
Abstract
Coupled tandem electrolyzer concepts have been predicted to offer kinetic benefits to sluggish catalytic reactions thanks to their flexibility of reaction environments in each cell. Here we design, assemble, test, and analyze the first complete low-temperature, neutral-pH, cathode precious metal-free tandem CO2 electrolyzer cell chain. The tandem system couples an Ag-free CO2-to-CO2/CO electrolyzer (cell-1) to a CO2/CO-to-C2+ product electrolyzer (cell-2). Cell-1 and cell-2 incorporate selective Ni-N-C-based and Cu-based Gas Diffusion Cathodes, respectively, and operate at sustainable neutral pH conditions. Using our tandem cell system, we report strongly enhanced rates for the production of ethylene (by 50%) and alcohols (by 100%) and a sharply increased C2+ energy efficiency (by 100%) at current densities of up to 700 mA cm−2 compared to the single CO2-to-C2+ electrolyzer cell system approach. This study demonstrates that coupled tandem electrolyzer cell systems can offer kinetic and practical energetic benefits over single-cell designs for the production of value-added C2+ chemicals and fuels directly from CO2 feeds without intermediate separation or purification.
Similar content being viewed by others


Introduction
In the light of intensified efforts to mitigate power and chemicals production from fossil sources and their substitution by renewable ones, electrocatalytic power-to-chemicals and power-to-fuels technologies are emerging as one of the future pillars of sustainable chemicals industry1. Recent years have seen a drastic increase in attention for the electrocatalytic CO2 reduction reaction (CO2RR), advancing the production of 2e− reduction products, such as CO and formic acid, towards commercialization2,3,4. In contrast, the electrocatalytic reaction process pathway towards C2+ hydrocarbons and oxygenates, in particular C2H4, Ethanol, Propanol, lacks similar technological advances5,6,7,8,9,10.
Following Hori’s pivotal work on CO2RR, establishing CO as key intermediate in hydrocarbons and oxygenates production, studies investigated CO as feedstock in what is referred to as the CO reduction reaction (CORR). CORR achieves a higher faradaic efficiency (FE) for the production of C2+ compounds, in part because some common C1 side-products of the CO2RR, such as CO or HCOO-, cannot be formed, but also due to more favorable kinetics and surface coverages of H* and CO*. Specifically, the production of oxygenates has been shown to be favored by CORR compared to CO2RR, especially under alkaline conditions that are only sustainable for CORR due to the invariable formation of (bi)carbonate from CO211,12,13,14,15,16,17,18,19.
The favorable kinetics of alkaline CORR compared to CO2RR for production of C2+ species has motivated researchers to try to find ways to separate off the CORR in a CO2RR electrolyzer. Tandem Cathode Concepts follow the idea to spatially decouple the initial 2e- reduction of CO2 to CO and the subsequent reduction steps of CO towards C2+ products. The tandem split can be realized at vastly different length scales and distinct configurations: (i) at the atomic scale by two neighboring active sites coupled by CO spillover, (ii) at the macroscopic electrode scale with macroscopically separated regions of distinct catalysts inside the same electrolyzer cell, and (iii) at the macroscopic electrolyzer cell scale forming a coupled process cascade20,21,22,23,24,25,26,27,28,29,30,31,32,33,34,35. While the idea of placing the active sites of CO source and sink inside the same electrode is an intriguing one, it has been shown to be inherently sensitive to the relative spatial catalyst distribution that is challenging to maintain over longer operation times considering the reported mobility of Cu catalysts under CO2RR36,37,38,39,40,41.
The present work sets out to explore the coupled tandem electrolyzer cell approach, i.e., two individual electrolyzers in series, for a significantly enhanced production of C2+ compounds from CO2 in a catalytic reaction cascade. We deliberately avoid the use of high-temperature technologies, harsh reaction conditions, and demonstrate the advantages by avoiding CO2 scrubbing steps to increase the practicality of the overall process, and make use of kinetic benefits for CO2/CO co-feeds agreeing with the presence of reactant- and product-specific reaction sites on Cu catalysts20,42. Accordingly, we deploy a noble metal-free single Ni atom site electrocatalyst (Ni-N-C) in a neutral-pH, near-ambient temperature, zero-gap setup for selective conversion of CO2 to CO-rich CO2/CO mixed streams. The CO2/CO mixed feed was then directly introduced to a second flow electrolyzer cell equipped with a Cu-catalyst for conversion of CO2/CO mixed feeds into C2+ products. We report strongly enhanced rates for the production of ethylene (~50%) and of alcohols (>= 100%) and a sharply increased C2+ energy efficiency (>=100%) in the tandem systems compared to the conventional single CO2 electrolyzer cell approach. To validate our conclusions and provide kinetic insight, we also conducted single-cell control experiments with controlled mixed CO2/CO co-feeds to explore the origin of the observed tandem effect. These experiments suggest that CO2 is being selectively scrubbed from the gas mixture by cathodically generated OH- in vicinity of the cathode thus locally increasing the effective CO concentration and CO surface coverage, mimicking the performance of CO-rich gas feeds.
Results and Discussion
Design of a low-temperature tandem electrolyzer cascade for the production of C2+ compounds
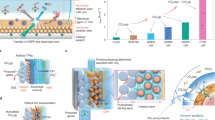
Figure 1a introduces the concept and the process design of the first low-temperature coupled tandem CO2 electrolyzer cell system developed and studied in this contribution.

a Concept of coupled Tandem CO2 Electrolyzer Cells highlighting the flows of gas (red) and liquid (blue) reactants and products. Additional information on the detailed reaction conditions can be found in Supplementary Fig. 2. b Photographs of the two electrolysis reactors used for coupled CO2-to-CO electrolysis and CO2/CO-to-C2+ electrolysis, with inserts showing an enlargement of the cells used in each case. c Detailed Process scheme of tandem cell set up with information on gas (red) and liquid (blue) flow directions and the most important components of the investigated tandem system.
The numbering of the cells refers to the order in which they were deployed in the tandem system electrolyzer cascade and is used as a denomination throughout this work. Cell-1, the first electrolyzer in the reaction cascade, converts CO2 to CO and is the point of entry for the CO2 feed. At the cathode Gas Diffusion Electrode (GDE) of cell-1, we use a previously developed noble metal-free single Ni atom site electrocatalyst (NiNC) deployed into a neutral anode-pH, zero-cathode-gap electrode design43. Since the stoichiometric CO2 ratio, λstoich, remains above one, the cathodic CO2 to CO conversion in cell-1 invariably results in mixed CO2/CO exit feeds, even if the cathode is operated at 100% faradaic CO efficiency. This mixed CO2/CO gas feed is directly used as input feed for a Cu-based electrolyzer “cell-2” to efficiently produce C2+ products, as schematically depicted in Fig. 1a. Note, that while within this work we refer to the commercial Cu catalysts as “Cu” for simplicity, XRD analysis showed that the material is partially oxidized and shows a presence of a Cu2O phase, see Supplementary Fig. 1.
Figure 1b shows photographs of our experimental setup with inserts of the individual electrolysis cells used during electrochemical characterization.
The detailed tandem cell process scheme is given in Fig. 1c. Note how cell-1 and cell-2 were connected in series by the CO2 gas flow to enable an enrichment with CO in the gas stream before entering cell-2. A more detailed discussion on the geometry of the individual cells and testing setup including operational parameters can be found in Supplementary Fig. 2.
Electrochemical CO2/CO mixed gas feed reduction
Prior to coupled tandem electrolyzer cell experiments, we studied how the presence of CO in the CO2 reactant gas feed affects the product production rates of a single Cu cathode-based CO2 electrolyzer.
Figure 2 displays the production rates of the four most prominent reaction products, i.e., hydrogen, ethylene, ethanol, and n-propanol as a function of CO mol% in the CO2/CO co-feed and of applied current density. While the absolute volumetric flow of the gas feed was kept constant at 50 mL min-1, we have varied its composition by adjusting the individual flow rates of CO2 and CO that were mixed prior to entering cell-2. Figure 2a shows a profound effect of CO in the co-feed on the production of hydrogen. For CO-lean feeds, an increase in CO raised H2 production, as well, before H2 production rates dropped for CO-rich co-feeds. The molar CO concentration associated with the maximum H2 production depended on the applied current density. While for low current densities such as 100 or 200 mA cm-2 the turning point was at 66 mol%CO, larger current densities showed the transition at 50 mol%CO (300 and 400 mA cm-2), or even at 34 mol%CO (500 to 700 mA cm-2). The single-maximum pattern of hydrogen production appears surprising, as the total carbon content in the electrolyte monotonically drops with more CO due to the vastly lower solubility of CO versus CO2. So, COx surface coverages should monotonically decline, as well. For the production rate of ethylene, shown in Fig. 2b, a distinctly different trend was apparent. Here, increasing CO mol% improved the ethylene production rate. Note that this improvement was most significant up to a co-feed composition of 50 mol%CO, beyond which it plateaued. The production rates of ethanol and n-propanol, Fig. 2c, d, showed unusually sharp enhancements with rising molar CO mol% in the co-feed. In contrast to the trends observed for ethylene, the beneficial effect on production rate was most pronounced past 50 mol%CO approaching pure CO gas feed. Production rates of other compounds such as formate, acetate and ally alcohol can be found in Supplementary Fig. 3. The trend for allyl alcohol generally agrees with the behavior observed for other alcohols, whereas formate production was suppressed by raising CO content in line with expectations.

Production rates of key products using a single electrolyzer cell as a function of CO mol per cent (mol%) in the CO2/CO co-feed, measured for various applied current densities in Cu-based cell-2. Production rates are shown for a hydrogen, b ethylene, c ethanol and d n-propanol. Electrolysis was conducted in single-cell experiments deploying only the Cu-based electrolyzer denoted as cell-2. The total volumetric flow of co-feed mixtures was kept constant at 50 mL min−1 throughout all experiments, while the CO concentration was gradually increased. Displayed values represent the average and error bars the standard deviation of at least 2 independent measurements.
To better appreciate the effect of CO in the feed, let us consider the kinetic enhancements of individual C2+ production rates on a relative scale to pure CO2 feeds: CO in the feed has the most significant effect on PrOH production, showing a 4x increase in rate, followed by a 2-3x increase of EtOH production and finally 2x increase in C2H4 production. This sharp enhancement of C2+ oxygenate production under CO-rich feed conditions is also reflected by the dramatically larger FE values obtained for pure CO feed (CORR) compared to pure CO2 feed (CO2RR), Supplementary Fig. 4. These enhancements can be explained mechanistically by enhanced cross-coupling CO dimerization rates on the surface of the Cu catalyst20. The combined presence of CO2 and CO gas in the feed offers new C-C coupling pathways between two adsorbed CO surface species derived from both CO2 and CO on two distinct, non-scrambling Cu surface sites, in line with the multi-site hypothesis on Cu catalysts20,42.
Kinetic analysis of CO2RR and CORR of CO2/CO feeds
To gain a better understanding of the origin of the CO dependence of the production rates, we tracked the applied cathode potentials as well as the CO consumption during co-feed experiments. Figure 3a shows IR-free cathode polarization curves obtained for experiments at varying CO feed concentrations. Evidently, cathode overpotentials (Ecathode, IR free) during electrolysis in pure CO2 and in small molar CO co-feeds of 10 mol% remained near −1.3 VSHE at a current density approaching 700 mA cm–2. In contrast, the required cathode potentials shifted to –1.4 VSHE for the case of a pure CO feed, possibly pointing to concentration overpotentials due to lower CO solubility.

a IR-corrected cathode polarization curves as a function of applied current density for various investigated co-feed compositions. b The net CO flux, COnet flux, defined as the difference in the molar CO influx, COin, and molar CO outflux, COout, plotted versus the applied current density and co-feed composition. c Apparent selectivity for CORR, denoted SCORR, as a function of applied current density and co-feed composition. SCORR was defined as the ratio between the electrocatalytic CO consumption rate (COconsumption = –COnet flux) and the theoretically required molar CO consumption rate to produce all C2+ species by CORR. Colored areas indicate regions controlled by the respective primary reactant used in the production of C2+ species, either CO or CO2, dependent on the absolute value of SCORR. Electrolysis was conducted in single-cell experiments deploying only the Cu-based electrolyzer denoted as cell-2. The total volumetric flow of co-feed mixtures was kept constant at 50 mL min−1 throughout all experiments, while the CO concentration was gradually increased. Displayed values represent the average and error bars the standard deviation of at least 2 independent measurements.
As our co-feeding experiments introduced two distinct reactants, i.e., CO2 and CO, we set out to quantify whether and how the shift in C2+ production rates with CO mol% correlated with a shift from predominantly CO2-controlled electroreduction to a predominantly CO-controlled one. For that purpose, we measured the CO outflux (area-normalized molar outflow rates) out of the electrolyzer at various co-feed compositions, Supplementary Fig. 5. Considering the large stoichiometric excess of reactants in the input feed, CO conversion was incomplete for all investigated co-feeds. Higher CO mol% in the feed led to higher molar CO outflux, COout, from the electrolyzer. However, the decrease in COout with larger current density was significantly more pronounced for CO-rich co-feeds, suggesting a larger relative consumption thereof. Taking the difference between the molar CO influx, COin, and the molar CO outflux during electrolysis, we derived the net CO flux, defined as COnet flux = (COout - COin), as function of applied current and co-feed composition, Fig. 3b. In this plot, positive and negative values of COnet flux denote a catalytic net production and net consumption of CO, respectively. Generally, both higher CO mol% in the feed and larger current density led to increasing CO consumption, i.e., more negative values of COnet flux. For CO mol% <= 50 in the feed, there appeared a critical current density, beyond which net CO production turned into net CO consumption (10, 33 and 50 mol%CO at 500, 200 and 100 mA cm-2, respectively). For CO mol% > 50, only net CO consumption was apparent over the entire range of current density. This data allowed us to estimate what fraction of the produced C2+ species can be accounted for by an exclusive CORR pathway. To do that, we considered the apparent CO reduction reaction selectivity, SCORR, defined as the ratio of the experimental CO consumption rate, COconsumption = –COnet flux and the theoretical CO demand that is required for the combined experimental C2+ production rate, according to:
where \({\nu }_{i}\) and \({\dot{n}}_{i}\) denote the number of carbon atoms in species i and its molar production rate, respectively.
Figure 3c plots SCORR versus the co-feed compositions and the applied current density. Both CO mol% in the feed and applied current density control SCORR, with SCORR approaching 100% with larger applied current density or with rising CO mol%. As expected, a pure CO feed showed a SCORR of 100% at all currents, agreeing with the conditions of CO being the only reactant. Importantly, sustained SCORR values near 100% were evident for 67 mol% CO feed and currents as low as 200 mA cm−2. This observation suggests that under conditions of 67 mol% CO and 200 mA cm-2 essentially all C2+ product formation can be accounted for by the reduction and dimerization of CO gas (prevalent CORR). This kinetic effect arises because the catalytically effective gas composition at the catalyst-electrolyte-interface differs from the bulk gas feed composition. It is a consequence of the preferential acid-base reaction of CO2 in CO2/CO co-feeds with catalytically generated OH−18,19. This acid-base reaction increases the effective CO concentration for a given CO2/CO co-feed, and this will become more severe at a larger current. Using SCORR, we split the catalytic co-feed reduction kinetics into a CO-controlled regime (SCORR > 50%) and a CO2-controlled one (SCORR < 50%), see colored regions in Fig. 3c, which deconvolutes the overall reduction reactivity.
Carbon Selectivity of C2+ production under CO2/CO co-feeds
Rising CO concentration in CO2/CO co-feeds sharply enhances the total production rates of C2+ species (Fig. 2). In part, this influence could be explained by the reduced number of electrons required to produce C2+ species from the CORR pathway, see Supplemental Discussion 1. However, charge stoichiometry falls short of the 2 to 4-fold experimental increase shown in Fig. 2. This is manifested in the single feed experiments CO2RR and CORR in Supplementary Fig. 4 that showed drastically larger FE values for C2+ species in CORR experiments: In particular, the FE values of alcoholic oxygenates showed sharp increase under CORR compared to CO2RR14,16. We hypothesize that the sharp rise in the C2+ production must originate from a change in surface catalytic selectivity associated with an enrichment of CO gas in the feed inside the electrolyzer, and associated with a changed surface coverage of CO at the Cu catalyst. Accurate calculation of faradaic product selectivity requires knowledge of the electron transfer numbers, which, however, are not known for co-feeding. This is why, for the purpose of a mechanistic discussion, we calculated a carbon selectivity of species i, CSi, (see Supplementary Fig. 6) defined as the ratio between the carbon atom flux into an individual C2+ species i and the total carbon atom flux into all C2+ products.
Here, \({\dot{n}}_{i}\) and \({\nu }_{i}\) denote the molar production rate of species i and the number of carbon atoms in species i, respectively. CSi reveals changes in mechanistic reaction pathways for different co-feeds and currents and can be compared to kinetic modeling results. More detailed information on CSi is given in the Supplemental Discussion 1.
In Supplementary Fig. 6, CSethylene showed a maximum at 33 CO mol% and decreased at higher CO mol% in the feed. Above 10 mol%CO, CSEtOH rose gradually by ca. 10 percentage points, and so did CSPrOH. Evidently, higher CO mol% favor formation of oxygenates. While the applied current density showed a minor effect on CSEthylene, the production of either alcohol was profoundly affected: Higher currents favor the production of EtOH over PrOH. Mechanistically, this can be rationalized by a lower surface coverage of CO that makes formation of C3 products less likely, as concluded in prior experiments of pure CO feeds at varying partial pressures12,13,44,45,46. A more detailed mechanistic discussion of the observed trends in CSi values under CO2/CO co-feeds is provided in the Supplemental Discussion 2.
Operation of a two-cell, tandem system for enhanced production rates and energy efficiency of C2+ compounds
After we had deconvoluted and rationalized the increase in overall C2+ production rates under controlled CO2/CO co-feed electrolysis, we moved to assemble, operate, and analyze the first complete low-temperature, coupled CO2 tandem electrolyzer cell system excluding intermediate processing steps. Unlike tandem catalyst designs20, tandem cell designs allow for individually optimized active sites and electrolysis conditions for each step. We generated CO-rich co-feeds in “Cell-1” using a neutral-pH, zero-gap electrolyzer with a Ni-N-C catalyst-based Gas Diffusion Electrode (GDE)43. The use of the CO2-to-CO electrolyzer as cell-1 allowed precise control over the resulting mixed CO2/CO exit gas composition (equivalent down-stream to the CO2/CO co-feed into cell−2) via two control parameters, that is the applied current density and the CO2 feed rate.
As operating conditions of cell-1, we selected 200 mA cm−2 and near-100% FECO including only minute amounts of H2 to achieve a controlled mixed of predominantly CO2/CO exit steam out of cell-1. Supplementary Fig. 7 shows how the FECO affects the absolute CO production rate as a function of the applied current density of cell-1. At the chosen operating point, cell-1 generates about 7.6 mlCO min-1. To ensure stable performance of cell-1, we monitored the experimental FECO and cell potential over the electrolysis time of 12 hours at 200 mA cm−2, shown in Supplementary Fig. 8. We thereby convinced ourselves that the FECO value remained at near-unity without changes in the cell potential. Since the CO2 flow rate into cell-1 controls the molar CO2/CO ratio of the exit steam, we have also investigated the effect of CO2 flow rate. We chose (i) a flow rate of 50 ml min−1 corresponding to the total flow rate of the co-feed tests above, and (ii) a reduced CO2 flow rate of 30 ml min−1 to enrich the exit flow in CO further. In cell-1 the CO2 flow of 50 mL min−1 was converted to a gas mixture (co-feed) of CO2 and CO with 18 mol% of CO and a total volumetric flow rate of 42 mL min−1, whereas a CO2 input of 30 mL min−1 resulted in an exit flow of 22 mL min−1 with 35 mol% of CO.
Figure 4 compares the catalytic production rates of the tandem system versus the reference single-cell CO2RR (using cell-2) as a function of the applied current density of cell-2. The investigated CO2 flow rates of 50 mL min−1 and 30 mL min−1 are denoted as Tandem-50 and Tandem-30, respectively, the reference is denoted as Single Cell. In good agreement with our previous experiments, hydrogen production slightly increased with more CO in the co-feed between cell-1 and cell-2, Fig. 4a. Tandem-30 showed a larger H2 production rate than Tandem-50, consistent with its larger CO mol% in the exit of cell-1. While Tandem-50 matched the H2 production rate of the single-cell experiment at 700 mA cm−2, H2 rate of Tandem-30 increased rapidly at larger current densities, most likely due to reactant transport limitation. CO production rates of Tandem-50/30 at 50 mA cm−2, Fig. 4b, are controlled by CO production of cell-1. CO rates of Tandem-30 declined steadily approaching the Single Cell, while CO rates of Tandem-50 showed a maximum at 200 mA cm−2 before declining. Ethylene rates, Fig. 4c, were significantly enhanced (up to 50% at 500 mA cm−2) for the Tandem case over the entire current density range. A similar behavior was seen in the EtOH production rates, Fig. 4d, with space-time yield enhancements of up to 100% at 700 mA cm−2. Likewise, production rates of C3 compounds, i.e., PrOH and AllylOH, Fig. 4e, f, showed more than 100% enhanced production rates at 700 mA cm−2 in tandem configuration, with Tandem-30 suffering from mass transport limitations.

Production rate of a hydrogen, b carbon monoxide, c ethylene, d ethanol, e n-propanol, and f allyl alcohol as a function of current density applied to cell-2. Values given in the legend correspond to the volumetric flow of CO2 introduced to the tandem system. In the tandem configuration, cell-1 has been continually operated at a current density of 200 mA cm−2 while the current density applied to cell-2 has been varied. For both cells the active geometric surface area is 5 cm2. The single-cell refers to CO2RR experiments using only cell-2 of the reaction cascade at a CO2 volume flow of 50 mL min−1. Displayed values represent the average and error bars the standard deviation of at least 2 independent measurements.
The experimental performance of the tandem systems are in good agreement with our CO2/CO co-feeds results. Tandems showed a sharp increase in C2+ production rates over the single-cell CO2RR experiments. While C2+ production rates of Tandem-30 were superior to Tandem-50, consistent with the larger CO concentration in the co-feed to cell-2, clear signs of mass transport limitation were apparent when exceeding a current density of 500 mA cm−2, reflected in a rising H2 rate and a declining C2+ rate. Note that the extent of the decrease in C2+ rates at large current density followed the order of PrOH/AllylOH>EtOH>C2H4 suggesting that C3 compounds are more susceptible to the insufficient reactant transport. This behavior fits well with our previous observation in single-cell experiments that showed that C3 production rates were particularly sensitive to CO depletion in the gas feed.
In order to provide a direct comparison of product productions in the tandem experiments (18 mol%CO for Tandem-50, and 35 mol%CO for Tandem-30) and the CO2/CO co-feed experiments, we overlaid their production rates in Supplementary Fig. 9. This revealed some unexpected, yet important kinetic discrepancies: In either tandem experiment, the observed production rates suggested a higher CO mol% in the co-feed to cell-2 than inferred from the overlaid co-feed single-cell experiments. This trend is also reflected in the C2+ product carbon selectivity comparisons in Supplementary Fig. 10. With larger current densities, this seeming CO-enrichment becomes progressively more pronounced. To explain this observation, we recall the lower actual tandem feed rates of 42 mL min−1 and 22 mL min−1 for Tandem-50 and Tandem-30, respectively, compared to 50 mL min−1 for the single cell case. This difference in available CO2 becomes clearer in a direct comparison of the molar reactant flow to cell-2 under the various conditions, see Supplementary Fig. 11. As a result of this, at a given OH- generation (given current density), the lower molar CO2 flow into cell-2 results in a larger share of CO2 being depleted. This enhances the effective CO concentration near the electrode, and results in a product pattern resembling a higher bulk mol%CO.
The technological application of the tandem concept ultimately requires an acceptable overall energy efficiency, henceforth denoted EE. Estimation of EE values in a tandem system requires power-in /power out consideration of both cells of the reaction cascade. Equation (3) defines the EE as the ratio between the rate of chemical energy stored in products based on their higher heating values (HHV) and the total electrical input power introduced to the electrolyzer system.
In Fig. 5, we compare the performance of the single-cell and tandem system experiments with particular focus on the electrical power consumption, energy efficiency, and system characteristics. Figure 5a deconvolutes the electrical input power, Pin, of the Tandem-50 system in terms of cell-1 and cell−2 in absolute and relative terms. As cell-1 was operated at constant current and cell voltage, absolute Pin of cell-1 remained constant, while Pin of cell-2 strongly increased. This has important implications for the relative Pin balance between the two cells: At low currents applied to cell-2, total Pin is clearly dominated by cell-1, later by cell−2 increasing to over 90% at 3.5 A. This is because of the higher applied currents and cell resistances of the 1-gap design of cell−2 compared to the zero-gap design of cell-1.

a Total input Power, Pin, as function of applied current to cell-2 deconvoluted in absolute and relative terms into the individual contributions of cell-1 and cell−2 during tandem experiments with CO2 input flow of 50 mL min−1. b Energy efficiency for C2H4, EtOH and n-PrOH based on their higher heating value (HHV) as a function of applied current for single-cell and tandem experiments. Tandem experiments are labeled in the legend with the respective input flow of CO2 (30 or 50 mL min−1). c, d Schematic illustrations of c the tandem electrolyzer cell system and d single-cell system with arrows indicating the flow of products and reactants, CO2, CO and C2+, as well as the distribution of electrical power across the coupled electrochemical cells. e, f Distribution profiles of total power input and concentrations of CO2, CO and C2H4 within the reaction cascade for e tandem experiments with 50 mL min−1 CO2 feed and f single-cell experiments. In the tandem configuration, cell-1 has been continually operated at a current density of 200 mA cm−2 while the current density applied to cell-2 has been varied. For both cells the active geometric surface area is 5 cm2. Displayed error bars represent the standard deviation of at least 2 independent measurements.
Figure 5b plots the EE of C2H4, EtOH and PrOH in Tandem-50 and Tandem-30 versus the single-cell design. While at low currents the single-cell system shows competitive C2+ EE values, at larger currents > 1 A tandem designs sharply outperform the single-cell system. In particular, the Tandem-30 experiments shows a strongly enhanced total absolute C2+ EE of up to 13%. We then analyzed the individual EE of C2H4, EtOH and PrOH as a function of total Pin for each electrolyzer design in Supplementary Fig. 12. The tandem systems showed relative enhancements in C2+ EE values of up to 100% over a broad operation range, in particular Tandem-50 at high C2+ productivities and large power inputs. At low power input, the productivity of the tandem system was dominated by cell-1 reflected in the high EE for CO and low EE for H2 in comparison to the single-cell system shown in Supplementary Fig. 13. In addition to the advantages for EE, we also observed a more efficient reactant utilization described by the single pass carbon efficiency, SPCE. When directly comparing the single-cell system to the tandem-system, the SPCE nearly doubled from 17% to 30–35% depending on the applied current, see Supplementary Fig. 14. The increase in SPCE was larger than can be expected by a simple consideration of the additional operation of cell-1 suggesting the inherent benefit of tandem operation. A more detailed discussion can be found in the Supplementary Information of this work.
Apart from the electrochemical performance differences, tandem electrolyzer systems also differ from a single-cell system in terms of some of their process characteristics. As the single-cell system deploys only one single reactor, the total input power, the CO2 consumption, and product generation occur at the same physical location, schematically depicted in Fig. 5d. This results in a single-stepped experimental concentration and power profile, as given for 700 mA cm−2 in Fig. 5f. By contrast, in the tandem system the total input power splits into two reactors and the conversion of CO2 occurs along a reaction cascade, schematically depicted in Fig. 5c. Consequentially, the experimental concentration and power profile at 700 mA cm−2 (cell-2), given in Fig. 5e, reveals two distinct steps.
In summary, we have designed, assembled, and analyzed the first full low-temperature tandem electrolyzer cell system designed for an efficient electrochemical CO2 reduction to C2+ products, such as Ethylene, EtOH, and PrOH. The tandem design consisted of two low-temperature electrolyzer cells in series, which enabled a catalytic reaction cascade of the reaction of CO2 to mixed CO2/CO streams in cell-1, coupled to the reaction of mixed CO2/CO feeds to C2+ products in cell-2. We demonstrated that the tandem system strongly enhanced production rates of C2+ compounds compared to the use of a conventional singe-cell system. We also provided evidence that the additional power input required to operate the tandem system was comperatively minor thanks to the drastically lower energetic demand of the CO2-to-CO electrolyzer cell-1. Consequentially, the proposed tandem system allowed for an increase in individual EE of C2+ species compared to the conventional single-cell system.
Methods
Electrochemical cell and electrode preparation
Depending on the target product of electrolysis, electrochemical tests were performed using two different cell geometries. In the case of CO2-to-CO reduction, a “zero-gap” cell was used shown in Supplementary Fig. 2a, referred to as cell-1. For the reduction of either CO2 or CO, or co-feed mixtures of the two to produce C2+ species a “one-gap” cell was used, shown in Supplementary Fig. 2b, referred to as cell-2.
Details on cell-1
The Ni-N-C catalyst used in this work was synthesized through thermal carbonization of a Ni-imidazolate according to a previously established procedure. In short, to an aqueous solution of Ni(NO3)2 and imidazole, a NaOH solution was added dropwise. After overnight stirring, the precipitate was filtered, washed, and freeze-dried. This catalyst precursor material was then carbonized by thermal treatment in N2 atmosphere at 800 °C. The crude product was acid-washed at 80 °C in H2SO4, followed by rinsing with water to pH-neutral and freeze-drying to obtain the final catalyst powder43. For the preparation of the cathode GDE, 55 mg as-prepared Ni-N-C catalyst, 195 mg Sustainion solution (Dioxide Materials, 5 wt% Sustainion in ethanol solution), 100 μL DI-water and 2900 μL isopropanol were mixed and sonificated using a sonifer horn for 15 mins. Afterwards, the prepared ink was sprayed coated at 60 °C onto 5 cm2 of the micro porous side of a commercial gas diffusion layer provided by DeNora (GDL2). As electrochemical cell, a membrane electrode assembly type electrolyzer setup was deployed for the CO2 to CO conversion. An exploded-view of the electrolyzer system is given in Supplementary Fig. 2a. The catalyst-coated cathode GDL, membrane (Sustainion membrane X37-50 RT, Dioxide material), and the anode (5 cm2, commercial IrO2-GDE supplied from Dioxide Materials) were assembled layer by layer including PTFE gaskets (thickness: 200 microns; window size: 5c m2) to guarantee for leak-tightness. Finally, all layers were compressed between the electrolyzer endplates, which served as flow fields and current collectors. For catalytic tests involving cell-1, an aqueous solution of 0.1 M KHCO3 electrolyte was used on the anode side only and constantly recycled at a volumetric flow rate of 20 mL min−1. On the cathode, a humidified stream of CO2 was introduced by a mass flow controller at a rate of 25 mL min−1. After the reaction in cell-1, the product stream, composed mainly of CO and unreacted CO2, was introduced as reactant gas feed to the cathode GDE of cell-2.
Details on cell-2
As anode material Ni foam (Fraunhofer IFAM, thickness of 0.45 mm) was placed inside the flow field of the anode Titanium endplate (Dioxide materials). For the preparation of the cathode GDE a sintered PTFE membrane (Elringklinger, thickness of 0.5 mm) was used as substrate and coated by spray-painting with a dispersion of commercial spherical Cu particles (Sigma-Aldrich, 40–60 nm) and PTFE particles (Sigma-Aldrich, 1 μm) in ethanol. The relative content of PTFE particles was adjusted to achieve a final loading of 20 wt.% in relation to the combined loading of Cu and PTFE particles on the substrate. The absolute loading of Cu particles on the PTFE substrate was controlled by the volume of the catalyst dispersion used during the spray-painting process and fixed at 3.0 mg cm−2 throughout the whole study. When assembling the electrolysis cell, the Cu-PTFE GDE is fixed to the cathode Titanium endplate with a conductive Cu tape to enable electronic contacting of the catalyst layer. Silicon gaskets of 0.5 mm in thickness and a cut-out window of 5 cm2 were used to expose 5 cm2 of the electrode as geometric active area and guarantee a leak-free operation during catalytic testing. An aqueous solution of 1.0 M KHCO3 with a volume of 500 mL each was deployed as anolyte and catholyte solution. In the cathode compartment, a custom-designed flow field composed of PEEK and a thickness of 1.0 mm was deployed. The anode and cathode compartments of the electrochemical cell were separated by an anion exchange membrane (Selemion, AMV).
Electrochemical setup
For electrochemical testing the electrolysis cell was embedded in the teststand depicted in Supplementary Fig. 2c that allowed careful control over pressure levels, as well as gas and electrolyte flow rates. Mass flow controllers (Bronkhorst) were used to accurately adjust the flow of all gases (N2, CO, CO2). The reactant gas feeds, CO2 and CO pure feeds or gas mixtures of the two, were introduced at a total constant rate of flow of 50 mL min−1. The anolyte and catholyte were constantly recirculated through the respective compartments of the cell by membrane pumps (KNF, SIMDOS 10) at a constant flow of 50 mL min−1. Additionally, a dead volume filled with gas is induced on the inlets and outlets of the membrane pumps to mitigate their inherent pulsation during operation. The cell was operated with an overpressure on the liquid side of the cathode GDE. Here, the differential pressure across the cathode was set to 100 mbar by adjusting the absolute pressure of the cathode gas side to 1.300 bar and the absolute pressure of the catholyte reservoir headspace to 1.400 bar by use of back pressure regulators (Bronkhorst).
Electrochemical measurement protocol
Electrochemical characterization for cell-2 was carried out according to a preset measurement protocol comprising three successive measurement techniques: Firstly, galvanostatic steps in current density, secondly cyclovoltammetry (CV) and finally galvanostatic electrochemical impedance spectroscopy (GEIS). All electrochemical data that show error bars represent the mean values and standard deviation obtained from at least two independent samples.
Galvanostatic current density steps
Here, after an initial equilibration time of 20 min at open circuit potential (OCP), the cathodic current density was increased stepwise as follows: 50, 100, 200, 300, 400, 500, 600 and finally 700 mA cm−2. After the 700 mA cm−2 current density step, the current was decreased again, retracing the previous steps in the opposite direction towards a final current density of 50 mA cm−2. During the whole procedure, each of the set current density steps was kept constant for a period of 20 min before moving towards the next value.
Cyclovoltammetry (CV)
After completion of the galvanostatic current density steps, CVs were conducted in a potential window of 100 mV (−0.600 to −0.700 VAg/AgCl). Within this potential window 5 CVs were conducted at a constant scan rate before increasing the scan rate and performing 5 CVs again. This procedure was performed sequentially for scan rates of 1, 2, 5, 10, 20, 50, 100, 200, 500, 700, 1000, 1500 and 2000 mV s−1.
Galvanostatic electrochemical impedance spectroscopy (GEIS)
In the final step, GEIS measurements were conducted at 1, 10, 20, 50, 100, 200, 300, 400, 500, 600, and 700 mA cm−2 of absolute cathodic current. The frequency was varied between 500 kHz and 100 mHz and the amplitude of the interference signal was set to 10% of the respective current density value at which the GEIS measurement was carried out.
Quantification of products
All data that show error bars represent the mean values and standard deviation obtained from at least two independent samples.
Gaseous products
After electrochemical reaction in the electrolyzer the outgoing gas stream was mixed with a constant flow of N2 as an internal standard (16 mL min−1) and CO2 (150 mL min−1) that has been purged through the headspace of the catholyte compartment in order to collect gaseous products that were released at the catholyte side during operation. This mixture of gases was introduced into a gas chromatograph (Shimadzu, GC 2014 series) in the following denoted as GC. The GC was equipped with a thermal conductivity detector (TCD) used in the quantification of H2 and N2 concentrations, as well as a flame ionization detector (FID) that was used for quantification of methane, ethylene and CO. For the detection of CO by the FID a methanizer has been used for the thermal reduction of CO to CH4 prior to its detection. Determination of the absolute volumetric flow after electrolysis has been conducted based on changes in the concentration of N2 standard as detected by the TCD.
Liquid products
During electrolysis, aliquots of the catholyte have been collected after 20 min of reaction time at constant current. These aliquots were analyzed for their content of alcohols by liquid injection gas chromatography (Shimadzu, GC 2010 series) equipped with an FID. Furthermore, a high-performance liquid chromatograph (Agilent, 1200 series) equipped with a refractive index detector has been deployed for quantification of carbonic acids and their respective salts.
Data availability
All data supporting the results of this study, are included in the published article or the associated Supplementary Information. Additional information will be made available upon reasonable request to the corresponding author.
References
Gür, T. M. Review of electrical energy storage technologies, materials and systems: challenges and prospects for large-scale grid storage. Energy Environ. Sci. 11, 2696–2767 (2018).
De Luna, P. et al. What would it take for renewably powered electrosynthesis to displace petrochemical processes? Science 364, eaav3506 (2019).
Krause, R. et al. Industrial application aspects of the electrochemical reduction of CO2 to CO in aqueous electrolyte. Chem. Ing. Tech. 92, 53–61 (2020).
Haas, T., Krause, R., Weber, R., Demler, M. & Schmid, G. Technical photosynthesis involving CO2 electrolysis and fermentation. Nat. Catal. 1, 32–39 (2018).
Masel, R. I. et al. An industrial perspective on catalysts for low-temperature CO2 electrolysis. Nat. Nanotechnol. 16, 118–128 (2021).
De Gregorio, G. L. et al. Facet-dependent selectivity of Cu catalysts in electrochemical CO2 reduction at commercially viable current densities. ACS Catal. 10, 4854–4862 (2020).
Dinh, C.-T. et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 360, 783 (2018).
Möller, T. et al. The product selectivity zones in gas diffusion electrodes during the electrocatalytic reduction of CO2. Energy Environ. Sci. 14, 5995–6006 (2021).
Zhong, M. et al. Accelerated discovery of CO2 electrocatalysts using active machine learning. Nature 581, 178–183 (2020).
García de Arquer, F. P. et al. CO2 electrolysis to multicarbon products at activities greater than 1 A cm−2. Science 367, 661–666 (2020).
Hori, Y., Murata, A., Takahashi, R. & Suzuki, S. Electroreduction of carbon monoxide to methane and ethylene at a copper electrode in aqueous solutions at ambient temperature and pressure. J. Am. Chem. Soc. 109, 5022–5023 (1987).
Li, J. et al. Constraining CO coverage on copper promotes high-efficiency ethylene electroproduction. Nat. Catal. 2, 1124–1131 (2019).
Wang, L. et al. Electrochemical carbon monoxide reduction on polycrystalline copper: effects of potential, pressure, and pH on selectivity toward multicarbon and oxygenated products. ACS Catal. 8, 7445–7454 (2018).
Jouny, M., Luc, W. & Jiao, F. High-rate electroreduction of carbon monoxide to multi-carbon products. Nat. Catal. 1, 748–755 (2018).
Verdaguer-Casadevall, A. et al. Probing the active surface sites for CO reduction on oxide-derived copper electrocatalysts. J. Am. Chem. Soc. 137, 9808–9811 (2015).
Li, C. W., Ciston, J. & Kanan, M. W. Electroreduction of carbon monoxide to liquid fuel on oxide-derived nanocrystalline copper. Nature 508, 504–507 (2014).
Rabinowitz, J. A., Ripatti, D. S., Mariano, R. G. & Kanan, M. W. Improving the energy efficiency of CO electrolysis by controlling Cu domain size in gas diffusion electrodes. ACS Energy Lett., 4098–4105 (2022).
Ma, M. et al. Insights into the carbon balance for CO2 electroreduction on Cu using gas diffusion electrode reactor designs. Energy Environ. Sci. 13, 977–985 (2020).
Eriksson, B. et al. Mitigation of carbon crossover in CO2 electrolysis by use of bipolar membranes. J. Electrochem. Soc. 169, 034508 (2022).
Wang, X. et al. Mechanistic reaction pathways of enhanced ethylene yields during electroreduction of CO2-CO co-feeds on Cu and Cu-tandem electrocatalysts. Nat. Nanotechnol. 14, 1063–1070 (2019).
Siahrostami, S., Bjorketun, M. E., Strasser, P., Greeley, J. & Rossmeisl, J. Tandem cathode for proton exchange membrane fuel cells. PCCP 15, 9326–9334 (2013).
Lin, Y.-R. et al. Vapor-fed electrolyzers for carbon dioxide reduction using tandem electrocatalysts: cuprous oxide coupled with nickel-coordinated nitrogen-doped carbon. Adv. Funct. Mater. 32, 2113252 (2022).
Liu, Y., Qiu, H., Li, J., Guo, L. & Ager, J. W. Tandem electrocatalytic CO2 reduction with efficient intermediate conversion over pyramid-textured Cu–Ag catalysts. ACS Appl. Mater. Interfaces 13, 40513–40521 (2021).
Gurudayal et al. Sequential cascade electrocatalytic conversion of carbon dioxide to C–C coupled products. ACS Appl. Energy Mater. 2, 4551–4559 (2019).
Lum, Y. & Ager, J. W. Sequential catalysis controls selectivity in electrochemical CO2 reduction on Cu. Energy Environ. Sci. 11, 2935–2944 (2018).
Zhang, J. et al. Tandem effect of Ag@C@Cu catalysts enhances ethanol selectivity for electrochemical CO2 reduction in flow reactors. Cell Rep. Phys. Sci. 3, 100949 (2022).
Akter, T., Pan, H. & Barile, C. J. Tandem electrocatalytic CO2 reduction inside a membrane with enhanced selectivity for Ethylene. J. Phys. Chem. C. 126, 10045–10052 (2022).
Ozden, A. et al. Cascade CO2 electroreduction enables efficient carbonate-free production of ethylene. Joule 5, 706–719 (2021).
Zhang, T., Li, Z., Zhang, J. & Wu, J. Enhance CO2-to-C2+ products yield through spatial management of CO transport in Cu/ZnO tandem electrodes. J. Catal. 387, 163–169 (2020).
She, X. et al. Tandem electrodes for carbon dioxide reduction into C2+ products at simultaneously high production efficiency and rate. Cell Rep. Phys. Sci. 1, 100051 (2020).
Han, H. et al. Selective electrochemical CO2 conversion to multicarbon alcohols on highly efficient N-doped porous carbon-supported Cu catalysts. Green. Chem. 22, 71–84 (2020).
Chen, C. et al. Cu-Ag tandem catalysts for high-rate CO2 electrolysis toward multicarbons. Joule 4, 1688–1699 (2020).
Gao, J. et al. Selective C–C coupling in carbon dioxide electroreduction via efficient spillover of intermediates as supported by Operando Raman Spectroscopy. J. Am. Chem. Soc. 141, 18704–18714 (2019).
Hoang, T. T. H. et al. Nanoporous Copper–Silver alloys by additive-controlled electrodeposition for the selective electroreduction of CO2 to Ethylene and Ethanol. J. Am. Chem. Soc. 140, 5791–5797 (2018).
Romero Cuellar, N. S. et al. Two-step electrochemical reduction of CO2 towards multi-carbon products at high current densities. J. CO2 Util. 36, 263–275 (2020).
Wang, M., Loiudice, A., Okatenko, V., Sharp, I. D. & Buonsanti, R. The spatial distribution of cobalt phthalocyanine and copper nanocubes controls the selectivity towards C2 products in tandem electrocatalytic CO2 reduction. Chem. Sci. 14, 1097–1104 (2023).
Ma, Y. et al. Confined growth of Silver–Copper Janus nanostructures with {100} facets for highly selective tandem electrocatalytic carbon dioxide reduction. Adv. Mater. 34, 2110607 (2022).
Simon, G. H., Kley, C. S. & Roldan Cuenya, B. Potential-dependent morphology of copper catalysts during CO2 electroreduction revealed by in situ atomic force microscopy. Angew. Chem. Int. Ed. 60, 2561–2568 (2021).
Arán-Ais, R. M. et al. Imaging electrochemically synthesized Cu2O cubes and their morphological evolution under conditions relevant to CO2 electroreduction. Nat. Commun. 11, 3489 (2020).
Vavra, J., Shen, T.-H., Stoian, D., Tileli, V. & Buonsanti, R. Real-time monitoring reveals dissolution/redeposition mechanism in copper nanocatalysts during the initial stages of the CO2 reduction reaction. Angew. Chem. Int. Ed. 60, 1347–1354 (2021).
Ma, S. et al. Electroreduction of carbon dioxide to hydrocarbons using bimetallic Cu–Pd catalysts with different mixing patterns. J. Am. Chem. Soc. 139, 47–50 (2017).
Lum, Y. & Ager, J. W. Evidence for product-specific active sites on oxide-derived Cu catalysts for electrochemical CO2 reduction. Nat. Catal. 2, 86–93 (2019).
Brückner, S. et al. Design and diagnosis of high-performance CO2-to-CO electrolyzer cells. ChemRxiv (2023). https://doi.org/10.26434/chemrxiv-2023-z6v6m.
Zhan, C. et al. Revealing the CO coverage-driven C–C coupling mechanism for electrochemical CO2 reduction on Cu2O nanocubes via Operando Raman Spectroscopy. ACS Catal. 11, 7694–7701 (2021).
Schreier, M., Yoon, Y., Jackson, M. N. & Surendranath, Y. Competition between H and CO for active sites governs copper-mediated electrosynthesis of hydrocarbon fuels. Angew. Chem. Int. Ed. 57, 10221–10225 (2018).
Huang, Y., Handoko, A. D., Hirunsit, P. & Yeo, B. S. Electrochemical reduction of CO2 using copper single-crystal surfaces: effects of CO* coverage on the selective formation of ethylene. ACS Catal. 7, 1749–1756 (2017).
Acknowledgements
The research leading to these results has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement no. 851441, SELECTCO2 (W.J., S.B. and P.S.). The research leading to these results has received funding from the European Union’s Horizon 2020 research and innovation program under grant agreement no. 101006701, ECOFUEL (T.M., M.F. and P.S.). The work leading to this publication was supported by the PRIME program of the German Academic Exchange Service (DAAD) with funds from the German Federal Ministry of Education and Research (BMBF) T.M.
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
T.M. and M.F. designed the electrochemical experiments, analyzed the results, and wrote the manuscript. S.B. and W.J. set up and conducted electrochemical experiments using “cell−1”. P.S. co-designed and co-wrote the manuscript. All authors participated in the discussion and evaluation of results.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Fengwang Li and the other anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Möller, T., Filippi, M., Brückner, S. et al. A CO2 electrolyzer tandem cell system for CO2-CO co-feed valorization in a Ni-N-C/Cu-catalyzed reaction cascade. Nat Commun 14, 5680 (2023). https://doi.org/10.1038/s41467-023-41278-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-41278-7
This article is cited by
-
Steering electroreduction of carbon dioxide to valuable C3+ products
Science China Materials (2024)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
