
Abstract
The lack of available protons severely lowers the activity of alkaline hydrogen evolution reaction process than that in acids, which can be efficiently accelerated by tuning the coverage and chemical environment of protons on catalyst surface. However, the cycling of active sites by proton transfer is largely dependent on the utilization of noble metal catalysts because of the appealing electronic interaction between noble metal atoms and protons. Herein, an all-non-noble W/WO2 metallic heterostructure serving as an efficient solid-acid catalyst exhibits remarkable hydrogen evolution reaction performance with an ultra-low overpotential of −35 mV at −10 mA/cm2 and a small Tafel slope (−34 mV/dec), as well as long-term durability of hydrogen production (>50 h) at current densities of −10 and −50 mA/cm2 in alkaline electrolyte. Multiple in situ and ex situ spectroscopy characterizations combining with first-principle density functional theory calculations discover that a dynamic proton-concentrated surface can be constructed on W/WO2 solid-acid catalyst under ultra-low overpotentials, which enables W/WO2 catalyzing alkaline hydrogen production to follow a kinetically fast Volmer-Tafel pathway with two neighboring protons recombining into a hydrogen molecule. Our strategy of solid-acid catalyst and utilization of multiple spectroscopy characterizations may provide an interesting route for designing advanced all-non-noble catalytic system towards boosting hydrogen evolution reaction performance in alkaline electrolyte.
Similar content being viewed by others

Introduction
As a clean and sustainable energy carrier, hydrogen is one of the most promising alternatives to traditional fossil fuels for addressing global energy crisis and environmental pollution1. One of the most economical and effective strategy of hydrogen production is electrocatalytic hydrogen evolution reaction (HER), which is driven by electricity from sustainable energies (i.e., solar and wind) without any emission of carbon dioxide, satisfying the mission of global carbon neutrality2,3. Alkaline HER process can avoid the acidic corrosion and dissolution issues of catalysts, and achieve high-purity hydrogen gas (>99.7%), showing an attractive and extensive application4,5. HER electrocatalysis usually demands the usage of noble-metal-based catalysts to lower the applied overpotentials, but the high cost and low reserve have restricted their widespread utilization. Moreover, compared to the direct proton-coupled electron reaction (\({{{{{{\rm{2H}}}}}}}^{\ast }+{{{{{{\rm{2e}}}}}}}^{-}\to {{{{{{\rm{H}}}}}}}_{2}+\ast\), where ∗represents the active site) in acidic electrolyte, it is worth noting that an additional water dissociation step (\({{{{{{\rm{H}}}}}}}_{2}{{{{{\rm{O}}}}}}+\ast+{{{{{{\rm{e}}}}}}}^{-}\to {{{{{{\rm{H}}}}}}}^{\ast }+{{{{{{\rm{OH}}}}}}}^{-}\), also known as Volmer step) is required to produce available protons before hydrogen generation in alkaline HER process6, while noble platinum catalyst is kinetically inefficient for the cleavage of H-OH bonds7, usually leading to two or three orders of magnitude lower activity of alkaline HER process than that in acids8. Therefore, the exploration of cost-effective catalysts towards efficiently breaking H-OH bonds for proton generation is valuable and significant in alkaline HER process.
Transition metal oxides have long been advocated as highly efficient HER catalysts due to their flexible chemical and electronic structures with fascinating physical and chemical properties9. In particular, tungsten (W), remarkable for its complex electronic structure featuring open d and f shells10, can form rich oxidation states ranging from +6 to 0 (WO3-x, 0 ≤ x < 3)11. Tungsten oxides with substoichiometric phases have been demonstrated as potential alternatives to commercial platinum catalyst in acidic HER process12,13,14, benefiting from these abundant oxygen vacancies that can afford substoichiometric WO3-x catalysts with favorable hydrogen adsorption energies and improved conductivity15. However, their alkaline HER activities have been rarely explored, because tungsten oxides featured with acidic-oxide property will be gradually dissolved in alkaline electrolyte16,17,18. Although first-row (3d) transition metals (Fe, Co, Ni) as additives have been proven to significantly improve the alkaline HER performance of tungsten oxides19, the introduction of foreign atoms probably obscures the catalytic mechanism, since the identification of doping state of low-atomic-number 3d metals among tungsten arrays is rather challenging20. Therefore, it is still unclear that whether tungsten-oxide-dominant materials can be employed as stable catalysts for highly-efficient and stable alkaline HER process, when the local chemical environment of W atoms is altered.
To this end, we have focused on a special form of W/WO2 metallic heterostructure. Unlike previously reported acid-like catalyst surface constructed by metal-oxide matrix supported noble-metals18,21, where metal oxides are expected to provide an acid-like catalyst surface under neutral or alkaline conditions, while noble metals are essentially required as active sites due to the energy-favorable interactions of noble-metal and produced protons on catalyst surface22. The W/WO2 metallic heterostructure can spontaneously construct a dynamic proton-concentrated surface only by tungsten-atom itself, thus enabling kinetically fast HER process in high-pH conditions, which can be understood in the viewpoint of solid-acid catalyst: (i) featured by unusual metallic and acidic-oxide properties, the WO2 component can serve as highly-active Lewis acid sites for the adsorption and cleavage of H2O molecules23,24 (proton generation), facilitating the formation of hydrogen tungsten bronze (HxWOy) intermediates25 (proton storage); (ii) the in-situ generated HxWOy intermediates are considered as Brønsted acid sites with reversible adsorption/desorption behaviors of protons26,27; (iii) considering the relatively sluggish hydrogen desorption kinetics of HxWOy intermediates, the introduction of zero-valence W (W0) sites can further accelerate the deprotonation kinetics of Brønsted acids for the cycling of active sites due to the optimized electronic interactions between W0 atoms and protons at the W/WO2 interface28 (proton donation and regeneration of active sites). In addition, compared to traditional semiconducting tungsten oxides, the metallic feature of WO2 component can also afford the tungsten-oxide matrix with improved alkaline leaching resistance (WO2 + OH− → WO42− + H2O), because the aggressive hydroxyl species from produced OH− intermediates and electrolyte can be rapidly repulsed from the electron-rich catalyst surface at the cathode29,30.
Herein, we report a feasible pyrolysis-reduction strategy to synthesize W/WO2 metallic heterostructure on Ni foam. Owing to the solid-acid sites with strong abilities of proton generation and reversible behaviors of hydrogen adsorption/desorption, a dynamic proton-concentrated surface is constructed on W/WO2 solid-acid catalyst, which enables the all-non-noble W/WO2 catalyst to show superior HER activity with an ultra-low overpotential of −35 mV at −10 mA/cm2 and a small Tafel slope (−34 mV/dec) in alkaline electrolyte. Moreover, the solid-acid catalyst is exceptionally stable in alkaline electrolyte, showing no significant activity degradation for hydrogen production at −10 and −50 mA/cm2 over 50 h. To the best of our knowledge, this is the first time that a tungsten-oxide-dominant material serves as a solid-acid catalyst with a remarkable HER performance in alkaline electrolyte, outperforming all tungsten/molybdenum oxides and most 3d-metal oxides reported to date.
Results
Morphological characterization of W/WO2 metallic heterostructure
W/WO2 metallic heterostructure was synthesized by a pyrolysis-reduction method using W18O49 nanowires (NWs) as parent materials (Supplementary Fig. 1). In brief, W18O49 NWs were homogeneously coated on the pre-treated Ni foam (Supplementary Fig. 2), followed by stirring in the mixture solution of polyethylene oxide-co-polypropylene oxide-co-polyethylene oxide (P123, carbon source), 2-amino-2-hydroxymethyl-propane-1,3-dio (Tris) and dopamine (DA, carbon source) for 24 h to coat organic carbon source over W18O49 materials. Afterwards, the desired W/WO2 metallic heterostructure was obtained by pyrolyzing the free-standing organic-tungsten precursor at 700 °C under a mixture of Ar/H2 (v/v = 8:1) atmosphere for 2 h (Supplementary Fig. 2), where the reduction reactions can be simplified as the equations (\({{{{{\rm{C}}}}}}+{{{{{{\rm{W}}}}}}}_{18}{{{{{{\rm{O}}}}}}}_{49}\to {{{{{\rm{W}}}}}}+{{{{{{\rm{WO}}}}}}}_{2}+{{{{{{\rm{CO}}}}}}}_{2}\uparrow\), \({{{{{{\rm{H}}}}}}}_{2}+{{{{{{\rm{W}}}}}}}_{18}{{{{{{\rm{O}}}}}}}_{49}\to {{{{{\rm{W}}}}}}+{{{{{{\rm{WO}}}}}}}_{2}+{{{{{{\rm{H}}}}}}}_{2}{{{{{\rm{O}}}}}}\uparrow\)). In the pyrolysis process, carbon can cause the reduction of parent high-valence W18O49 parent materials for the generation of desired W and WO2 products. In the following alkaline HER process, the produced graphite carbon layers not only improve the electrocatalytic charge transfer, but also alleviate the alkaline-leaching rate of inner W/WO2 materials. Meanwhile, the possibly formed tungsten-carbide by-products (e.g., WC, W2C) are excluded by X-ray photoelectron spectroscopy (XPS) and Raman characterizations (Supplementary Fig. 3). For comparison, WO2 nanorods and W NPs were also synthesized under different conditions, respectively (seen in the method section).
The crystal structure of W/WO2 heterostructure was verified by X-ray diffraction (XRD) pattern. As shown in Supplementary Fig. 4, besides of the predominant characteristics of underlying Ni foam, the XRD pattern of WO2 sample reveals a set of characteristic signals at 25.8°, 37.1°, 52.9°, and 59.7°, which are indexed to the (011), (−211), (220), and (031) facets of monoclinic structured WO2 phase (JCPDS No. 32–1393), respectively, while metallic W NPs present a simple cubic phase with lattice parameters of a = b = c = 3.16 Å (JCPDS no. 89–2767). W/WO2 metallic heterostructure shows hybrid signals containing WO2 and W phases, indicating the coexistence of two types of tungsten-based phases. The overall morphology of W/WO2 product was characterized by scanning electron microscopy (SEM). From low-magnification SEM image, the Ni skeletons are closely coated by W/WO2 products (Supplementary Fig. 5a, b), and the high-magnification SEM image and corresponding energy dispersive X-ray spectra (EDS) show one-dimensional (1D) nanorod structure of the as-obtained W/WO2 materials with radial lengths over 500 nm (Supplementary Fig. 5c, d), indicating the 1D nanostructure of tungsten-oxide precursor is still retained after high-temperature pyrolysis treatment. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) can allow us to observe the hybrid structure of W/WO2 metallic heterostructure at atomic level. As shown in Fig. 1a, high density of nanoparticles with sizes less than 10 nm are dispersed on tungsten-based nanorods, and two sets of lattice fringes with spacing values of 0.22 and 0.34 nm can be seen from the high-magnification STEM image (Fig. 1b). Combining the analysis of corresponding fast Fourier transform (FFT) pattern (inset in Fig. 1b), we can conclude that the observed lattice fringes should be attributed to the (110) and (011) facet of supported W NPs and underlying WO2 matrix, respectively. Meanwhile, the homogeneous distribution of C, O, and W elements over the entire W/WO2 metallic heterostructure is visualized by the STEM-EDS mapping images (Fig. 1c). In addition, the morphology of WO2 nanorods and W NPs were also examined. Figure 1d shows a typical carbon-encapsulated WO2 nanorod, and the corresponding high-magnification STEM image and FFT pattern evidence the single-crystalline phase of WO2 species (Fig. 1f). In contrast, numerous W NPs with lateral dimension below 5 nm are immobilized on carbon matrix (Supplementary Fig. 6, Fig. 1f, g), indicating the 1D W18O49 precursors have been disintegrated into ultrasmall W NPs under high-temperature reducing conditions. Therefore, based on above crystal structure and morphological characterizations, three types of tungsten-based catalysts (W, WO2, and W/WO2) are indeed obtained (Fig. 1h).

Low-magnification STEM images of a W/WO2, d WO2, and f W NPs. High-resolution STEM images of b W/WO2, e WO2, and g W NPs, with the corresponding FFT patterns in the insets. c STEM-EDS mapping of W/WO2 showing the homogeneous distribution of C (blue), O (red) and W (green) elements. h Structural illustrations of W/WO2 (blue and deep blue), WO2 (blue) and W NPs (deep blue) embedded on carbon matrix.
Structural characterizations of W/WO2 metallic heterostructure
X-ray photoelectron spectroscopy (XPS) and X-ray adsorption spectroscopy (XAS) were performed to gain insight into the subtle change of the local chemical and electronic structures for the above three tungsten-based materials in. As shown in Fig. 2a, W and WO2 samples exhibit W0 and W4+ signals at low (31.4 eV for W0 4f7/2 and 33.5 eV for W0 4f5/2) and high (32.7 eV for W4+ 4f7/2 and 34.8 eV for W4+ 4f5/2) binding energies after deconvolution, respectively. In addition to the intrinsically characteristic peaks, the inevitable surface high-valence oxidation species (35.5 eV for W6+ 4f7/2 and 37.6 eV for W6+ 4f5/2) are also observed on W and WO2 samples31. As expected, W/WO2 sample contains features of both W and WO2 species at the same binding energies after deconvolution, thus demonstrating the coexistence of W and WO2 components in W/WO2. The normalized X-ray absorption near-edge structure (XANES) profiles of W L3-edge reveal that the white line intensity of W/WO2 sample is much higher than that of metallic W NPs, but slightly lower relative to that of WO2 counterpart (Fig. 2b), indicating the average oxidation state of tungsten atoms in W/WO2 is between 0 and 4 + . Also, the whole W L3-edge XANES profile of W/WO2 extremely resembles that of WO2 rather than metallic W, further suggesting that W atoms of WO2 and W/WO2 materials may have similar chemical state32. Correspondingly, from the viewpoint of coordinated oxygen atoms, the O K-edge near edge X-ray absorption fine structure (NEXAFS) spectroscopy can give additional information on the chemical structures of W atoms in above-mentioned tungsten species (Fig. 2c). One can see that both WO2 and W/WO2 samples exhibit split peaks located at energies ranging from 530 to 535 eV, where the peak at low energy (530.7 eV) originates from the overlapping band between W5d and O2p orbitals, corresponding to the W-O bonds, while the superoxide (O2-) species neighboring to surface oxygen vacancies should be responsible for another one at high energy (532.2 eV)33. Comparing to the O K-edge NEXAFS spectroscopy of WO2 counterpart, W/WO2 metallic heterostructure exhibits decreased intensity of W-O bonds but increased signal of O2- species, indicating the formation of rich oxygen vacancies. Meanwhile, such an enrichment of oxygen vacancies in W/WO2 metallic heterostructure is also resolved by electron spin resonance (ESR) spectroscopy (Supplementary Fig. 7), which exhibits a strong and symmetrical ESR signal at g = 2.002, manifesting rich unpaired electrons trapped on vacancies around W centers34. Further, the subtle difference of local coordination structure in W, WO2, and W/WO2 samples were discriminated by the Fourier-transformed extended X-ray absorption fine structure (FT-EXAFS) spectra (R-space) (Fig. 2d). As can be seen, W sample exhibits a distinct peak at 2.6 Å, corresponding to the W-W scattering path, while a predominant peak at approximately 1.6 Å is presented on WO2 sample, which can be assigned to the W-O coordination. Meanwhile, we also observe a weak but still visible W-W coordination at 2.4 Å on WO2 sample, which is almost absent in stoichiometric WO3 reference (Supplementary Fig. 8), demonstrating the metallic property of WO2 phase. As expected, the R-space profile of W/WO2 heterostructure exhibits the combined features of W and WO2 counterparts. However, comparing to WO2 sample, W/WO2 sample exhibits a relatively weak W-O coordination but significantly increased intensity of W-W scattering path, implying the introduction of W NPs probably breaks the homogeneity of local W-O coordination structure, and further enhance the metallic property of WO2 matrix. Wavelet transform (WT)-EXAFS with high-resolution in both k and R spaces can be used to visually examine the atomic configuration of W atoms. In line with the FT-EXAFS analysis, the WT-EXAFS contour plot of W/WO2 sample shows an increased intensity of W-W scattering path centered at about k = 14.3 Å−1 relative to that of WO2 sample, further suggesting the increased metallic feature. Moreover, W/WO2 sample exhibits intensity maximum at k = 5.6 Å−1 for W-O coordination, which is definitely different from WO2 sample (k = 6.3 Å−1), suggesting the local W-O bonding configuration of WO2 matrix has been altered due to the introduction of W NPs. Finally, the electronic structures of three tungsten-based materials were also examined by the electron localization function (ELF) calculations. Unlike the uniform electronic structures of W and WO2 counterparts, W/WO2 model exhibits distinctly modified electronic structures at the interface, where the coexistence of electron localized (O atoms in WO2) and delocalized (W0 atoms in W metal) states can afford W/WO2 heterostructure with synergistic effort for kinetically fast water dissociation and hydrogen desorption steps in alkaline HER process.

a W 4 f core-level XPS spectra. b W L3-edge XANES spectra. c O K-edge NEXAFS spectra. d R space profiles of W (light blue), WO2 (blue), and W/WO2 (red) extracted from the corresponding W L3-edge EXAFS spectra. e WT-EXAFS plots. f The calculated ELF images of W, WO2, and W/WO2. Green to red indicates the gradually increased electron localization.
Evaluation of alkaline HER electrocatalysis using W/WO2 catalyst
The HER performance of free-standing W/WO2 solid-acid catalyst was evaluated in 1.0 M KOH electrolyte using a typical three-electrode setup, where bare carbon coated Ni foam (C@Ni), W NPs, WO2, and PtRu/C catalysts were also measured under the same conditions for comparison. As shown in Fig. 3a, bare C@Ni sample exhibits a negligible alkaline HER activity, although W and WO2 catalysts exhibit enhanced alkaline HER activities with decreased overpotentials at −10 mA/cm2 (W: ƞ10 = −183 mV; WO2: ƞ10 = −106 mV), their activities are still inferior to other W-/Mo-based catalysts and conventional 3d-metal (Fe, Co, Ni) oxides reported to date (Fig. 3c and Supplementary Table 1). Encouragingly, the as-obtained W/WO2 solid-acid catalyst displays a superior alkaline HER activity with value of ƞ10 as low as −35 mV, excelling all previously reported tungsten oxide catalysts and most state-of-the-art non-noble oxide catalysts (Fig. 3c and Supplementary Table 1), and even being comparable to commercial PtRu/C (−11 mV). In addition, the alkaline HER activity of the physical mixture of W and WO2 (W + WO2) was also examined under the same conditions, where the values of ƞ10 and Tafel slope are determined to be −153 mV and −135 mV/dec, respectively, and the markedly improved Tafel slope may be attributed to the synergistic effect of WO2 and W components for water dissociation and hydrogen desorption steps in alkaline HER process. However, the alkaline HER activity of W + WO2 is still inferior to W/WO2 catalyst, suggesting the W/WO2 interfaces constructed by chemical bonds can improve the alkaline HER activity intrinsically. The turnover frequency (TOF) plot of W/WO2 solid-acid catalyst has been calculated at overpotentials of 0 ~ 0.4 V (Fig. 3b, calculated detail seen in method section), where W/WO2 solid-acid catalyst exhibits a high TOF value of 0.013 s−1 at overpotential of −100 mV, nearly 4.6- and 15.1-fold higher than WO2 and W counterparts, demonstrating the high intrinsic activity of W/WO2 solid-acid catalyst. Further, the accelerated reaction kinetics of alkaline HER process on W/WO2 solid-acid catalyst was revealed by Tafel slope. W/WO2 solid-acid catalyst exhibits a sharply decreased Tafel slope (−34 mV/dec) relative to those of W (−170 mV/dec) and WO2 (−190 mV/dec) counterparts (Fig. 3d), even being comparable to the value of commercial PtRu/C catalyst (−27 mV/dec). This result indicates the high energy barrier of additional water dissociation has been substantially weakened, and the concomitant hydrogen production follows the fastest kinetics of Tafel pathway due to the coverage of rich protons on W/WO2 catalyst surface. Moreover, the HER performance of W, WO2, and W/WO2 powders were also examined using the rotating disk electrode technique in 1 M KOH electrolyte (Supplementary Fig. 9). As can be seen, W/WO2 catalyst still exhibits a remarkable alkaline HER activity with a low overpotential (−60 mV) at −10 mA/cm2 and a small Tafel slope (−54 mV/dec), which are more excellent than those of W and WO2 counterparts, and still excelling most previously reported metal oxides (Fig. 3c).

a polarization (LSV) curves of C@Ni (wine red), W (light blue), WO2 (deep blue), W + WO2 (green), W/WO2 (red) and commercial PtRu@C (black) catalysts. b TOF plots of W, WO2, and W/WO2 catalysts at overpotentials of 0 ~ 0.4 V. c Comparison of overpotentials (10 mA/cm2) and Tafel slopes of W/WO2 solid-acid catalyst and previously reported excellent transition-metal-oxide based HER catalysts in alkaline electrolyte. d Tafel plots of C@Ni (wine red), W (light blue), WO2 (deep blue), W + WO2 (green), W/WO2 (red) and commercial PtRu@C (black) catalysts. e The determination of CdI by plotting the current density variation (Δj) against the scan rates (10–100 mV s−1). f Nyquist plots (the inset shows the equivalent circuit for the simulation) and g the corresponding Bode phase plots of W/WO2 solid-acid catalyst with the increase of applied overpotentials at 0 (black), −10 (red), −20 (deep blue), −30 (green), −40 (purple), −50 (light purple), and −60 (orange) mV. h LSV curves of W/WO2 solid-acid catalyst before (deep blue) and after (red) 1000 CVs. i Chronopotentiometry measurements of W/WO2 solid-acid catalyst at current densities of −10 (black), −50 (blue), and −200 (red) mA/cm2, and the red dashed circle indicates the extraction of electrolyte for ICP detection.
In order to better understand the proton coverage on W/WO2 catalyst surface, double-layer capacitance (CdI) and electrochemical impedance spectroscopy (EIS) of W/WO2 catalyst were examined, with W and WO2 counterparts as references. The larger value of CdI usually implies the higher electrochemical surface area (ECSA) for alkaline HER process. The CdI value of catalyst can be extracted from the cyclic voltammetry (CV) curves with different scan rates at non-Faradaic voltage windows (Supplementary Fig. 10). The CdI value of W/WO2 catalyst is determined to be up to 87.5 mF/cm2, nearly 3.7- and 2.1-fold enhancement than those of W and WO2 counterparts, respectively (Fig. 3e), suggesting the construction of W/WO2 metallic heterostructure can provide rich active sites for the adsorption of H2O reactants and reaction intermediates in alkaline HER process35. The electrochemical impedance spectroscopy (EIS) measurements were then performed to track the accumulation of protons deriving from the activation of H2O reactants on W/WO2 catalyst surface36. All Nyquist plots of W/WO2 samples were simulated by a double-parallel equivalent circuit model in accordance with previous reports36,37, where R1 represents the uncompensated solution resistance (Rs), the first parallel components (constant phase element (CPE) and R2) indicate the charge transfer resistance caused by the adsorption and activation of water molecules at low frequencies38,39, and the second parallel ones of R2 and Cφ are attributed to the hydrogen adsorption resistance and pseudo-capacitance at high frequencies, respectively (Fig. 3f and Supplementary Table 2). As expected, all electrochemically treated W/WO2 samples exhibit the similar R1 value (~4.0 Ω), and the small values of R2 for all W/WO2 catalysts suggest the fast charge transfer kinetics between catalyst surface and H2O molecules. In particular, R2 decreases to 4.4 Ω sharply with a negligible water diffusion resistance at applied overpotential of −20 mV, indicating the adsorption and activation of water molecules on the W/WO2 catalyst surface can be achieved under low overpotentials. Further, we also notice that R3 and Cφ are largely overpotential-dependent, where W/WO2 catalysts exhibit significantly decreased R3 with increased Cφ when increasing the applied overpotentials, in particular, the value of R3 can be as low as approximately 1.8 Ω, while Cφ is up to 0.017 F at an overpotential of −30 mV, suggesting the hydrogen adsorption resistance is very small, and the pseudo-capacitance of proton coverage is very large on W/WO2 catalyst surface under low overpotentials. In contrast, both W and WO2 counterparts show a sluggish alkaline HER kinetics with large values of Rct and high negative phase angles on the Nyquist and Bode plots, respectively (Supplementary Fig. 11), suggesting a sluggish HER kinetics in an alkaline environment.
Besides of high electrocatalytic activity, catalyst durability is another significant concern for practical application. In order to evaluate the durability of the W/WO2 solid-acid catalyst, the as-obtained catalyst was firstly performed by 1000 cyclic voltammograms (CVs) in the voltage window from 0 to −0.2 V (versus RHE) with a scan rate of 100 mV/s. As can be seen, the HER polarization curve of W/WO2 almost overlaps with the original one after 1000 potential cycles (Fig. 3h), suggesting the stable proton-coupled electron redox activity of W/WO2 solid-acid catalyst in alkaline electrolyte. A prolonged chronopotentiometry measurement was further applied to evaluate the long-term durability of the catalysts at current densities of −10, −50 mA/cm2. It can be seen that the superior alkaline HER activity is well retained on W/WO2 solid-acid catalyst after continuous hydrogen production for more than 50 h. The relatively good stability of W/WO2 solid-acid catalyst in alkaline electrolyte is also confirmed by the inductively coupled plasma-optical emission spectroscopy (ICP-OES). After long-term hydrogen production, the initial electrolyte, used electrolyte, and refreshed electrolyte (indicated by red dashed circle) shows no significant increase in the concentrations of dissolved W species with values of 7.5 × 10−6, 9.8 × 10−6, and 6.3 × 10−7 mol/L, respectively. In particular, the rather low concentration (6.3 × 10−7 mol/L) of dissolved W species in the refreshed electrolyte directly suggests the structural robustness of W/WO2 catalyst after long-term hydrogen production in alkaline electrolyte. In addition, we also evaluate the stability of W/WO2 catalyzing alkaline HER process at a current density of −200 mA/cm2 (the lower limit for industrial water electrolysis). No significant activity loss can be observed on W/WO2 catalysts after long-term hydrogen production, suggesting the good catalytic stability at industrial current density. For the characterization of used catalyst, a rough catalyst surface with rich defects is observed on the used W/WO2 solid-acid catalyst (Supplementary Fig. 12), suggesting the insertion of hydrogen may alter the local chemical structure of W/WO2 solid-acid catalyst. Meanwhile, the enhanced signal of oxygen vacancies detected by ESR and O K-edge NEXAFS demonstrates that the proton-coupled electron reaction of HER process might have caused a slight reduction of underlying WO2 matrix (Supplementary Fig. 13). Therefore, based on the comprehensive evaluations of alkaline HER activity and stability on W/WO2 materials, the significantly improved structural robustness of our well designed W/WO2 composites in high-pH solutions can be attributed to the following two reasons: (i) W/WO2 heterostructures have intrinsically good oxidation resistance with co-existence of metal and oxide features and the protection of surface carbon layers19,29,40,41, meanwhile, the strong chemical and electronic interactions of W and WO2 components within W/WO2 may further improve the structural robustness in alkaline solutions42,43; (ii) unlike the naturally alkaline leaching under circuit potential condition, the negative potentials at cathode can provide rich electrons to avoid the oxidation and dissolution of low-valence tungsten species during alkaline HER process.
Multiple spectroscopy characterizations discovering electrocatalytic mechanism
Comprehensive characterizations were performed to gain insight into the origin of significantly enhanced alkaline HER activity on W/WO2 metallic heterostructure. Firstly, the deuterated-effect-induced inferior alkaline HER activity suggests that the fast HER kinetics of water dissociation has been lowered on W/WO2 catalyst surface because of the larger zero-point energy of OD* relative to the OH* intermediates (difference of ~1400 cal) (Supplementary Fig. 14a)44. Besides, we also observe that the deuterated effect has more negative influence on the Tafel slope of W/WO2 catalyst relative to W and WO2 counterparts (Supplementary Fig. 14b), because the sluggish kinetics of water dissociation hampers the proton coverage on W/WO2 catalyst surface, which severely lowers subsequent hydrogen desorption kinetics. Secondly, near ambient pressure X-ray photoelectron spectroscopy (NAP-XPS) measurement was performed to investigate the adsorption and cleavage of H2O molecules on W, WO2, and W/WO2 catalyst surface under a water pressure of 0.1 mbar (Fig. 4a), with the core-level XPS spectroscopy recorded under an ultrahigh vacuum (UHV) condition as a reference8. For the referenced O 1 s XPS profile of W/WO2 sample under an UHV condition. It exhibits two peaks at 530.3 and 531.3 eV after deconvolution (Fig. 4b), corresponding to the lattice oxygen (W-O) species and oxygen vacancies (Ov), respectively45. After introducing 0.1 mbar H2O molecules (Supplementary Fig. 15), W/WO2 metallic heterostructure exhibits a vanished Ov signal but significantly enhanced concentrations of W-OH (532.3 eV) and H2O (533.3 eV) species at higher binding energies (Supplementary Table 3), indicating Ov have served as the adsorption sites of H2O molecules, and the cleavage of H-OH bonds is easily proceeded on W/WO2 catalyst surface. Also, such an accelerated water dissociation kinetics of W/WO2 catalyst has been confirmed by the obvious decrease of low-valence tungsten species in the corresponding W 4 f core-level XPS spectra (Fig. 4c). In contrast, W and WO2 counterparts display inferior activities of water dissociation kinetics (Supplementary Fig. 16 and Supplementary Table 3), in particular, only an extremely weak W-OH signal is observed on W catalyst surface after exposure in 0.1 mbar H2O atmosphere, because W0 (W) sites with more filled d orbitals have weaker electrostatic affinity to electron-rich H2O molecules than W4+ (WO2) atoms46.

a Schematic illustration of the in situ NAP-XPS measurement under 0.1 mbar H2O atmosphere. b O 1 s and c W 4 f XPS spectra of W/WO2 recorded under UHV and 0.1 mbar H2O conditions, respectively. d TOF-SIMS profiles of W/WO2 reference (black) and electrochemically treated sample (red), and the inset shows the enlarged signals ranging from 18.9 to 19.1, where the fluorine (F, m/z = 19.00) signal may be originated from the inevitable contamination of fluorine-containing sealing ring in the analysis chamber during Bi3+ cations impacting the sample, while the H3O+ species can be responsible for the higher signal at m/z = 19.02. e 2D distribution images of H3O+ species on W/WO2 only soaked in alkaline electrolyte and f W/WO2 after alkaline HER process. g REELS spectra, h 1H MAS NMR, and i Py-IR spectra of W/WO2 catalysts after electrochemical treatments by the gradually increased overpotentials, where soaked, 0, -10, -20, and -30 mV are represented by deep blue, pink, orange, light blue, and red colors, and the black color stands for commercial H2WO4 sample.
Finally, with the clear elucidation of proton production on W/WO2 catalyst surface in water dissociation step, the coverage and chemical environment of produced protons determine the activity of subsequent hydrogen generation on W/WO2 solid-acid catalyst surface36. Time-of-flight secondary ion mass spectroscopy (TOF-SIMS) is very sensitive to the examine the hydrogen species after alkaline HER process. In order to eliminate the interference of adsorbed H2O molecules and OH- anions (electrolyte), W/WO2 sample only soaked in KOH electrolyte was selected as a reference. We can observe that the electrochemically treated W/WO2 catalyst exhibits markedly increased signals of hydrogen-containing intermediates, i. e., OH, H2O, H3O+, in particular, the sharply increased H3O+ signal directly suggests the formation of proton-concentrated catalyst surface21 (Fig. 4d). In order to visually display the distribution of H3O+ on the electrochemically treated W/WO2 catalyst surface, two-dimensional (2D) image analysis of the soaked and electrochemically treated W/WO2 samples are also provided in Fig. 4e, f. Obviously, one could notice that the concentration of H3O+ species on the electrochemically treated W/WO2 catalyst is much higher than that of the only soaked counterpart, indicating the achievement of proton-concentrated catalyst surface on W/WO2 solid-acid catalyst. Multiple spectroscopy characterizations were further performed to analyze the property of the produced protons on the used W/WO2 catalyst surface treated by increased applied overpotentials (ƞ=0, −10, −20, −30 mV), with W/WO2 sample only soaked in KOH electrolyte and commercial tungstic acid (H2WO4) as the references. The reflection electron energy loss spectroscopy (REELS) can break the limitation of conventional XPS technique for the detection of H element. As shown in Fig. 4g, the REELS plot of W/WO2 soaked sample only exhibits a predominantly elastic peak, while an obvious H signal neighboring to elastic peak is observed on commercial H2WO4 sample47,48,49, indicating the hydrogen concentration of W/WO2 soaked sample is lower than the detecting limitation (molar ratio: ~20%) of REELS technique. As expected, all electrochemically treated W/WO2 catalysts exhibited gradually enhanced H signals when increasing the applied overpotentials (0 to −30 mV), indicating a proton-concentrated surface could be constructed on W/WO2 metallic heterostructure under an ultra-low overpotential, which is in good consistency with the analysis of EIS measurement. Note that the major contributor of detected H signal should be attributed to W/WO2 species rather than underlying carbon matrix, as evidenced by the REELS measurements of bare carbon materials after treatments under increased overpotentials (Supplementary Fig. 17). Solid-state 1H magic-angle-spinning nuclear magnetic resonance (1H MAS NMR) spectra exhibits two broad bands at approximately −1.3 and 3.4 ppm on soaked W/WO2 sample (Fig. 4h), in which the peak at lower chemical shift can be attributed to the mixture of terminated hydroxyl (W-OHT and/or C-OHT) species on WO2 and carbon matrix, while the higher one originates from the lattice hydroxyl (W-OHL) species in WO2 matrix. Similar to REELS characterization, all electrochemically treated W/WO2 samples exhibit significant increased signals of W-OHT and W-OHL species under increasing overpotentials, indicating the increased coverage of protons over W/WO2 catalyst surface. Meanwhile, we also notice that the proportion of W-OHT/W-OHL decreases with the increase of applied potentials, which may be attributed to the continuously increased coverage of produced protons. The increasing negative potentials can result in more water molecules being adsorbed on W/WO2 catalyst surface, and the water molecules rapidly react with electrons for the cleavage of H-OH bonds, then the produced H* intermediates insert into the lattice of tungsten oxides (W-OHL) for constructing proton-concentrated catalyst surface with electronic and chemical environments approaching to those of H2WO4 materials. While the terminated electron-rich hydroxyl species, such as the produced OH* intermediates and chemically/physically adsorbed OH- molecules (W-OHT) cannot follow the increasing tendency because of the enhanced Coulomb effect at cathode. In addition, 1H MAS NMR profiles collected at overpotentials of −10, −20, and −30 mV become extremely similar to that of commercial H2WO4 reference (Supplementary Fig. 18), demonstrating the chemical environment of protons on W/WO2 catalyst surface is approaching to commercial H2WO4 reference. All Py-IR spectroscopy of electrochemically treated samples exhibit typical adsorption peaks in the wavenumber range of 1400 ~ 1650 cm−1, where peaks at 1450, 1577, and 1613 cm−1 are ascribed to the chemisorption of pyridine at surface Lewis acid sites (L), while peaks at 1540 and 1638 cm−1 indicate the presence of Brønsted acid sites (B), and the signal at 1494 cm−1 contains contribution of Lewis acid and Brønsted acid sites (Fig. 4i and Supplementary Fig. 19). Impressively, the electrochemically treated W/WO2 catalysts also exhibited enhanced concentrations of Brønsted acid sites with the increase of applied overpotentials (Supplementary Table 4), indicating the produced protons of water dissociation mainly serve as Brønsted acids with reversible behaviors of hydrogen adsorption and desorption on W/WO2 solid-acid catalyst surface26,27.
DFT calculations
Such a significant enhancement of alkaline HER activity of W/WO2 solid-acid catalyst was also elucidated by first-principle density functional theory (DFT) calculations, with simulations of W and WO2 models as references (Fig. 5a, Supplementary Fig. 20). Specifically, more detailed front, side and top structures of W/WO2 interface were also calculated in Supplementary Fig. 21. WO2 (−0.33 eV) and W/WO2 (−0.32 eV) materials exhibit more negative adsorption energies of H2O molecules than that of W sample (−0.14 eV) (Fig. 5b), which suggests the WO2 and W/WO2 catalyst surfaces are beneficial for the adsorption and activation of H2O reactants50,51. As expected, W exhibits a sluggish kinetics of prior water dissociation step with energy barrier (ΔGH2O) up to 0.84 eV, while the activation energy barrier of H2O molecules can be sharply reduced to 0.06 eV on WO2 model (Fig. 5b), indicating oxygen-vacancy-rich WO2 is generally effective for the cleavage of H-OH bonds. Encouragingly, the energy barrier of water dissociation is further decreased to 0.02 eV on W/WO2 catalyst surface (Supplementary Table 5), excelling most previously reported noble metal alloy catalysts52,53. With an ultra-low energy barrier of water dissociation benefiting the formation of available protons on W/WO2 catalyst surface, the following proton detachment will determine the reaction rate of alkaline HER process, where hydrogen adsorption free energy (ΔGH) of H* intermediates is a good descriptor to evaluate the HER activity, and the optimum ΔGH is usually around thermoneutral value (0)22. In contrast to the calculations of water dissociation, W catalyst exhibits a lower hydrogen adsorption energy (−0.51 eV) than that of WO2 catalyst (−1.24 eV) (Fig. 5c), implying zero-valence W sites are more beneficial for hydrogen desorption. Accordingly, the calculated ΔGH (−0.41 eV) decreases sharply when coupling W and WO2 components as heterostructures (Fig. 5c), indicative of significantly improved hydrogen desorption kinetics on W/WO2 catalyst surface. Encouragingly, we observe that the ΔGH value (−0.27 eV) of W/WO2 is extremely closer to thermoneutral value (0) after the W/WO2 interface inserted by H+ atoms (W/HxWOy) (Fig. 5d, e), indicating the neighboring Brønsted acid site can further improve the hydrogen desorption kinetics on W/WO2 catalyst surface. Moreover, the hydrogen desorption activities of W/WO2 interface can be further tuned by the amounts of neighboring Brønsted acid site (Fig. 5e), demonstrating the coverage of Brønsted acids also plays a vital role in the hydrogen desorption step, which agrees well with aforementioned experimental characterizations (Figs. 3a, e, f and 4d, e, f).

a Schematic pathway for alkaline HER process on W/WO2 interface, where H2O molecule undergoes water adsorption, activated H2O adsorption, produced OH and H adsorption in alkaline HER process. The corresponding calculated free energy diagrams for b water adsorption and dissociation, and c hydrogen desorption steps on W/WO2 interface, W (110) and WO2 (01-1) facets, respectively. d H desorption (white) models and e the corresponding calculated free energies tuned by continuously increased neighboring Brønsted acid sites (pink). Panel a and d are created by VESTA software54.
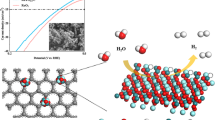
Based on above comprehensive characterizations in solid experimental evidences and theoretical simulations, a well-defined dynamic proton-concentrated surface is constructed on W/WO2 solid-acid catalyst, where the dynamic feature of proton-concentrated surface facilitates the proton transfer for the cycling of active sites on catalyst surface (Fig. 6). First, the oxygen-vacancy-rich WO2 component mainly serves as highly active Lewis acid sites for the adsorption and cleavage of H2O molecules (proton generation). Second, protons can be enriched on W/WO2 catalyst surface under ultra-low overpotentials due to the strong hydrogen storage ability of HxWOy intermediates (Brønsted acid sites, proton storage). Finally, the appealing electronic interaction between zero-valence W atoms and protons accelerates the deprotonation kinetics of Brønsted acids for the cycling of active sites (proton donation and regeneration of active sites).

The cleavage of water reactants is proceeded on WO2 surface for the generation of protons (light blue surface), where the aggressive OH- species can be rapidly repulsed from the electron-rich catalyst surface (orange layer), while the highly active protons prefer to transfer to the interface of WO2 (light blue) and W (deep blue) components for the proton recombination and release of hydrogen gas.
Discussion
In summary, a facial pyrolysis-reduction method is used to prepare free-standing W/WO2 solid-acid catalyst on Ni foam. Owing to the in situ constructed solid-acid catalyst surface with reversible adsorption/desorption behaviors of protons in alkaline HER process. The well-designed W/WO2 heterostructure catalyzing hydrogen production follows the kinetically fast Volmer-Tafel pathway with an ultra-low overptential at −10 mA/cm2 and a small Tafel slope (−34 mV/dec), as well as a long-term electrocatalytic stability (>50 h) in alkaline electrolyte, outperforming all tungsten/molybdenum oxide catalysts and most of 3d-metal oxides reported to date. Moreover, multiple spectroscopy characterizations combined with DFT calculations discover that the dynamic proton-concentrated surface can be constructed on W/WO2 metallic heterostructure under low overpotentials, enabling W/WO2 catalyzing alkaline hydrogen production to follow kinetically fast Volmer-Tafel pathway. Our strategy of solid-acid catalyst and utilization of multiple spectroscopy characterizations may provide a valuable route for designing advanced all-non-noble catalytic system towards boosting HER performance in alkaline electrolyte.
Methods
Chemicals
Tungsten chloride (WCl6 99%, Aladdin, USA), Polyvinylpyrrolidone (PVP, Macklin, China), polyethylene oxide-co-polypropylene oxide-co-polyethylene oxide (P123 98%, Adamas Reagent, China), dopamine hydrochloride (DA 98%, Aladdin, USA), and 2-amino-2-hydroxymethyl-propane-1,3-diol (Tris 98%, Aladdin, USA).
Preparation of W18O49 NWs
In a typical procedure, a mixture of WCl6 (500 mg) and PVP (20 mg) precursors was dissolved in absolute ethanol (50 mL), and a homogeneous yellow solution was obtained after stirring for 30 min. Afterwards, the obtained solutions were transferred into a 100 mL Teflon-lined autoclave and heated at 180 °C for 24 h. The resultant blue W18O49 product was collected after purification and centrifugation.
Preparation of W18O49@Ni foam
The Ni foam (2 cm × 4 cm) was treated by 0.1 M HCl solutions, and washed with absolute ethanol and distilled water before drying at 60 °C. A total of 50 mg W18O49 powder was re-dispersed into 2 mL ethanol by sonication, and the homogeneous ink was dropped onto the pre-treated Ni foam (2 cm × 4 cm). In order to better lock the W18O49 precursor, Ni foam supported W18O49 powder was pressed into a thinner foil structure (Supplementary Fig. 3).
Preparation of W18O49@PDA@Ni foam
The fabricated W18O49@Ni foam was immersed into mixture solutions (250 mL) of DA (100 mg), P123 (160 mg), and Tris (40 mg), and W18O49@PDA@Ni foam was obtained after stirring for 24 h. In addition, W18O49 powder was also added into the same mixture solutions containing DA, P123, and Tris components with continuous stirring for 24 h. After that, W18O49@PDA powder was collected by purification and centrifugation.
Preparation of W/WO2 metallic heterostructure
Both W/WO2 powder and free-standing electrode were prepared in accordance with the following thermal treatment. The desired W/WO2 metallic heterostructure was obtained by pyrolyzing the W18O49@PDA@Ni foam (W18O49@PDA powder) precursor at 700 °C under a mixture of Ar/H2 (v/v = 8:1) atmosphere for 2 h. For comparison, power and free-standing types of WO2 sample were synthesized at 700 °C under a pure Ar atmosphere for 2 h, while W sample was collected at high temperature of 750 °C under a mixture of Ar/H2 (v/v = 8:1) atmosphere for 2 h. The total loading mass of supported materials (W/WO2 and C, 32 mg) can be determined by the mass difference of bare Ni foam (2 × 4 cm, 250 mg) and Ni foam supported W/WO2 materials (2 × 4 cm, 282 mg). The loading mass of tungsten element was 3.2 mg/cm2 in accordance with the ICP-MS analysis (W: 79.1 wt%).
Characterization
XRD pattern was recorded on a Bruker AXS D8 Advance X-ray diffractometer with a Cu Ka radiation target (40 V, 40 A). TEM characterization was performed using an FEI Tecnai G2F20 microscope. Atomic-level HAADF-STEM images and the corresponding STEM-EDS elemental maps were measured on an FEI Titan Themis Z 3.1 equipped with a SCOR spherical aberration corrector and a monochromator. The probe convergence angle was 80 mrad, and camera length was 115 mm in the STEM mode. The loading mass of W atoms in W/WO2 materials and the dissolved W species were measured by the ICP-OES measurements (Agilent ICP-OES730). O K-edge NEXAFS spectra was performed at the Catalysis and Surface Science End-station at the BL11U beamline in the National Synchrotron Radiation Laboratory (NSRL) in Hefei, China. W L3-edge X-ray adsorption spectra including XANES and EXAFS profiles were collected at BL11B station at the Shanghai Synchrotron Radiation Facility (SSRF).
For NAP-XPS characterization, in situ NAP-XPS measurements were conducted on a SPECS NAP-XPS instrument, where the photon source is the monochromatic X-ray source of Al Kα (1486.6 eV), and the overall spectra resolution is Ag 3d5/2, <0.5 eV FWHM at 20 kcps@UHV. The dried sample was transferred to the analysis chamber of XPS, and then W 4 f, C 1 s, O 1 s XPS profiles were simultaneously recorded under the pressure of 1 × 10−9 mbar. After introducing water molecules, the corresponding XPS signals were collected under the water atmosphere with a pressure of 1 × 10−1 mbar.
For TOF-SIMS characterization, TOF-SIMS measurement was performed on a TOF-SIMS 5–100 instrument (ION-TOF GmbH) using a 30-keV Bi3+ as analysis beam for negative and positive polarity measurements. The analysis beam current, raster size, and incident angles of all beam are 0.7 pA, 50 × 50 μm2, and 45°, respectively.
For REELS characterization, the electrochemically treated materials were dried, and then transferred to the analysis chamber for REELS characterization using ESCALAB Xi equipment (Thermo Scientific). In detail, approximately 1000 eV electrons were incident on the surface of samples, the XPS analyzer would investigate the elastic and non-elastic scattering, corresponded to the elastic signal and energy loss peak (H loss). In our measurements, all parameters, such as the total acquisition time, energy step size, number of energy steps, and number of scans were set to be 2 mins, 0.02 eV, 1001, and 4, respectively.
For NMR characterization, 1H Magic angle spinning nuclear magnetic resonance (MAS NMR) spectra was collected on a Bruker AVANCE NEO 400 MHz Wide Bore spectrometer operating at a magnetic field of 9.40 T. The chemical shifts for 1H MAS NMR spectra was referenced to tetramethy (TMS). 1H MAS NMR spectra was acquired at a spinning rate of 15 kHz with a π/2 pulse width of 3.5 μs and s recycle of 5 s.
For Py-IR characterization, Py-IR measurements were performed on a Nicolet 380 FT-IR instrument (Thermo Co., USA). The catalyst sample was pretreated at 353 K for 2 h under a vacuum condition, and background spectra were recorded in the ranging of 1700–1400 cm−1 before adsorbing pyridine molecules. After reaching the adsorption equilibrium, the catalyst sample was retreated at 353 K for 4 h under a vacuum condition for the complete remove of physically adsorbed molecules, and the Py-IR spectra of pyridine chemisorption was obtained.
Evaluation of HER performance
Electrochemical measurements were conducted on a CHI760E electrochemical station (Shanghai Chenhua Co., China) using a standard three-electrode system in 1 M KOH electrolyte, where free-standing W/WO2 electrode (0.5 cm × 1.0 cm, area immersed in electrolyte is 0.5 cm × 0.5 cm), Ag/AgCl electrode, and a carbon rod were used as working, reference, and counter electrodes, respectively. For the fabrication of free-standing electrode for the physical mixture of W and WO2 powders (tungsten loading mass: 3.2 mg/cm2), W (5 mg) and WO2 (5 mg) were dispersed into in a mixture of 800 µL ethanol, 170 µL water, and 30 µL nafion, followed by sonication treatment at least 30 min for preparation of the homogeneous catalyst ink. Then, 200 µL of the mixture catalyst ink was dropped onto the pre-treated Ni and pressed into a thinner foil for the following electrocatalytic measurement. For the HER measurement of commercial PtRu/C catalyst, 3.2 mg of PtRu/C powder was dispersed in a mixture of of 800 μL ethanol, 170 μL water, and 30 μL Nafion, followed by sonication for at least 30 min to form a homogeneous catalyst ink. Then, all the catalyst ink was dropped onto the pre-treated Ni foam and pressed into a thinner foil for further measurement. For the preparation of W, WO2, W/WO2 catalyst ink, 5 mg of the powder materials is dispersed in the mixture of 800 µL ethanol, 170 µL water, and 30 µL nafion, followed by sonication treatment at least 30 min for the preparation of homogeneous catalyst ink. Then, 20 µL of the catalyst ink was dropped on the polished glassy carbon rotating disk electrode for electrocatalytic measurements at 1600 rpm. All potentials were recorded against the Ag/AgCl electrode and calibrated with respect to a reversible hydrogen electrode (RHE) in accordance with the Eq. (1):
TOF calculations
The H2 conversion efficiencies of W, WO2, and W/WO2 can be evaluated from the TOF values, which were obtained according to the following Eq. (2):
where j (A) is the current at a given overpotential, 2 is the number of electrons consumed to form 1 mol H2, F represents the Faraday constant (96500 C/mol), n (mol) is the number of moles of loaded metals, which can be evaluated based on the analysis of ICP-OES measurements (W wt%: W (80.8%), WO2 (78.7%), W/WO2 (79.1%))。
DFT calculations
All computations were performed by applying the plane-wave-based DFT method as implemented in the Vienna Ab Initio Simulation Package (VASP) and periodic slab models. The projector augmented wave potentials with the Perdew–Burke–Ernzerhof form of the exchange-correlation functional were employed in all the simulations with the energy cut-off of 450 eV, and the long-range van der Waals (vdW) interaction was described with the DFT-D3 method. The k-point meshes were generated using the VASPKIT tool with the grid separation of 0.04 Å−1 for the geometry optimizations and self-consistent field energy calculations, in which the total energy convergence and interaction force were set to be 10−6 eV and 10−2 eV/Å, respectively.
Based on the experimental TEM analysis, the structures of W and WO2 substrates were built by cleaving the (110) plane of bulk W and (01-1) bulk WO2, respectively. A vacuum region of 15 Å was set along the z direction to avoid the interaction between periodic images. Since the lattice parameters of WO2 (01-1) plane were 7.38 Å and 5.58 Å, a 2 × 2 supercell was built as the WO2 substrate for constructing the W/WO2 interface. The (110) plane of W with the lattice parameter of 2.70 Å was enlarged to build a 3 × 4 supercell, which was then cleaved (100) surface to build a W slab whose (110) and (110) surfaces were exposed. The W/WO2 interface was constructed by placing the W slab on the WO2 substrate. The lattice mismatch between W slab (10.8 Å) and WO2 substrate (11.2 Å) was only about 3%. The structures of constructed W/WO2 interface with front, side, and top images can be observed in Supplementary Fig. 21.
For evaluating the H2O dissociation energy barrier, the transitional state was located using the Nudged Elastic Band method. The free energy of adsorbed H (ΔGH) on surfaces is expressed as Eq. (3):
where ΔEH is the hydrogen adsorption energy, ΔEZPE and ΔS are the zero point energy difference and the entropy difference between the adsorbed state and the gas phase, respectively, and T is the system temperature (298.15 K).
Data availability
Data reported herein have been deposited in the Figshare database, and are accessible through https://doi.org/10.6084/m9.figshare.23578770. Source data are provided with this paper.
References
Zhang, J. et al. Efficient hydrogen production on MoNi4 electrocatalysts with fast water dissociation kinetics. Nat. Commun. 8, 15437 (2017).
Yu, Z. Y. et al. Clean and affordable hydrogen fuel from alkaline water splitting: past, recent progress, and future prospects. Adv. Mater. 33, 2007100 (2021).
Luo, Y. T. et al. Recent advances in design of electrocatalysts for high-current-density water splitting. Adv. Mater. 34, 2108133 (2022).
Wang, J. et al. Non-noble metal-based carbon composites in hydrogen evolution reaction: fundamentals to applications. Adv. Mater. 29, 1605838 (2017).
Zhang, L. et al. Cable-like Ru/WNO@C nanowires for simultaneous high-efficiency hydrogen evolution and low-energy consumption chlor-alkali electrolysis. Energy Environ. Sci. 12, 2569–2580 (2019).
Zhu, J. et al. Recent advances in electrocatalytic hydrogen evolution using nanoparticles. Chem. Rev. 120, 851–918 (2020).
Subbaraman, R. et al. Enhancing hydrogen evolution activity in water splitting by tailoring Li+-Ni(OH)2-Pt interfaces. Science 334, 1256–1260 (2011).
Dinh, C. T. et al. Multi-site electrocatalysts for hydrogen evolution in neutral media by destabilization of water molecules. Nat. Energy 4, 107–114 (2019).
Zhu, Y. L. et al. Metal oxide-based materials as an emerging family of hydrogen evolution electrocatalysts. Energy Environ. Sci. 13, 3361–3392 (2020).
Goya, P. et al. W for tungsten and wolfram. Nat. Chem. 3, 336 (2011).
Chen, Z. G. et al. Coordination-controlled single-atom tungsten as a non-3d-metal oxygen reduction reaction electrocatalyst with ultrahigh mass activity. Nano Energy 60, 394–403 (2019).
Wu, R. et al. Metallic WO2-carbon mesoporous nanowires as highly efficient electrocatalysts for hydrogen evolution reaction. J. Am. Chem. Soc. 137, 6983–6986 (2015).
Li, Y. H. et al. Local atomic structure modulations activate metal oxide as electrocatalyst for hydrogen evolution in acidic water. Nat. Commun. 6, 8064 (2015).
Jing, S. Y. et al. Carbon-encapsulated WOx hybrids as efficient catalysts for hydrogen evolution. Adv. Mater. 30, 1705979 (2018).
Zheng, T. T. et al. Conductive tungsten oxide nanosheets for highly efficient hydrogen evolution. Nano Lett. 17, 7968–7973 (2017).
Pan, J. B. et al. A high-performance electrochromic device assembled with hexagonal WO3 and NiO/PB composite nanosheet electrodes towards energy storage smart window. Sol. Energy Mater. Sol. Cells 207, 110337 (2020).
Can, F. et al. Tungsten-based catalysts for environmental applications. Catalysts 11, 703 (2021).
Chen, J. D. et al. Reversible hydrogen spillover in Ru-WO3-x enhances hydrogen evolution activity in neutral pH water splitting. Nat. Commun. 13, 5382 (2022).
Liu, J. C. et al. Optimizing hydrogen adsorption by d-d orbital modulation for efficient hydrogen evolution catalysis. Adv. Energy Mater. 12, 2103301 (2022).
Li, S. et al. Oxygen-evolving catalytic atoms on metal carbides. Nat. Mater. 20, 1240–1247 (2021).
Tan, H. et al. Engineering a local acid-like environment in alkaline medium for efficient hydrogen evolution reaction. Nat. Commun. 13, 2024 (2022).
Greeley, J. et al. Computational high-throughput screening of electrocatalytic materials for hydrogen evolution. Nat. Mater. 5, 909–913 (2006).
Jiang, X. L. et al. The heterostructure of Ru2P/WO3/NPC synergistically promotes H2O dissociation for improved hydrogen evolution. Angew. Chem. Int. Ed. 60, 4110–4116 (2021).
Rasmussen, M. J. et al. Role of tungsten modifiers in bimetallic catalysts for enhanced hydrodeoxygenation activity and selectivity. Catal. Sci. Technol. 10, 414–423 (2020).
Park, J. Y. et al. Investigation of the support effect in atomically dispersed Pt on WO3-x for utilization of Pt in the hydrogen evolution reaction. Angew. Chem. Int. Ed. 58, 16038–16042 (2019).
Fu, J. Y. et al. Modulating the dynamics of Brønsted acid sites on PtWOx inverse catalyst. Nat. Catal. 5, 144–153 (2022).
Mehdad, A. et al. Effect of steam and CO2 on ethane activation over Zn-ZSM-5. Catal. Sci. Technol. 8, 358–366 (2018).
Wang, F. H. et al. Robust porous WC-based self-supported ceramic electrodes for high current density hydrogen evolution reaction. Adv. Sci. 9, 2106029 (2022).
He, C. Y. et al. A highly sensitive and stable SERS substrate using hybrid tungsten dioxide/carbon ultrathin nanowire beams. J. Mater. Chem. C 6, 3200–3205 (2018).
Chen, Z. G. et al. Thermal migration towards constructing W-W dual-sites for boosted alkaline hydrogen evolution reaction. Nat. Commun. 13, 763 (2022).
Lu, X. F. et al. Ultrafine dual-phased carbide nanocrystals confined in porous nitrogen-doped carbon dodecahedrons for efficient hydrogen evolution reaction. Adv. Mater. 31, 1900699 (2019).
Wei, Y. S. et al. Fabricating dual-atom Iron catalysts for efficient oxygen evolution reaction: a heteroatom modulator approach. Angew. Chem. Int. Ed. 59, 16013–16022 (2020).
Wang, X. Y. et al. Atomic-scale insights into surface lattice oxygen activation at the spinel/perovskite interface of Co3O4/La0.3Sr0.7CoO3. Angew. Chem. Int. Ed. 58, 1–7 (2019).
Zhang, N. et al. Oxide defect engineering enables to couple solar energy into oxygen activation. J. Am. Chem. Soc. 138, 8928–8935 (2016).
Chen, Z. G. et al. Eutectoid-structured WC/W2C heterostructures: a new platform for long-term alkaline hydrogen evolution reaction at low overpotentials. Nano Energy 68, 104335 (2020).
Li, J. Y. et al. Ethylene-glycol ligand environment facilitates highly efficient hydrogen evolution of Pt/CoP through proton concentration and hydrogen spillover. Energy Environ. Sci. 12, 2298–2304 (2019).
Damian, A. et al. Ni and Ni Mo hydrogen evolution electrocatalysts electrodeposited in a polyaniline matrix. J. Power Sources 158, 464–476 (2006).
Chen, W. et al. Deciphering the alternating synergy between interlayer Pt single-atom and NiFe layered double hydroxide for overall water splitting. Energy Environ. Sci. 14, 6428–6440 (2021).
Alexandria, R. C. et al. Electrochemical impedance spectroscopy of metal oxide electrodes for energy applications. ACS Appl. Energy Mater. 3, 66–98 (2020).
Li, Z. C. et al. Native Ligand carbonization renders common platinum nanoparticles highly durable for electrocatalytic oxygen reduction: annealing temperature matters. Adv. Mater. 34, 2202743 (2022).
Ma, Z. et al. NbOx nano-nail with a Pt head embedded in carbon as a highly active and durable oxygen reduction catalyst. Nano Energy 69, 104455 (2020).
Yuan, L. P. et al. Molecularly engineered strong metal oxide-support interaction enables highly efficient and stable CO2 electroreduction. ACS Catal. 10, 13227–13235 (2020).
Zeng, X. et al. Mesoporous TiO2 nanospheres loaded with highly dispersed Pd nanoparticles for pH-universal hydrogen evolution reaction. Mater. Today Nano 6, 100038 (2019).
Minamimoto, H. et al. In-situ observation of isotopic hydrogen evolution reactions using electrochemical mass spectroscopy to evaluate surface morphological effect. Electrochim. Acta 304, 87–93 (2019).
Qian, K. C. et al. Directional oxygen activation by oxygen-vacancy-rich WO2 nanorods for superb hydrogen evolution via formaldehyde reforming. J. Mater. Chem. A 7, 14592–14601 (2019).
Yan, J. Q. et al. Single atom tungsten doped ultrathin α-Ni(OH)2 for enhanced electrocatalytic water oxidation. Nat. Commun. 10, 2149 (2019).
Rico, V. J. et al. Determination of the hydrogen content in diamond-like carbon and polymeric thin films by reflection electron energy loss spectroscopy. Diam. Relat. Mater. 16, 107–111 (2006).
Yubero, F. et al. Identification of hydrogen and deuterium at the surface of water ice by reflection electron energy loss spectroscopy. Appl. Phys. Lett. 95, 084101 (2009).
Ren, B. et al. Thermal stability of hydrogenated diamond films in nitrogen ambience studied by reflection electron energy spectroscopy and X-ray photoelectron spectroscopy. Appl. Surf. Sci. 388, 565–570 (2016).
Zhang, Z. C. et al. Evoking ordered vacancies in metallic nanostructures toward a vacated barlow packing for high-performance hydrogen evolution. Sci. Adv. 7, eabd6647 (2017).
Wang, L. Q. et al. Rapid complete reconfiguration induced actual active species for industrial hydrogen evolution reaction. Nat. Commun. 13, 5785 (2022).
Li, G. W. et al. Dirac nodal arc semimetal PtSn4: An ideal platform for understanding surface properties and catalysis. Angew. Chem. Int. Ed. 58, 13107–13112 (2019).
Chen, L. W. et al. Structurally ordered intermetallic Ir3V electrocatalysts for alkaline hydrogen evolution reaction. Nano Energy 81, 105636 (2021).
Momma, K. et al. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 44, 1272–1276 (2011).
Acknowledgements
Y.C. would like to acknowledge the supported from the National Key R&D Program of China (Nos. 2022YFA1503801 and 2022YFA1503802), the National Natural Science Foundation of China (22172190), the CAS Project for Young Scientists in Basic Research (No. YSBR-022), and the Young Cross Team Project of CAS (No. JCTD−2021-14). Z.C. would like to acknowledge the support from the National Natural Science Foundation of China (No. 22109171). W.G. would like to acknowledge the support from the National Natural Science Foundation of China (No. 12004324), Science and Technology Project of Jiangxi Province (No. 20202ACBL211005), and Natural Science Foundation of the Jiangsu Higher Education Institutions of China (No. 22KJA140003). The authors are grateful for the technical support of Nano-X from Suzhou Institute of Nano-Tech and Nano-Bionics, Chinese Academy of Sciences (SINANO), in particular, we thank R.H. and Z.L. for their help in the measurements and analysis of REELS and TOF-SIMS, respectively. We also thank Q.X., J.H., H.D., and J.Z. for their help in Near Edge X-ray Absorption Fine Structure (O K-edge NEXAFS) performed at the Catalysis and Surface Science End-station at the BL11U beamline in the National Synchrotron Radiation Laboratory (NSRL) in Hefei, China. We thank J.W. and R.G. for their help in the measurements and analysis of X-ray adsorption spectra (W-L3 edge XAS) characterization at BL11B station in the Shanghai Synchrotron Radiation Facility (SSRF).
Author information
Authors and Affiliations
Contributions
Y.C. conceived the project and designed the experiments. Z.C., S.H., G.Y., C.Z., X.F., and Y.L. performed the synthesis and characterization of catalysts and the electrocatalytic measurements. Z.C. R.G., and J.W. performed the X-ray adsorption experiments and analyzed the raw data. Z.C. and W.G. carried out the DFT calculations. Z.C. and Y.C. wrote the paper. All authors discussed the results and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Xiaoming Sun and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, Z., Gong, W., Wang, J. et al. Metallic W/WO2 solid-acid catalyst boosts hydrogen evolution reaction in alkaline electrolyte. Nat Commun 14, 5363 (2023). https://doi.org/10.1038/s41467-023-41097-w
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-41097-w
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
