS may significantly reduce the cell voltage of OWS for low-energy-consumption hydrogen production, meanwhile, the OER is inhibited in the coupling system that could avoid many drawbacks brought by oxygen. Moreover, the azofurazan EMs are obtained at the anode through green and mild electrooxidation pathway.
With this idea in mind, a low-energy-consumption coupling system was constructed in our work by replacing OER with thermodynamically more favorable electrooxidation of 3,4-diaminofurazan (DAF) to synthesize 3,3’-diamino-4,4’-Azofurazan (DAAzF) EMs. The platinum single atoms/nanoparticles (Pt1,n) supported on the tungsten disulfide nanosheets and loaded on the 3D conductive carbon cloth (CC@WS2/Pt1,n) was prepared as high-efficiency HER catalysts. The Pt nanoparticles (Pt NPs) loaded near the Pt single atoms (Pt SAs) may induce the variation of electronic metal-support interaction (EMSI) to regulate the charge redistribution between the neighboring Pt NPs/SAs and support, which was closely related to the catalytic activity29. It is noted that single atoms are not the only active sites in many single-atom catalysts, where the nanoclusters exist as well due to the extremely high surface free energy of single atoms, which is usually overlooked30. Hence, the formation of Pt1,n on the substrate is energetically more favored and more stable than pure Pt SAs under long-term harsh alkaline working condition31. The only single atoms active sites may also not exhibit their overwhelming advantages in multi-step catalytic process (e.g., alkaline HER)32,33,34. Compared with carbon-based substrates, the transition metal dichalcogenides (TMDs) supported metal single atoms/nanoparticles could adjust the electronic structure through both anchoring atom and the adjacent transition metal atoms with higher atomic number, which provided more flexible local coordination environment to regulate the catalytic activity35. The core anchoring chalcogen (S) and the adjacent transition metal (W) can synergistically regulate the d-orbital state electronic structure of metal single atoms/nanoparticles through EMSI. As a result, the adsorption energy of intermediates on active sites could be optimized and thus has influence on the catalytic activities36. As for one of the important candidates in TMDs, the metallic WS2 achieved high catalytic activity for HER compared with other TMDs because the basal plane of metallic WS2 was also catalytically active than natural MoS237,38. The periodic density functional theory calculations also indicated that the catalytic activity of WS2 was predicted to be comparable to or even better than MoS239. The as-prepared CC@WS2/Pt1,n can be directly used as self-supporting electrode for superior alkaline HER. DFT calculations revealed the facile energy barrier of water dissociation and H intermediates (H*) contributed by the synergistic effect among Pt NPs, Pt SAs, and WS2 substrate, resulting in high alkaline HER performance. The as-prepared CC@WS2/Pt1,n cathode integrated with anode electrode (copper oxide nanowires) to construct two-electrode coupling system, which can produce H2 with an ultralow cell voltage in contrast to traditional OWS, realizing low-energy-consumption hydrogen production. Meanwhile, green electrosynthesis of DAAzF was achieved to avoid hazardous synthetic condition of traditional strategy (Fig. 1).

a Traditional pathway to synthesize DAAzF EMs and the corresponding drawbacks. b Green electrochemical pathways to synthesize DAAzF EMs and coupled with hydrogen production and corresponding advantages.
Results
Preparation and structural characterizations of cathodic CC@WS2/Pt1,n catalyst
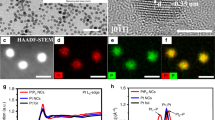
The cathode CC@WS2/Pt1,n catalysts was synthesized based on the hydrothermal reaction to produce WS2 nanosheets that was supported on the CC (CC@WS2 NSs), followed by the electrochemical cyclic voltammetry (CV) method at a constant potential range to homogeneously distribute Pt NPs and Pt SAs (Pt1,n) on the CC@WS2 NSs (Fig. 2a). Scanning electron microscope (SEM) shows that dense of cross-linking WS2 NSs are uniformly supported on the conductive CC substrate (Supplementary Fig. 1). Transmission electron microscopy (TEM), high-resolution TEM (HRTEM), and elemental mapping indicated the uniform distribution of W and S elements on the WS2 NSs (Supplementary Fig. 2). After the electrochemical deposition of Pt1,n, negligible differences in morphology were observed in SEM indicating the sub-nanometer clusters or single atoms of Pt in WS2/Pt1,n (Fig. 2b). Because the HER performance depends largely on the content of Pt1,n, if not otherwise stated, the CC@WS2/Pt1,n mentioned in the following sections was the optimal Pt content (2.0 mL Pt4+, 10 mg mL–1 in the electrolyte). To further verify the structure of Pt, aberration-corrected high-angle annular dark-field (AC-HAADF-STEM) was performed and shown in Fig. 2c, d. The prepared Pt with the nanoparticles and single atoms (within white dotted circle) forms were dispersed uniformly on the surface of corrugate WS2 NSs. The average size of Pt NPs was determined to 2.6 nm (Supplementary Fig. 3), and the content of Pt1,n in WS2/Pt1,n was 0.106 mgPt cm–2 as determined by the inductively coupled plasma mass spectrometer (ICP-MS). The average lattice spacing of 0.232 nm can be indexed to the Pt (111) plane, indicating the successfully synthesis of Pt NPs without any impurities (Fig. 2e)40. Besides, the elemental mapping of the WS2/Pt1,n showed Pt1,n along with W and S were homogeneously distributed on the WS2 NSs (Fig. 2f–i). Notably, the concordant coexistence of Pt NPs and Pt SAs on the WS2 substrate was attributed to the strong coupling effect among these three active components in virtue of EMSI, which could synergistically promote alkaline HER (as discussed in the electrochemical tests and theoretical calculations section).

a Schematic illustration of synthetic procedure for CC@WS2/Pt1,n. b SEM, c TEM, d AC-HAADF-STEM images of CC@WS2/Pt1,n (scale bar: 200 nm for b, 50 nm for c, 2 nm for d). e Corresponding intensity profiles along the line of d. f–i HAADF-STEM image and elemental mapping of Pt, W, and S of CC@WS2/Pt1,n (scale bar: 10 nm for f).
The X-ray diffraction (XRD) patterns showed a broad peak located at around 21.6° was indexed to CC substrate, the diffraction peaks around 14.3° and 32.7° can be assigned to (002) and (100) planes of hexagonal WS2 (JCPDS no. 08–0237), respectively. After the electrodeposition of Pt1,n on the WS2 NSs, two new peaks presented at around 39.8° and 46.2° were attributed to the metallic Pt (JCPDS no. 04–0802), which was well consistent with the TEM analysis and further supporting the successful synthesis of Pt NPs on the WS2 NSs (Fig. 3a). We also investigated the impact of different Pt1,n content in CC@WS2/Pt1,n toward XRD patterns (Supplementary Fig. 4). The content of Pt1,n in CC@WS2/Pt1,n could be well controlled by adjusting the volume of Pt4+ in electrolyte. No apparent diffraction peaks related to Pt were observed if the volume of Pt4+ lower than 1.0 mL, suggesting the low content and small size of Pt on WS2. The diffraction peak intensity increased with the volume of Pt4+ up to 3.0 mL, indicating the high-loading and aggregation of Pt NPs (Supplementary Fig. 5). In order to further verify whether the phase transition of WS2 occurred after the deposition of Pt1,n, the Raman spectra of CC@WS2 and CC@WS2/Pt1,n were performed. As shown in Fig. 3b, two strong features located at 352 and 416 cm–1 in both CC@WS2 and CC@WS2/Pt1,n corresponded to E12g and A1g of 2H phase of WS2, respectively41. Some weak peaks (J1, J2) at low frequency region were typically metastable 1 T phase of WS2. Notably, the relative intensities of A1g/E12g increased from 0.96 of CC@WS2 to 1.04 of CC@WS2/Pt1,n, indicating the dopant of Pt1,n on WS2 could induce lattice strain of in-plane W–S phonon mode, featuring a larger number of edge-terminated active sites for promoting HER42,43. To reveal the electronic structure of CC@WS2/Pt1,n, X-ray photoelectron spectroscopy (XPS) were carried out and shown in Fig. 3c and Supplementary Fig. 6. The survey XPS spectrum of CC@WS2/Pt1,n demonstrated the existence of Pt, W, and S elements on the surface. The high-resolution W 4 f spectrum of CC@WS2 showed main peaks located at 32.1 and 34.1 eV were attributed to 4f7/2 and 4f5/2 of W4+ in WS2, respectively. While two other weak peaks at 35.2 and 37.8 eV were assigned to 4f7/2 and 4f5/2 of W6+, resulting from the inevitable surface oxidation when exposing to the air11,44. The high-resolution S 2p spectrum of CC@WS2 can be divided into two peaks of 161.6 and 162.8 eV, which were ascribed to S2– 2p3/2 and S2– 2p1/2 in WS2, respectively45. After the deposition of Pt1,n, the peaks of W 4 f and S 2p shifted to lower position, respectively, indicating the strong electronic configuration between Pt1,n and WS2. Therefore, the schottky contact and abundant interface charge between these two components were formed46. Compared with standard Pt 4 f signal of commercial Pt/C, where the metallic Pt were detected at the location of 71.7 (4f7/2) and 75.0 eV (4f5/2) with partial surface oxidation, two new peaks positioned at 73.0 (4f7/2) and 76.8 eV (4f5/2) were attributed to the formation of Pt SAs (Ptδ+, 2 <δ < 4) in CC@WS2/Pt1,n, which was consistent with AC-HAADF-STEM analysis47 (Fig. 3c).

a XRD and b Raman spectra of CC@WS2/Pt1,n and CC@WS2. c High-resolution XPS spectra of Pt 4 f signal for CC@WS2/Pt1,n and CC@Pt/C. d W L3-edge XANES spectra and e FT-EXAFS spectra of CC@WS2/Pt1,n and references samples. f, g Pt L3-edge XANES spectra and h FT-EXAFS spectra of CC@MoS2/Pt1,n with different Pt content and references samples. i The fitting of FT R-space Pt L3-edge EXAFS of CC@MoS2/Pt1. j WT for the k3-weighted EXAFS of CC@MoS2/Pt1 and reference samples.
To further reveal the local atomic coordination environments of WS2 and Pt1,n active sites in CC@WS2/Pt1,n, X-ray absorption spectroscopy (XAS) was conducted and shown in Fig. 3d–j. First, the X-ray absorption near edge structure (XANES) spectra was performed to explore the oxidation state of the samples. The white line intensity of XANES at the 5d transition metals (e.g., W, Pt) was reliable indicator to their electronic structures (the measured intensity was relevant to the transition of 2p3/2 to 5d3/2 or 5d5/2) because the unoccupied states above Fermi level was crucial to 5d character of W and Pt48. Figure 3d demonstrated that the white line intensity at the W L3-edge for both CC@WS2/Pt1,n and CC@WS2 located between those of WO3 and W foil reference samples, suggesting that the valence states of W atoms in CC@WS2/Pt1,n and CC@WS2 were between 0 and +6. Noticeably, the white line intensity of CC@WS2/Pt1,n was lower than that of CC@WS2, indicating the strong electronic configuration between Pt1,n and WS2 in CC@WS2/Pt1,n. This result was in good consonance with XPS analysis. Furthermore, the corresponding Fourier-transformed (FT) k3-weighted extended X-ray absorption fine structure (EXAFS) spectra revealed that both CC@WS2/Pt1,n and CC@WS2 showed one prominent peak at 2.0 Å was associated to the W–S path. Another weak peak located at 2.7 Å was attributed to the W–W path. No other W–O interactions at ~1.3 Å could be detected in both CC@WS2/Pt1,n and CC@WS2, demonstrating the pure phase of WS2. In addition, the decrease of intensity at the W–S and W–W shells of CC@WS2/Pt1,n in contrast to CC@WS2 indicated that the deposition of Pt1,n could activate inert basal plane of WS2 and expose more catalytic activated edge sites with dangling bonds for adsorption/desorption of intermediates. The electronic structure of Pt1,n was further explored by the normalized Pt L3-edge XANES spectra. It was noted that the energy of Pt L3-edge was closed to that of W L3-edge, it is difficult to analyze the coordination environment and electronic structure of Pt1,n on the WS2 substrate49. Instead, the MoS2 NSs prepared by the same method as WS2 was the substrate for the deposition of Pt1,n, the structural characterizations of MoS2/Pt1,n were displayed in supplementary Fig. 7. As shown in Fig. 3f, g, the white line-peak intensity of CC@MoS2/Pt1,n with different Pt contents located between Pt foil and PtO2 reference samples, demonstrating the positive charged Pt1,n owing to the strong EMSI leading to the electron transferred from Pt1,n to the substrate, which agreed well with XPS analysis. Moreover, the white line-peak intensity was positive correlation to the ratio of Pt SAs in Pt1,n, where the Pt-0.1 (the Pt4+ content in the electrolyte was 0.1 mL) showed the highest intensity. The white line-peak intensity then decreased and gradually approached to Pt foil with the increase of Pt4+ content to 3.0 mL. This result indicated that the higher concentration of Pt4+ in the electrolyte could lead to the smaller content of Pt SAs and some of Pt SAs were agglomerated to Pt NPs during the electrodeposition of Pt1,n. In order to further reveal the fine coordination environment of Pt1,n, the FT-EXAFS was performed and shown in Fig. 3h. the absence of Pt–Pt contribution at 2.5 Å and the only dominant peak located at 1.9 Å (Pt–S) for Pt-0.1 suggested the atomic dispersed of Pt SAs without Pt NPs in Pt1,n and the Pt SAs were coordinated with S sites of MoS250. In addition, the attenuation of Pt–S peaks and the rise of Pt–Pt peaks confirmed the more Pt SAs were converted into Pt NPs with the increase of Pt4+ content during the preparation of Pt1,n. The fitting results of FT-EXAFS for Pt-0.1 exhibited a coordination number (CN) of 3.1 and 1.1 in the Pt–S and Pt–Mo shells, respectively, suggesting that the Pt SAs coordinated with S at the Mo-top sites of MoS2 (Fig. 3i and Supplementary Table 1)36. The wavelet transform (WT)-EXAFS can directly reflect the k and R spaces structure information. The WT of FT-EXAFS spectra for Pt-0.1 and Pt-2.0 were shown in Fig. 3g. The WT contour plots of Pt-0.1 displayed only one intensity at ~5.1 Å–1, corresponding to the Pt-light atoms bonding, further confirming the atomic dispersion of Pt in Pt-0.151. Apart from the Pt–S region, Pt–Pt bonds were detected in Pt-2.0 at the k-space of 10.8 Å–1, strongly confirming the coexistence of Pt SAs and Pt NPs49. Taken together with XAS, AC-HAADF-STEM and XPS analysis, it could be evidenced that Pt1,n composed of Pt SAs and Pt NPs under a higher Pt4+ electrolyte concentration (>0.1 mL), and the Pt SAs were well stabilized by the coordination with S atoms on the WS2 substrate.
Electrochemical cathodic HER performance
The catalytic performance of the CC@WS2/Pt1,n was investigated by a three-electrode system in 1.0 M KOH. For comparison, the CC@WS2/Pt1,n with different Pt contents, CC@WS2, and commercial 20% Pt/C supported on CC (CC@Pt/C, Supplementary Fig. 8) were also measured under the same condition. The HER performance of CC@WS2/Pt1,n was firstly optimized by adjusting various Pt4+ contents (0.1–3.0 mL) in the electrolyte during the electrodeposition of Pt1,n on CC@WS2 (Fig. 4a). At a current density of 100 mA cm–2, the overpotential for CC@WS2/Pt1,n−0.1, 0.5, 1.0, 2.0, and 3.0 were 191.5, 80.0, 73.0, 60.4, and 66.9 mV, respectively. It was observed that the CC@WS2/Pt1,n−0.1 showed insufficient activity, whereas a significant improvement in the HER activity was observed when the Pt4+ contents higher than 0.1 mL and the 2.0 mL exhibited the best HER performance, indicating the simple Pt SAs sites may not promote the multi-step alkaline HER, the introduction of Pt NPs could induce combined effects of Pt NPs and Pt SAs to provide multiple active sites to accelerate HER activity. The HER polarization curves of different controlled samples were shown in Fig. 4b. The LSV curves of CC@WS2 showed low activity of 315 mV to deliver current density of 10 mA cm–2 (η10), Upon the introduction of Pt1,n, the drastic HER activity enhancement in the CC@WS2/Pt1,n was obtained with a low η10 of 27.1 mV, which was even lower than those of commercial CC@Pt/C (30 mV) and most of the reported state-of-the-art Pt-based HER catalysts at 10 and 100 mA cm–2 (Fig. 4c and Supplementary Fig. 9, Tables 2 and 3). In order to further discuss the synergistic effect of Pt NPs and Pt SAs toward HER, the pure Pt NPs supported on the CC@WS2 (CC@WS2/Pt NPs) was prepared by chemical reduction method (Supplementary Figs. 10 and 11). The LSV curves of CC@WS2/Pt NPs was performed and the CC@WS2/Pt1,n was set here for comparison. The η10 of CC@WS2/Pt NPs (66.0 mV) was significantly higher than that of CC@WS2/Pt1,n (27.1 mV), indicating the combined effect between Pt NPs and Pt SAs for promoting HER (Supplementary Fig. 12). As both Pt NPs and Pt SAs were present in CC@WS2/Pt1,n, potassium thiocyanide (KSCN) and ethylenediaminetetraacetic acid disodium (EDTA) were used as poisoning agents to further help differentiate their actual contributions to HER. Note that the EDTA was dominantly coordinated with Pt SAs while SCN– readily adsorbed onto both Pt SAs and Pt NPs. Thus, EDTA will block and inactivate of Pt SAs sites, and KSCN can deactivate all the Pt sites34,52. As shown in Supplementary Fig. 13, with the addition of 10 mM KSCN in the electrolyte, the η10 value shifted negatively to >180 mV, while the much smaller shift (ca. 11 mV) was observed when 10 mM EDTA was appended to the electrolyte. The different poisoning effect of KSCN and EDTA indicated that both Pt SAs and Pt NPs contributed to the HER, and the latter likely played a dominant role. This result agreed with the theoretical calculations in the following section. To investigate the kinetic behavior and possible reaction pathway for alkaline HER on CC@WS2/Pt1,n, the Tafel plots and the corresponding Tafel slopes of each sample were fitted and shown in Fig. 4d. The Tafel slopes of CC@WS2/Pt1,n (27.5 mV dec–1) was much lower than that of CC@Pt/C (118.5 mV dec–1), suggesting the fast kinetic rate of Volmer–Tafel pathway with the rate-determining step (RDS) of Tafel that dominated the reaction on CC@WS2/Pt1,n53. In addition, the electrochemical impedance spectroscopy (EIS) was performed and shown in Fig. 4e. CC@WS2/Pt1,n displayed the smallest charge transfer resistance (Rct = 0.96 Ω) value compared those of CC@Pt/C (2.64 Ω) and CC@WS2 (143.4 Ω), indicating the incorporation of Pt1,n on CC@WS2 can indeed enhance the HER kinetics. The apparent activity of the catalysts were closely associated with the electrochemical active surface area (ECSA), which can be calculated by the electrochemical double-layer capacitance (Cdl). The Cdl was obtained from the CV profiles over a non-Faradic potential range at various scan rates (Supplementary Fig. 14). The calculated Cdl of CC@WS2/Pt1,n was 226.0 mF cm–2, much higher than those of CC@WS2 (205.5 mF cm–2) and CC@Pt/C (160.1 mF cm–2). Such a high Cdl of CC@WS2/Pt1,n implied an enriched exposed active sites for facilitating the electrochemical HER activity (Fig. 4f).

a LSV curves of CC@WS2/Pt1,n with different Pt4+ content in the electrolyte. b The LSV curves of optimal CC@WS2/Pt1,n (mass loading: 0.106 mgPt cm–2) and the control samples of CC@WS2 and CC@Pt/C. c The comparison of overpotential at the 10 mA cm–2 for the prepared CC@WS2/Pt1,n and the reported Pt-based catalysts. d Tafel plots of CC@WS2/Pt1,n and CC@Pt/C. e, f Nyquist plots and the calculated Cdl for the CC@WS2/Pt1,n, CC@WS2, and CC@Pt/C, respectively. g Mass activities of CC@WS2/Pt1,n and CC@Pt/C on the basis of Pt content. h TOFs of CC@WS2/Pt1,n and CC@Pt/C. i, j LSV curves before and after 2000 CV tests for CC@WS2/Pt1,n and CC@Pt/C. k Chronopotentiometry tests of CC@WS2/Pt1,n and CC@Pt/C. All the tests were measured in 1.0 M KOH solution (pH = 14).
The intrinsic activity of the catalysts was another crucial criteria for evaluating HER performance. The ECSA-normalized LSV current density for CC@WS2/Pt1,n and CC@WS2 were conducted. The CC@WS2/Pt1,n still showed the highest specific activity with a low overpotential of 70.2 mV than that of commercial CC@Pt/C (105.6 mV) at 10 µA cm−2 (Supplementary Fig. 15). The mass activity normalized to the loading mass of Pt in CC@WS2/Pt1,n.was conducted to evaluate the economic efficiency of Pt. As displayed in Fig. 4g, at an overpotential of 80 mV, the calculated mass activity of CC@WS2/Pt1,n was 1.11 A mgPt–1, which were 37 times larger than that of commercial CC@Pt/C (0.03 A mgPt–1). In addition, the turnover frequency (TOF) was calculated to further reveal the intrinsic activity of the samples. Assuming all the Pt sites were all participated in a reaction, the TOF value of CC@WS2/Pt1,n was 1.16 s–1 at an overpotential of 80 mV, which was ~39 times higher than that of CC@Pt/C (0.03 s–1). Therefore, the obtained specific activity, mass activity, and TOF calculation demonstrated the significantly enhanced intrinsic activity of CC@WS2/Pt1,n, delivering substantial hydrogen production. For a practical HER electrocatalyst, the stability and long-term durability of CC@WS2/Pt1,n were further analyzed by the cycling performance and chronopotentiometry (CP) tests. As depicted in Fig. 4i, j, the activity of CC@WS2/Pt1,n exhibited negligible degradation after 2000 CV, whereas the activity of CC@Pt/C displayed significant deterioration after 2000 CV cycles. Furthermore, the CP measurements were performed and confirmed superior long-term stability of CC@WS2/Pt1,n, where the potential showed ignorable degradation after 100-hour tests under continuous working condition at 10 mA cm–2. As a sharp contrast, the apparent activity degradation fluctuation of curve was observed in CC@Pt/C (Fig. 4k). The AC-HAADF-STEM images of CC@WS2/Pt1,n after the CP test were conducted, the dispersion of Pt NPs and Pt SAs on WS2 NSs remain nearly unchanged, confirming the stable structure of CC@WS2/Pt1,n after long-term stability test, which may due to the strong EMSI effect between Pt1,n and WS2 substrate to control the dispersion and stabilization of Pt1,n with high surface free energy (Supplementary Fig. 16)54. The chronoamperometric (CA) response (at 100 mV) of the CC@WS2/Pt1,n showed negligible activity decay within 24 hours, the corresponding LSV curves after the CA test was almost the same as that of the initial state, indicating the superior stability of CC@WS2/Pt1,n electrode (Supplementary Fig. 17). Moreover, the CP test at high current density of –100 mA cm–2 was performed and shown in Supplementary Fig. 18. The result indicated that the CC@WS2/Pt1,n could also maintain long-term stability for at least 100 h, which was due to the in-situ growth of binder-free CC@WS2/Pt1,n catalysts that can accelerate charge transfer and mass diffusion, promote the exposed active sites, enhance reaction kinetics, and stability55.
Theoretical insights of the synergistic HER mechanism
Density functional theory (DFT) calculations were carried out to reveal the underlying mechanism of alkaline HER on WS2/Pt1,n. According to the AC-HAADF-STEM and EXAFS results, the structural models of pristine WS2, Pt atom located at the top site of W on WS2 (WS2/Pt SA), and Pt13 nanoclusters loaded on the WS2 (WS2/Pt NC) were built (Fig. 5a–c). Since Pt NPs with an average size of 2.6 nm in our work is nearly impossible for DFT calculations, the Pt13 cluster was adopted for simplicity50,56,57,58,59,60. According to the calculated charge density distributions, the increased charge densities could be observed at the WS2/Pt SA and WS2/Pt NC interfaces, which implied the strong EMSI and synergistic effect between Pt1,n and WS2 substrate in hybrid. Additionally, the electron transfer occurred at the interfaces, leading to reduced electron density around the Pt atom. To deeply understand the enhancement origin of HER, the projected density of states (PDOS) were conducted. As displayed in Fig. 5g, the d-band center (εd) is an effective descriptor to directly associate the adsorption properties of surface intermediates and electronic structure of catalysts. The calculated εd of Pt-d orbitals for WS2/Pt SA (–2.41 eV) was more far from to the Fermi level than that of WS2/Pt NC (–1.70 eV), meaning the more moderate interaction between the adsorbed molecules (e.g., H*) and Pt sites of WS2/Pt SA61. On the other hand, the enhanced adsorption of intermediates (e.g., OH* in alkaline HER) on WS2/Pt NC intended to promote Volmer reaction during alkaline HER. It was noted that the metal sites may not the only active catalytic centers in some catalysis reactions, the p-band center (εp) of non-metal sites needed to be revisited and together with εd to synergically regulate electronic structures62. In addition, the εp can also be considered as a reliable descriptor for H* adsorption. As shown in Fig. 5h, the εp trend of S-p orbitals states was WS2 > WS2/Pt SA > WS2/Pt NC, indicating the more strengthened adsorption of H* on the S sites of WS2/Pt SA and WS2/Pt NC compared to WS2, which could be in concert with the H* adsorption free energy analysis (see Fig. 5i)63. The free energy of H* (ΔGH*) is an important descriptor to evaluate the HER activity, and highly-efficient HER electrocatalysts should possess moderate ΔGH* near zero. Before the calculation, an ab initio molecular dynamics (AIMD) simulation of WS2/Pt NC was conducted to further prove the stability of the constructed Pt NC on WS2 substrate (Supplementary Fig. 19). As displayed in Fig. 5i and Supplementary Fig. 20, the ΔGH* of WS2/Pt NC were calculated to –0.416 and –0.463 eV on the top and side sites of Pt NC, respectively, indicating the strengthened H* adsorption on Pt NC may limit the desorption of H* and blocked the active sites for subsequent reaction. The ΔGH* was reduced to –0.05 eV at the adsorption sites of Pt SA in WS2/Pt SA, indicating the most thermoneutral adsorption of H* (Tafel step). The S sites in both pristine WS2 and WS2/Pt SA showed the ΔGH* values of 2.069 and 1.648 eV, respectively with more weak adsorption for H* (Supplementary Table 4)64,65. It was noted that the |ΔGH*| on Pt SA of WS2/Pt SA was even lower than that of benchmark Pt (111), indicating the well EMSI between Pt SA and WS2 substrate for more thermoneutral adsorption of H*66. The energy barrier of water dissociation (Volmer step) is a crucial descriptor to evaluate intrinsic catalytic activity of alkaline HER as the Volmer step is generally considered as RDS of alkaline HER process67. As illustrated in Fig. 5j and Supplementary Fig. 21, the water dissociation energy barrier on Pt NC-top sites (0.91 eV) was much smaller related to those of Pt NC-top (1.69 eV) and Pt SA sites (2.05 eV), verifying the accelerated Volmer process on the Pt NC-top sites that dominated the reaction. The above calculation results combined with experimental design demonstrated that the promoted alkaline HER performance could be attributed to the synergistic effect in WS2/Pt1,n, is that, the water dissociation kinetic of Volmer step could be promoted on top site of Pt NC, and the obtained H* were spilled and immigrated to adjacent Pt SAs sites of WS2 for hydrogen production (Tafel step) through fast kinetic rate of Volmer–Tafel pathway (Fig. 5k).

a–c The optimized structures of WS2, WS2/Pt SA, and WS2/Pt NC. d–f The 2D differential charge density distributions of WS2, WS2/Pt SA, and WS2/Pt NC. Red and blue area represents electron accumulation and depletion, respectively. PDOS of g Pt 5d for WS2/Pt SA and WS2/Pt NC, h S 3p for WS2, WS2/Pt SA, and WS2/Pt NC. i H* adsorption and j water adsorption/dissociation free energy on different sites of WS2, WS2/Pt SA, and WS2/Pt NC. k Schematic illustration of the alkaline HER mechanism on WS2/Pt1,n.
Low-potential anodic DAF oxidation
The low-potential anodic DAF oxidation reaction (DAFOR) is key to our low-energy-consumption hydrogen production system. DAF was used as model substrate for low-potential anodic oxidation reaction. The copper foam supported copper oxide nanowires (CF@CuO NWs) as the catalysts were prepared by a two-step facile method (Fig. 6a). Typically, the copper foam supported copper hydroxide nanowires (CF@Cu(OH)2 NWs) was fabricated by anodic oxidation process, as confirmed by XRD, SEM, and TEM (Supplementary Figs. 22 and 23). The as-formed CF@Cu(OH)2 NWs was then calcined under argon to obtain CF@CuO NWs. The XRD patterns of CF@CuO NWs in Fig. 6b showed three dominant peaks located at 43.3, 50.5, and 74.1° were attributed to the CF support. The remaining diffraction peaks were well matched with CuO (JCPDS no. 45–0937 and 78–0428). Figure 6c present the SEM images of CF@CuO NWs, the cross-linking nanowires were uniformly and densely grown on CF. The TEM images clearly showed nanowires morphology of CuO with the lattice distances of 0.245 and 0.253 nm, corresponding to the (111) and (002) planes of CuO, respectively. The HAADF-STEM and elemental mapping indicated the homogeneous distribution of Cu and O elements throughout the whole nanowire (Supplementary Fig. 24). The high-resolution XPS spectra of Cu 2p for CF@CuO NWs possessed two main peaks at 933.6 and 953.4 eV can be assigned to 2p3/2 and 2p1/2 of Cu2+ in CuO68,69. The shake-up satellites peaks were found at 941.0, 943.6, and 961.9 eV, respectively. High-resolution O 2p spectra consisted of two peaks located at 529.5 and 531.2 eV were attributed to the lattice oxygen (Cu–O–Cu) and adsorbed oxygen in –OH groups, respectively (Supplementary Fig. 25)69. Electrocatalytic DAFOR was conducted in a typical three-electrode system using CF@CuO NWs as working electrode, Hg/HgO (1.0 M KOH filler) and Pt wire were reference and counter electrodes. Figure 6d present LSV curves of the CF@CuO NWs in 1.0 M KOH with various concentration of DAF. We found that the DAF oxidation activity was highly dependent on the concentration of DAF. Impressively, the anodic current became gradually obvious when the DAF concentrations changing from 0.02 to 0.20 M. The optimal working potential was 1.23 and 1.44 V vs. RHE at the DAF concentration of 0.20 M to achieve 10 and 100 mA cm–2 (Fig. 6e). The low-potential DAFOR showed a promising alternative to traditional OER. To discuss the various catalysts for this reaction, the LSV curves of pristine copper foam (CF) and CF@Cu(OH)2 NWs in 0.20 M DAF + 1.0 M KOH electrolyte were carried out and showed in Fig. 6f. Compared with CF@CuO NWs, the activity attenuation was observed on the CF and CF@Cu(OH)2 NWs catalysts, which may due to the different adsorption capacity of DAF on the catalysts. In view of this, the adsorption energies (Eads) of DAF on CF, CF@Cu(OH)2, and CF@CuO were calculated (Supplementary Fig. 26). The Eads of DAF (N sites of furazan ring) bonding with Cu sites of CuO (–1.57 eV) was the lowest compared with those of CuO (N sites of amino bonding with Cu sites of CuO) (–1.34 eV), pristine CF (–1.32 eV), and Cu(OH)2 (–1.48 eV), implying that the Cu sites of CuO as the main adsorption and reaction active site were preferred to bond with N sites of furazan ring in DAF for DAF oxidation70. The DAF oxidation mechanism was investigated by the addition of 2,2,6,6-tetramethylpiperdine-1-oxyl (TEMPO) and isopropanol free radical (•OH) scavengers in the electrolyte71,72. The polarization curves of DAFOR on CF@CuO NWs in 0.20 M DAF + 1.0 M KOH with and without the addition of TEMPO or isopropanol (Pt film as working electrode to eliminate the poison effect of isopropanol to CuO electrode) were carried out (Supplementary Fig. 27). A significant decrease of current density was detected when TEMPO was added to the electrolyte. In addition, the current intensity was also suppressed after the addition of isopropanol. These results indicated that the DAF oxidation mechanism may involve the oxidative coupling of DAF into DAAzF initiated by the •OH free radical. In presence of applied voltage, the hydroxide anion was converted into •OH free radical, which was acted as oxidant for oxidative coupling of DAF into DAAzF EMs.

a Schematic illustration of anodic catalysts of CF@CuO NWs. b XRD pattern and c SEM images of CF@CuO NWs (scale bar: 2 μm and 1 μm for low and high-resolution SEM images, respectively). d LSV curves of DAF oxidation reaction at the different concentration of DAF in 1.0 M KOH medium. e Corresponding working potential at the constant current density of 10 and 100 mA cm–2. f LSV curves of DAF oxidation reaction on different catalysts in 0.20 M DAF + 1.0 M KOH (pH = 14) media.
Two-electrode coupling system performance
To establish an energy-efficient system that benefitted from the DAFOR, we assembled an two-electrode DAFOR||HER electrolyser that used CC@WS2/Pt1,n and CF@CuO NWs as cathode and anode catalysts, respectively. The electrolytes contained 1.0 M KOH and 1.0 M KOH + 0.20 M DAF for cathode and anode, respectively (Fig. 7a). Coupling this low-potential DAFOR with a cathodic HER enables a co-production system, with hydrogen production at the cathode side, while the more valuable DAAzF EMs than oxygen was synthesized from DAF starting material. Figure 7b present the LSV curves of two-electrode electrolyser for our novel DAFOR||HER coupling system and conventional OWS on CF@CuO NWs||CC@WS2/Pt1,n catalysts. Remarkably, it only required the cell voltage of 1.26 V to reach the current density of 10 mA cm–2, whereas conventional OWS required much higher cell voltage of 1.67 V to obtain the same current density. This result clearly indicated energy-efficient advantage of DAFOR than OER. The superior activity of CF@CuO NWs||CC@WS2/Pt1,n outperformed the electrolyzer employing CF||CC@Pt/C, CF@Cu(OH)2 NWs||CC@WS2/Pt1,n, and CF||CC@WS2/Pt1,n (Fig. 7c). Compared to the recenly reported OWS on noble metal-based catalysts, the DAFOR-assisted OWS proposed in our work showed significantly reduced cell voltage (Fig. 7d and Supplementary Table 5). To investigate the stability of our coupling system, the CP test lasting for 40 h was performed. As shown in Fig. 7e, a few activity attenuation was observed after continuous operation for 40 h. Furthermore, inspection of the structural and morphological evolution of anode catalyst after CP test by XRD and SEM showed that the CuO phase was well maintained. Surprisingly, the CuO nanowires were aggregated into brush-like morphology (Supplementary Fig. 28). It was speculated that the activity attenuation was associated with a decrease in the ECSA because of the morphological evolution of CF@CuO but did not affect the feasibility of DAFOR to synthesize DAAzF EMs, as evidenced by the nuclear magnetic resonance (NMR) analysis in the following section. During the CP test, the hydrogen gas was observed on the surface of CC@WS2/Pt1,n. At the anode side, the color of electrolyte changed from colorless from yellow-orange, preliminarily supporting the successful synthesis of DAAzF EMs. The obtained anodic electrolyte was then rotarily evaporated and the product was characterized by 13C NMR (Fig. 7g). The position located at δ = 151.1 and 156.2 ppm were ascribed to –C–NH2 and –C–N=N in furazan ring, respectively73. Additionally, the fourier transform infrared spectroscopy (FTIR) of DAAzF EMs exhibited the vibrational peak at 1497 cm–1 was attributed to the v(N=N) in DAAzF EMs (Supplementary Fig. 29)74. In addition, the Raman spectra was performed to further support the formation of DAAzF EMs and involved the N–N oxidative coupling of DAF mechanism during the synthesis of DAAzF (Supplementary Fig. 30). Furthermore, only one intense exothermic peak located at 327.1 °C was detected in differential scanning calorimetry (DSC) spectrum (Supplementary Fig. 31). These results confirmed the pure phase of DAAzF with high energy for the application of energetic materials. The faradic efficiency (FE) of the cathodic HER and anodic DAFOR were calculated and shown in Supplementary Fig. 32. The results confirmed a FE of 98.5% for HER and optimal FE of 94.6% for DAFOR at 10 mA cm–2. From the perspective of energy-efficiency, the electricity input of our coupling system and conventional OWS were evaluated (Fig. 7h). The conventional OWS required an high electricity input of 3.98 and 4.76 kWh per m3 of H2 at the current density of 10 and 100 mA cm–2, and the consumption of electricity significantly increased at higher current densities, in which the typical electricity input of ~5 kWh per m3 of H2 was required for conventional OWS at 100 mA cm–25. Impressively, because of the low-cell voltage for the DAFOR||HER coupling system, the electrolyzer only required an electricity input of 3.01, 3.35, and 3.70 kWh per m3 of H2 at the current density of 10, 50, 100 mA cm–2, respectively, which was much lower than that of conventional OWS. In view of the low-carbon economy and sustainability, the DAFOR-assisted OWS system provided an energy-efficient, low-cost, and safe strategy for hydrogen production.

a Schematics of the proposed electrocatalytic coupling system. b Comparison of LSV curves for conventional OWS and DAF oxidation-coupled OWS system. c Comparison of LSV curves of DAF oxidation-coupled OWS on various catalysts. d Comparison of the cell voltage required to reach 10 mA cm–2 for the coupling system in our work and reported OWS on noble metal-based catalysts. e CP curves of the coupling system recorded at 10 mA cm–2 for 40 h. The electrochemical tests were measured in 1.0 M KOH (pH = 14) with or without the addition of 0.2 M DAF. f Optical image of our two-electrode coupling system before and after the CP test. g 13C NMR spectrum of the as-synthesized DAAzF EMs (inset: optical image of DAAzF EMs powder). h Electricity consumed for hydrogen production in conventional OWS and our coupling system using various catalysts.
Discussion
In summary, a novel strategy for the concurrent and highly-efficient production of hydrogen and value-added DAAzF EMs was proposed by using CC@WS2/Pt1,n and CF@CuO NWs as cathodic and anodic catalysts, respectively. The low-potential DAFOR in our coupling system was able to realize the low-cell-voltage hydrogen production, offering the current density of 10 mA cm–2 at the cell voltage of as low as 1.26 V, which was 410 mV lower than that of conventional OWS. Consequently, this coupling system only required electricity input of 3.01 and 3.70 kWh per m3 of H2 at 10 and 100 mA cm–2, respectively, significantly lower than that of conventional OWS. DFT calculations revealed that the Volmer step in cathodic alkaline HER was promoted on the Pt NCs sites of WS2/Pt1,n, while the Pt SAs sites exhibited more thermoneutral H* intermediates adsorption, realizing the synergistic effect of WS2/Pt1,n to promote Volmer–Tafel kinetic in alkaline HER. The anodic DAFOR mechanism involved the oxidative coupling of DAF triggered by •OH free radicals for the synthesis of DAAzF. Apart from the energy-saving hydrogen production, our novel coupling system avoided the traditional hazardous synthetic condition of DAAzF EMs and realized the green and sustainable pathway to electro-synthesize DAAzF EMs. Our work reported a novel alternative reaction of DAFOR to integrate with OWS for energy-saving hydrogen production, and provided a sustainable approach for the production of DAAzF EMs, offering a promising avenue for the development of green electrosynthesis of valuable chemicals coupled hydrogen production systems.
Methods
Synthesis of CC@WS2 NSs
CC@WS2 NSs was prepared by one-pot hydrothermal reaction. Typically, 0.90 mmol sodium tungstate dihydrate (Na2WO4·2H2O) and 5.25 mmol thioacetamide (C2H5NS) were mixed into 15 mL distilled water and stirred for 30 min. Subsequently, 0.15 g oxalic acid (C2H2O4) was added to the above aqueous solution and stirred to form a clear solution. A piece of CC (1 cm × 3 cm) was washed with acetone and ethanol several times to clean the surface of CC. After that, the treated CC was immersed into the above solution and transferred to 25 mL Teflon-lined stainless-steel autoclave. The mixture was heated to 200 °C for 24 h. After cooling down to room temperature, the obtained CC@WS2 NSs was washed with distilled water and ethanol, then dried at 60 °C.
Synthesis of CC@WS2/Pt1,n
The as-prepared CC@WS2 NSs was used as substrate to support Pt1,n catalysts. The CC@WS2/Pt1,n was prepared by electrochemical depositions of Pt1,n on CC@WS2 via a cycle voltammetry method. The electrodeposition was carried out in a standard three-electrode system. The self-supporting CC@WS2 was working electrode, a Hg/HgO (1.0 M KOH) was used as reference electrode, and a high-purity graphite rod was used as counter electrode. The 1.0 M KOH with a certain amount of chloroplatinic acid hydrate solution (H2Cl6Pt·xH2O, 10 mg mL–1) was the electrolyte. The electrodeposition was conducted from –0.6 to –1.5 V vs. Hg/HgO with a sweeping rate of 50 mV s−1 for 200 cycles. The content of Pt1,n on CC@WS2 NSs could be controlled by adjusting the volume of H2Cl6Pt solution in the electrolyte. For comparison, various volumes of H2Cl6Pt (0.1, 0.5, 1.0, 2.0, and 3.0 mL) were set here to investigate the optimal HER performance.
Synthesis of CC@WS2/Pt NPs
The Pt NPs supported on the CC@WS2 was prepared by one-pot hydrothermal reaction. Ethanol was used as the solvent to dissolve certain amount of H2Cl6Pt solution. One piece of CC@WS2 NSs (1.0 cm × 0.5 cm) was loaded at the bottom of the autoclave and heated to 180 °C for 6 h. The final CC@WS2/Pt NPs product was cooled to room temperature and washed with distilled water several times.
Synthesis of CC@MoS2/Pt1,n
The MoS2 nanosheets supported CC (CC@MoS2 NSs) was firstly prepared by one-pot hydrothermal reaction. Typically, the pre-treated (washed by acetone, ethanol several times) CC substrate (1 × 3 cm2) was immersed in the 15 mL aqueous solution containing 1.0 mmol Na2MoO4⋅2H2O and 4.0 mmol thiourea. The mixture was then transferred to autoclave and heated at 200 °C for 24 h. After cooling down to the room temperature, the obtained CC@MoS2 NSs was used as the support to deposit Pt1,n. The CC@MoS2/Pt1,n was prepared by the same electrochemical CV methods as CC@WS2/Pt1,n except for the change of WS2 to MoS2.
Synthesis of CC@Pt/C
To prepared the Pt/C ink, the commercial 20% Pt/C powder (4 mg) was dispersed in 2.0 mL ethanol containing 50 μL Nafion solution (DuPont, D520, 5%). The suspension was sonicated for 30 min and then dripped on the CC (1.0 cm × 0.5 cm) and dried at 60 °C overnight.
Synthesis of CF@Cu(OH)2 NWs and CF@CuO NWs
The CF@Cu(OH)2 NWs was prepared by a in-situ oxidative etching method at room temperature. Before the reaction, the CF substrate (1 cm × 0.5 cm) was pre-treated by dilute hydrochloric acid and acetone to remove the surface oxides and organic matters. Then the cleaned CF was immersed in the mixed aqueous solution containing 0.13 M ammonium persulfate ((NH4)2S2O8) and 2.67 M NaOH and soaked for 30 min. The obtained CF@Cu(OH)2 NWs was rinsed with ethanol and distilled water for several times and dried at 60 °C for 1 h. The as-prepared CF@Cu(OH)2 NWs was then calcined in muffle furnace at 300 °C for 1 h under air atmosphere to harvest CF@CuO NWs.
Synthesis of DAF substrate material
The DAF substrate material was synthesized by the two steps from commercial glyoxal as the previous literature75. Typically, diaminoglyoxime was firstly synthesized by immersing glyoxal in hydroxylamine hydrochloride (4 equivalents) and sodium hydroxide (4 equivalents) in water at 90 °C. Then, the obtained diaminoglyoxime (23.6 g, 0.2 mol) was dissolved in aqueous potassium hydroxide (2 M, 80 mL) and transferred to a stainless-steel reactor to heat at 170 °C for 2 h. After that, the stainless-steel reactor was cooled at 0~5 °C for 2 h, the obtained white crystals were treated by suction filtrating and vacuum drying to yield DAF substrate material.
Material characterizations
XRD patterns were obtained from an X-ray diffractometer (Bruker D8 Advance). The morphology of the samples were obtained using Hitachi, SU8010. TEM images and elemental mappings were conducted by FEI Talos F200X. Atomic-resolution HAADF-STEM images of Pt1,n were obtained using FEI Themis Z. The electronic structure of the samples were investigated by XPS (Thermo Scientific Nexsa). Raman spectroscopies of the samples were performed at Thermo Fisher DXR2 Xi acquired by a green laser of 532 cm−1. The DSC was conducted on a Q2000/Q600-TA instruments under N2 atmosphere over the heating rate of 10 °C min–1. The 13C NMR spectrum was carried out on a Bruker Advance 400 in D2O at 25 °C. FTIR spectra was acquired on a Bruker Vertex 70 instrument. The content of Pt1,n in the sample was determined by the ICP-MS (Agilent 7800). Raman spectra were obtained on a Thermo Fisher-Dxr 2Xi. The X-ray absorption spectra were conducted at the beamline 1W1B of the Beijing Synchrotron Radiation Facility (BSRF, operated at typical energy of the storage ring of 2.5 GeV). The X-ray absorption data was collected in the mode of fluorescence for Pt and W L-edges. The least-squares EXAFS fitting was performed using the ARTEMIS module of Demeter package.
Electrochemical characterizations
The cathodic HER performance of the samples were conducted in 1.0 M KOH electrolyte on a CHI 760E electrochemistry workstation. The as-prepared self-supporting samples were used as working electrode. The high-purity graphite rod and Hg/HgO (1.0 M KOH filler) were counter and reference electrodes, respectively. All potentials were referenced to RHE by Eq. (1):
The LSV curves were carried out with a scan rate of 2 mV s−1. CV tests were performed at the non-faradic potential range of 0.124~0.224 V vs. RHE with the scan rates of 2.5, 5, 10, 20, and 50 mV s−1. EIS spectra were obtained in the frequency range of 0.1~100,000 Hz at the overpotential of 200 mV. The stability tests were performed by repeating the CV scan from 0.324~–0.576 V vs. RHE for 2000 cycles. The ECSA of the samples were calculated based on the Eq. (2):
Where the Cdl is double-layer capacitance, the Cs is the a specific capacitance (0.04 mF cm–2)76. CP test was performed at a constant current density of –10 mA cm–2. Mass activities of the Pt1,n in the samples were calculated by the Eq. (3):
Where the J is measured current density of Pt, the mPt is content of Pt in the samples. The TOF of the samples were calculated based on the Eq. (4):
Where I is measured current of polarization curve. The F is the Faraday constant (96485 C mol–1), the factor 2 is the number of transferred electrons after the formation of one hydrogen molecule. The n is the number of active sites77. The anodic DAFOR performance was conducted in a three-electrode system. The as-prepared CF, CF@Cu(OH)2 NWs, and CF@CuO NWs self-supporting catalysts were used as the working electrodes. The Pt wire and Hg/HgO (1.0 M KOH filler) were counter and reference electrodes, respectively. The electrolyte contained 1.0 M KOH with various volume of Pt4+ solution (10 mg mL−1, 0.1, 0.5, 1.0, 2.0, and 3.0 mL). The LSV curves were carried out with a scan rate of 2 mV s−1 over a potential range of 0.724 ~ 1.724 V vs. RHE. The two-electrode coupling system performance were performed in a H-type electrolyzer. The as-prepared Cu based catalysts were used as anode electrodes. The CC@WS2/Pt1,n and CC@Pt/C were cathode electrodes. The electrolyte for anode and cathode were 1.0 M KOH + 0.2 M DAF and 1.0 M KOH, respectively. A piece of anion-exchange membrane (Fumasep FAA-3-50) was used to divide anode and cathode. The LSV curves of the samples were conducted with a scan rate of 2 mV s−1. The FE of the HER and DAFOR were calculated based on the following Eq. (5):
Where n is the mole value of substrate, the theoretical produced amount of the hydrogen and DAAzF were calculated by the Eq. (6):
Where Q is the charge passed during the reaction, F is Faradic constant (96485 C mol–1), n is the number of electron transferred after the formation of each product molecule (n = 2 and 4 for HER and DAFOR, respectively). The electricity input (W) of the coupling system per m3 of H2 produced (kWh per m3 H2) was calculated by the Eq. (7):
Where the n is the number of electrons transferred for H2 production (n = 2), U is the applied cell voltage in the two-electrode cell, Vm is the molar volume of H2 at normal temperature and pressure (22.4 mol L−1), F is Faraday constant (96485 C mol−1).
Computational method
The DFT calculations were partially performed via Dmol3 package of Materials Studio program78. The generalized gradient approximation of Perdew–Burke–Ernzerhof exchange correlation function was used. A periodic supercell (4 × 4) and with a vacuum region of 15 Å were added to avoid the interaction between WS2 and its periodic image. An energy convergence criterion of 10–5 Hartree was set for geometrical optimization. For the construction of cathodic WS2/Pt SA and WS2/Pt NC catalysts, one Pt atom or Pt13 clusters was supported on the surface of WS2 (002) plane. To evaluate the adsorption capacity of DAF on anodic catalysts, one DAF molecule was loaded on the pristine Cu (100), CuO (111), and Cu(OH)2 (002) planes, respectively. 2 × 2 × 1 Monkhorst–Pack k-points were sampled for the geometrical optimization.
The transition state for water dissociation step (Volmer) on the Pt sites was determined by the climbing image nudged elastic band method79. The adsorption Gibbs free energy of hydrogen atom (ΔGH*) was calculated by Vienna ab initio simulation package (VASP) code80 via the Eq. (8):
Where the \({E}_{{{{{{\rm{surf}}}}}}+{{{{{\rm{H}}}}}}}\) and \({E}_{{{{{{\rm{surf}}}}}}}\) represent the total energies before and after the adsorption of H atom, \({E}_{{{{{{{\rm{H}}}}}}}_{2}}\) was the total energy of hydrogen gas molecule. ΔEZPE, T, and ΔS are the difference in the zero-point energy, temperature, and the difference in entropy, respectively. The effect of implicit solvation was considered by using VASPsol software for simulating the H2O solvent environment and the dielectric constant was set to ε = 80 for water81.
Data availability
All data are available from the corresponding authors upon reasonable request. Source data are provided with this paper.
References
Li, Z. et al. Alcohols electrooxidation coupled with H2 production at high current densities promoted by a cooperative catalyst. Nat. Commun. 13, 147 (2022).
Xiao, Z. et al. Bifunctional Co3S4 nanowires for robust sulfion oxidation and hydrogen generation with low power consumption. Adv. Funct. Mater. 33, 2212183 (2023).
Liu, Y. et al. Manipulating dehydrogenation kinetics through dual-doping Co3N electrode enables highly efficient hydrazine oxidation assisting self-powered H2 production. Nat. Commun. 11, 1853 (2020).
Liu, T. et al. Interfacial electron transfer of Ni2P-NiP2 polymorphs inducing enhanced eectrochemical properties. Adv. Mater. 30, 1803590 (2018).
Wang, T. et al. Combined anodic and cathodic hydrogen production from aldehyde oxidation and hydrogen evolution reaction. Nat. Catal. 5, 66–73 (2022).
Rausch, B., Symes, M. D., Chisholm, G. & Cronin, L. Decoupled catalytic hydrogen evolution from a molecular metal oxide redox mediator in water splitting. Science 345, 1326–1330 (2014).
Du, J. et al. Electrochemical hydrogen production coupled with oxygen evolution, organic synthesis, and waste reforming. Nano Energy 104, 107875 (2022).
Wang, H. Y., Sun, M. L., Ren, J. T. & Yuan, Z. Y. Circumventing challenges: design of anodic electrocatalysts for hybrid water electrolysis systems. Adv. Energy Mater. 13, 2203568 (2023).
Yan, D., Mebrahtu, C., Wang, S. & Palkovits, R. Innovative electrochemical strategies for hydrogen production: From electricity input to electricity output. Angew. Chem. Int. Ed. 62, e202214333 (2023).
You, B. & Sun, Y. Innovative strategies for electrocatalytic water splitting. Acc. Chem. Res. 51, 1571–1580 (2018).
Li, J. et al. Elucidating the critical role of ruthenium single atom sites in water dissociation and dehydrogenation behaviors for robust hydrazine oxidation-noosted alkaline hydrogen evolution. Adv. Funct. Mater. 32, 2109439 (2022).
Li, Y. et al. Partially exposed RuP2 surface in hybrid structure endows its bifunctionality for hydrazine oxidation and hydrogen evolution catalysis. Sci. Adv. 6, eabb4197 (2020).
Sun, F. et al. Energy-saving hydrogen production by chlorine-free hybrid seawater splitting coupling hydrazine degradation. Nat. Commun. 12, 4182 (2021).
Geng, S. K. et al. Nickel ferrocyanide as a high-performance urea oxidation electrocatalyst. Nat. Energy 6, 904–912 (2021).
Lu, X. F., Zhang, S. L., Sim, W. L., Gao, S. & Lou, X. W. Phosphorized CoNi2S4 yolk-shell spheres for highly efficient hydrogen production via water and urea electrolysis. Angew. Chem. Int. Ed. 60, 22885–22891 (2021).
Fan, L. et al. High entropy alloy electrocatalytic electrode toward alkaline glycerol valorization coupling with acidic hydrogen production. J. Am. Chem. Soc. 144, 7224–7235 (2022).
Li, G. et al. Dual hydrogen production from electrocatalytic water reduction coupled with formaldehyde oxidation via a copper-silver electrocatalyst. Nat. Commun. 14, 525 (2023).
Han, G., Li, G. & Sun, Y. Electrocatalytic dual hydrogenation of organic substrates with a Faradaic efficiency approaching 200%. Nat. Catal. 6, 224–233 (2023).
Li, Y., Wei, X., Chen, L., Shi, J. & He, M. Nickel-molybdenum nitride nanoplate electrocatalysts for concurrent electrolytic hydrogen and formate productions. Nat. Commun. 10, 5335 (2019).
Sheremetev, A. B. et al. A practical anodic oxidation of aminofurazans to azofurazans: An environmentally friendly route. RSC Adv. 5, 37617–37625 (2015).
Yount, J. & Piercey, D. G. Electrochemical synthesis of high-nitrogen materials and energetic materials. Chem. Rev. 122, 8809–8840 (2022).
Yamamoto, K., Kuriyama, M. & Onomura, O. Anodic oxidation for the stereoselective synthesis of heterocycles. Acc. Chem. Res. 53, 105–120 (2020).
Tang, S., Liu, Y. & Lei, A. Electrochemical oxidative cross-coupling with hydrogen evolution: a green and sustainable way for bond formation. Chem 4, 27–45 (2018).
Yuan, Y., Yang, J. & Lei, A. Recent advances in electrochemical oxidative cross-coupling with hydrogen evolution involving radicals. Chem. Soc. Rev. 50, 10058–10086 (2021).
Wallace, L. Electrochemical methods for synthesis of energetic materials and remediation of waste water. Green Energ. Mater. 259–280 (2014).
Lyalin, B. V. et al. Coupling of aminofuroxans into azofuroxans under the action of electrogenerated hypohalites. Mendeleev Commun. 28, 518–520 (2018).
Florenoe, T. M. Polarography of aromatic azo compounds. I. Effect of substituents on the electroreduction of azo compounds. Aust. J. Chem. 18, 609–618 (1965).
Li, J. et al. Green electrosynthesis of 5,5′-azotetrazolate energetic materials plus energy-efficient hydrogen production using ruthenium single-atom catalysts. Adv. Mater. 34, 2203900 (2022).
Gao, M. et al. Mutual-modification effect in adjacent Pt nanoparticles and single atoms with sub-nanometer inter-site distances to boost photocatalytic hydrogen evolution. Chem. Eng. J. 446, 137127 (2022).
Liu, J. et al. High performance platinum single atom electrocatalyst for oxygen reduction reaction. Nat. Commun. 8, 15938 (2017).
Sun, M. et al. Overwhelming the performance of single atoms with atomic clusters for platinum-catalyzed hydrogen evolution. ACS Catal. 9, 8213–8223 (2019).
Zhang, L. et al. Exploring the dominant role of atomic- and nano-ruthenium as active sites for hydrogen evolution reaction in both acidic and alkaline media. Adv. Sci. 8, 2004516 (2021).
He, Q. et al. Synergic reaction kinetics over adjacent ruthenium sites for superb hydrogen generation in alkaline media. Adv. Mater. 34, 2110604 (2022).
Su, P. et al. Exceptional electrochemical HER performance with enhanced electron transfer between Ru nanoparticles and single atoms dispersed on a carbon substrate. Angew. Chem. Int. Ed. 60, 16044–16050 (2021).
Yang, J., Li, W., Wang, D. & Li, Y. Electronic metal–support interaction of single-atom catalysts and applications in electrocatalysis. Adv. Mater. 32, 2003300 (2020).
Shi, Y. et al. Electronic metal-support interaction modulates single-atom platinum catalysis for hydrogen evolution reaction. Nat. Commun. 12, 3021 (2021).
Lukowski, M. A. et al. Highly active hydrogen evolution catalysis from metallic WS2 nanosheets. Energy Environ. Sci. 7, 2608–2613 (2014).
Tao, B., Unwin, P. R. & Bentley, C. L. Nanoscale variations in the electrocatalytic activity of layered transition-metal dichalcogenides. J. Phys. Chem. C. 124, 789–798 (2020).
Tsai, C., Chan, K., Abild-Pedersen, F. & Nørskov, J. K. Active edge sites in MoSe2 and WSe2 catalysts for the hydrogen evolution reaction: a density functional study. Phys. Chem. Chem. Phys. 16, 13156–13164 (2014).
Gong, L. et al. Marriage of ultralow platinum and single-atom MnN4 moiety for augmented ORR and HER catalysis. ACS Catal. 13, 4012–4020 (2023).
Liu, Q. et al. Stable metallic 1T-WS2 nanoribbons intercalated with ammonia ions: The correlation between structure and electrical/optical properties. Adv. Mater. 27, 4837–4844 (2015).
Anjum, M. A. R., Jeong, H. Y., Lee, M. H., Shin, H. S. & Lee, J. S. Efficient hydrogen evolution reaction catalysis in alkaline media by all-in-one MoS2 with multifunctional active sites. Adv. Mater. 30, 1707105 (2018).
Ding, W. et al. Highly ambient-stable 1T-MoS2 and 1T-WS2 by hydrothermal synthesis under high magnetic fields. ACS Nano 13, 1694–1702 (2019).
Ji, L. et al. Facilitating electrocatalytic hydrogen evolutionviamultifunctional tungsten@tungsten disulfide core-shell nanospheres. J. Mater. Chem. A 9, 9272–9280 (2021).
Yao, Y. et al. Graphdiyne-WS2 2D-nanohybrid electrocatalysts for high-performance hydrogen evolution reaction. Carbon 129, 228–235 (2018).
Geng, S. et al. Activating interfacial S sites of MoS2 boosts hydrogen evolution electrocatalysis. Nano Res. 15, 1809–1816 (2022).
Pan, Y. et al. Boosting the performance of single-atom catalysts via external electric field polarization. Nat. Commun. 13, 3063 (2022).
Zhang, T. et al. Pinpointing the axial ligand effect on platinum single-atom-catalyst towards efficient alkaline hydrogen evolution reaction. Nat. Commun. 13, 6875 (2022).
Ma, T. et al. Crystalline lattice-confined atomic Pt in metal carbides to match electronic structures and hydrogen evolution behaviors of platinum. Adv. Mater. 34, 2206368 (2022).
Jiang, H. et al. Single atom catalysts in Van der Waals gaps. Nat. Commun. 13, 6863 (2022).
Zhang, H. et al. Dynamic traction of lattice-confined platinum atoms into mesoporous carbon matrix for hydrogen evolution reaction. Sci. Adv. 4, eaao6657 (2018).
Lu, B. et al. Ruthenium atomically dispersed in carbon outperforms platinum toward hydrogen evolution in alkaline media. Nat. Commun. 10, 631 (2019).
Tian, X., Zhao, P. & Sheng, W. Hydrogen evolution and oxidation: mechanistic studies and material advances. Adv. Mater. 31, 1808066 (2019).
Kuang, P. et al. Pt single atoms supported on N-doped mesoporous hollow carbon spheres with enhanced electrocatalytic H2-evolution activity. Adv. Mater. 33, 2008599 (2021).
Chandrasekaran, S. et al. Developments and perspectives on robust nano- and microstructured binder-free electrodes for bifunctional water electrolysis and beyond. Adv. Energy Mater. 12, 2200409 (2022).
Piotrowski, M. J., Piquini, P. & Da Silva, J. L. F. Density functional theory investigation of 3d, 4d, and 5d 13-atom metal clusters. Phys. Rev. B 81, 155446 (2010).
Zhang, R. et al. Single-atomic platinum on fullerene C60 surfaces for accelerated alkaline hydrogen evolution. Nat. Commun. 14, 2460 (2023).
Wang, Z. et al. PtSe2/Pt heterointerface with reduced coordination for boosted hydrogen evolution reaction. Angew. Chem. Ed. 60, 23388–23393 (2021).
Jin, H., Ha, M., Kim, M. G., Lee, J. H. & Kim, K. S. Engineering Pt coordination environment with atomically dispersed transition metal sites toward superior hydrogen evolution. Adv. Energy Mater. 13, 2204213 (2023).
Wang, D., Liu, Z. P. & Yang, W. M. Proton-promoted electron transfer in photocatalysis: key step for photocatalytic hydrogen evolution on metal/titania composites. ACS Catal. 7, 2744–2752 (2017).
Wu, G. et al. In-plane strain engineering in ultrathin noble metal nanosheets boosts the intrinsic electrocatalytic hydrogen evolution activity. Nat. Commun. 13, 4200 (2022).
Cheng, F. et al. Accelerated water activation and stabilized metal-organic framework via constructing triangular active-regions for ampere-level current density hydrogen production. Nat. Commun. 13, 6486 (2022).
Wang, Y. et al. Optimized electronic structure and p-band centre control engineering to enhance surface absorption and inherent conductivity for accelerated hydrogen evolution over a wide pH range. Phys. Chem. Chem. Phys. 22, 14537–14543 (2020).
Shi, X. et al. Rapid flame doping of Co to WS2 for efficient hydrogen evolution. Energy Environ. Sci. 11, 2270–2277 (2018).
Wang, H. et al. Super-hybrid transition metal sulfide nanoarrays of Co3S4 nanosheet/P-doped WS2 nanosheet/Co9S8 nanoparticle with Pt-like activities for robust All-pH hydrogen evolution. Adv. Funct. Mater. 32, 2112362 (2022).
Zhao, Y. et al. Modulating Pt-O-Pt atomic clusters with isolated cobalt atoms for enhanced hydrogen evolution catalysis. Nat. Commun. 13, 2430 (2022).
Tang, T. et al. Synergistic electrocatalysts for alkaline hydrogen oxidation and evolution reactions. Adv. Funct. Mater. 32, 2107479 (2022).
He, D. et al. Design and construction of three-dimensional flower-like CuO hierarchical nanostructures on copper foam for high performance supercapacitor. Electrochim. Acta 210, 639–645 (2016).
Gu, Z., Li, G., Hussain, N., Tian, B. & Shi, Y. 3D freestanding CuO@copper foam as an anode for potassium ion batteries. Appl. Surf. Sci. 592, 153323 (2022).
Yang, S. et al. Construction of synergistic Ni3S2-MoS2 nanoheterojunctions on Ni foam as bifunctional electrocatalyst for hydrogen evolution integrated with biomass valorization. Small 18, 2201306 (2022).
Meng, Y., Gnanamani, E. & Zare, R. N. Direct C(sp3)-N bond formation between toluene and amine in water microdroplets. J. Am. Chem. Soc. 144, 19709–19713 (2022).
Korpanty, J., Parent, L. R. & Gianneschi, N. C. Enhancing and mitigating radiolytic damage to soft matter in aqueous phase liquid-cell transmission electron microscopy in the presence of gold nanoparticle sensitizers or isopropanol scavengers. Nano Lett. 21, 1141–1149 (2021).
Chavez, D., Hill, L., Hiskey, M. & Kinkead, S. Preparation and explosive properties of azo- and azoxy-furazans. J. Energ. Mater. 18, 219–236 (2000).
Chellappa, R. S. et al. Intermolecular stabilization of 3,3′-diamino-4,4′-azoxyfurazan (DAAF) compressed to 20 GPa Raja. J. Phys. Chem. A 118, 5969–5982 (2014).
Zelenin, A. K. & Trudell, M. L. A two‐step synthesis of diaminofurazan and synthesis of N‐monoarylmethyl and N,N′‐diarylmethyl derivatives. J. Heterocycl. Chem. 34, 1057–1060 (1997).
Gao, Y. et al. Rhodium nanocrystals on porous graphdiyne for electrocatalytic hydrogen evolution from saline water. Nat. Commun. 13, 5277 (2022).
Wei, J. et al. In situ precise anchoring of Pt single atoms in spinel Mn3O4 for a highly efficient hydrogen evolution reaction. Energy Environ. Sci. 15, 4592–4600 (2022).
Qi, K. et al. Single-atom cobalt array bound to distorted 1T evolution catalysis MoS2 with ensemble effect for hydrogen. Nat. Commun. 10, 5231 (2019).
Wang, P. et al. Precise tuning in platinum-nickel/nickel sulfide interface nanowires for synergistic hydrogen evolution catalysis. Nat. Commun. 8, 14580 (2017).
Kresse, G. & Furthmu, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B Condens Matter 54, 11169 (1996).
Mathew, K., Sundararaman, R., Letchworth-Weaver, K., Arias, T. A. & Hennig, R. G. Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways. J. Chem. Phys. 140, 084106 (2014).
Acknowledgements
This work has received funding from the National Natural Science Foundation of China (52101230 (J.L.), 22102007 (M.X.)), the Shaanxi Key Science and Technology Innovation Team Project (2022TD-33 (Ha.M.)), the Shaanxi Innovation Capability Support Program-Young Science and Technology Star Project (2023KJXX-036 (J.L.)), the Shaanxi Key Laboratory of Special Fuel Chemistry and Material (SPCF-SKL-2021-0004 (J.L.)), the Young Elite Scientists Sponsorship Program by Xi’an Association for Science and Technology (959202313084 (J.L.)), and the Fundamental Research Funds for the Central Universities (buctrc202112 (M.X.)). The authors thank professor Bingbing Suo, professor Haiyan Zhu, and Mr. Jiezhen Xia for the help of VASP calculations, analysis, and discussions. The authors are thankful for the support of the BSRF (Beijing Synchrotron Radiation Facility) during the XAFS measurements at the beamline 1W1B, 4B7A, 4B7B, 4B9A, 4B9B.
Author information
Authors and Affiliations
Contributions
J.L. and Ha.M. conceived the idea. J.L., Y.M., Co.Z., and N.L. synthesized and characterized the materials, and tests the electrochemical performances. J.L. and Ch.Z. performed the theoretical calculations. J.L. and Hu.M. performed the TEM characterizations. J.L. and Z.G. performed the 13C NMR and FTIR characterizations. J.L. and M.X. performed the XAS spectra. J.L. drew the schematic illustrations, wrote the manuscript, and arranged the text layout. Ha.M. and J.Q. revised the manuscript and supervised the work. All the authors discussed the results and review the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Yijin Kang and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Li, J., Ma, Y., Zhang, C. et al. Green electrosynthesis of 3,3’-diamino-4,4’-azofurazan energetic materials coupled with energy-efficient hydrogen production over Pt-based catalysts. Nat Commun 14, 8146 (2023). https://doi.org/10.1038/s41467-023-43698-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-43698-x
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
