
Abstract
Transition metal-catalyzed C–H bond functionalization is an important method in organic synthesis, but the development of methods that are lower cost and have a less environmental impact is desirable. Here, a Cu-catalyzed asymmetric C(sp2)–H arylation is reported. With diaryliodonium salts as arylating reagents, a range of ortho-arylated P-chiral phosphonic diamides were obtained in moderate to excellent yields with high enantioselectivities (up to 92% ee). Meanwhile, enantioselective C-3 arylation of diarylphosphine oxide indoles was also realized under similar conditions to construct axial chirality.
Similar content being viewed by others
Introduction
Transition metal-catalyzed C–H bond functionalization has become an important methodology for organic synthesis in terms of atom economy and step economy1,2,3,4. Over the past several decades, elegant and efficient palladium-, rhodium-, iridium-, and ruthenium-catalyzed transformations for the stereoselective construction of C–C and/or C–X bonds through C–H activation have been well exploited5,6,7,8. In these well-established methodologies, two general strategies have been followed to achieve enantiocontrol: (1) enantio-determining activation of enantiotopic C–H bonds by chiral transition metal complexes; (2) unbiased C–H bond scission followed by stereochemistry-generating incorporation of coupling partners. To realize those transformations, many chiral ligands and catalysts have been developed to achieve excellent selectivity9,10,11,12,13. On the other hand, a cost-effective and environmentally benign method to selectively activate inert C–H bonds is still desirable in this field.
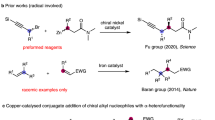
First-row metals are more earth-abundant and less toxic than precious 4d and 5d metals. Recently, the utilization of these 3d metals to activate omnipresent C–H bonds, especially enantioselectively, has gained increasing interest from chemists14,15,16,17,18,19,20,21,22,23,24,25,26,27,28. Hou and coworkers developed chiral cyclopentadienyl scandium complexes to catalyze enantioselective transformations, including C–H addition of pyridines to olefins16, C–H alkylation of imidazoles with 1,1-disubstituted alkenes17 and C–H alkenylation of ferrocenes with alkynes18. In 2017, Butenschön revealed an iron-catalyzed arylation of ferrocenamide with moderate enantioselectivities using (R,R)-chiraphos as the ligand19. Cramer and coworkers reported nickel-catalyzed asymmetric cyclization of pyridone derivatives with N-heterocyclic carbenes (NHCs) as the chiral source20. Despite significant achievements in the literature, inexpensive 3d metal-catalyzed asymmetric C–H activation is still in its infancy. Furthermore, alkenes and alkynes are the most commonly used coupling partners in reports to date, and examples of C–H arylation are quite scarce. Palladium, rhodium, and iridium are commonly used transition metals in directed enantioselective C–H arylation reactions (Fig. 1a).

a Directed enantioselective ortho-C–H arylation catalyzed by 4d transition metals. b Cu-catalyzed asymmetric C(sp3)–H functionalization. c Cu-catalyzed asymmetric C(sp2)–H functionalization. d This work: Cu-bisoxazoline-catalyzed asymmetric arylation. NFSI N-fluorobenzenesulfonimide, DTBP 2,6-di-tert-butylpyridine.
Copper, which constitutes approximately 0.01% of the Earth’s crust, has three main oxidation states ranging from 0 to +3, making it more properly a 3d metal in transformations involving radicals or two-electron transfer29,30. Recently, Liu and others developed a Cu-catalyzed asymmetric C(sp3)–H functionalization involving radicals with impressive enantioselectivities (Fig. 1b)31,32. Cu-catalyzed C–H arylation of heteroarenes with aryl iodides was first reported by Daugulis, Miura, and Ackermann33,34,35,36. Since then, Cu-catalyzed C–H functionalization has made considerable progress in the construction of C–C and C–X bonds37,38,39,40,41,42,43. For example, Yu reported a Cu-catalyzed ortho-arylation of carboxamides directed by oxazoline-based bidentate groups in 201444. Although research on Cu-catalyzed C–H functionalization, including arylation, alkylation, nitrogenation, halogenation, and so on, has surged in recent years, asymmetric transformations remain poorly developed. In 2016, Sawamura’s group published their work on Cu-catalyzed asymmetric allyl phosphate substitution by azoles to construct all-carbon quaternary stereocenters (Fig. 1c)45. A designed chiral NHC ligand was used to generate excellent branch regioselectivity and enantiocontrol, but C–H bond cleavage was not the enantio-determining step. Dai and Yu attempted Cu-mediated diastereoselective C–H thiolation of ferrocenes utilizing a chiral oxazoline bidentate directing group in 201846.
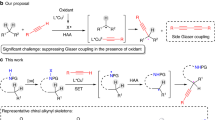
In 2008, Gaunt found that diaryliodonium salts could serve as arylating reagents in the copper-catalyzed C–H arylation of indoles, and an acetyl moiety on the N atom switched the regioselectivity from C3 to C247. The author proposed that a highly electrophilic aryl–Cu(III) intermediate was essential to the reactivity. Several groups extended this methodology, and diaryliodonium salts were proven to be powerful arylating reagents for the C–H bond functionalization of arenes48,49,50,51,52,53,54,55. While chiral bisoxazolines (BOXs) are common ligands in copper-catalyzed transformations56,57,58, we questioned whether asymmetric Cu-catalyzed C–H bond arylation could be achieved by the combination of chiral bisoxazolines and diaryliodonium salts.
In this work, we develop a Cu/bisoxazoline-catalyzed asymmetric C(sp2)–H arylation with diaryliodonium salts, and high stereoselectivities were obtained for P-chiral phosphonic diamides and axially chiral phosphorus-containing indoles (Fig. 1d).
Results and discussion
Optimization of reaction conditions
We commenced our investigations with symmetric phosphonic diamide a1 and [PhIMes]SbF6 with CuI as the catalyst (Table 1; see the SuppIementary Information for more details). Initially, various bisoxazoline ligands were screened, and commercially available chiral BOX L1 generated desired ortho-arylated product b1 in 50% yield with weak enantiocontrol (10% ee, Entry 1). Next, phenyl-substituted spiro-BOXs L2–L5 were examined (Entries 2–5). Cyclopropane- and cyclobutane-backboned ligands L2 and L3 yielded similar results, with moderate ee values, while cyclopentane-backboned spiro-BOX L4 showed low reactivity. The enantioselectivity increased to 75% when the ring size was increased to a cyclohexane structure (Entry 5). A dramatic increase in enantioselectivity from 75 to 90% was observed when 2,3-dihydro-1H-phenalene-backboned BOX L6 was used, which may be attributed to the additional rigidity of the backbone compared to that of L5 (Entry 6). Unfortunately, modifications of the phenyl substituent of L6 did not result in positive effects on selectivity (Entries 7–9). In addition, ligand L10 with a seven-membered ring backbone was also tested and exhibited inferior enantioselectivity (69% ee, Entry 10). Under the same conditions, CuCl resulted in slightly better enantioselectivity than CuI (91%, Entry 11). 2,6-Di-tert-butylpyridine (DTBP) is a general acid side-product scavenger in Gaunt’s works with diaryliodonium salts;47,51 hence, it was also applied in this reaction to improve the yield, but to our surprise, the reaction was severely suppressed (Entry 12). We hypothesized that DTBP might abstract an anion, which facilitates H atom dissociation during C–H bond activation, from the system, so NaOTf as an external anion source was added (Entries 13–15). After investigating the molar ratio of NaOTf, 0.5 mol% was determined to be the best value, affording the product in 97% yield and 91% ee (Entry 14). Thus, we established an optimal catalyst system: CuCl (10 mol%)/L6 (15 mol%) with 1.5 equiv. of DTBP and 0.5 mol% NaOTf in dichloromethane.

Substrate scope
With the optimal conditions in hand, we examined the substrate scope of this enantioselective ortho-C(sp2)-H arylation reaction (Fig. 2). Initially, unsubstituted P-tert-butyl-N,N’-diphenyl phosphonic diamine was subjected to the standard conditions, but only trace product was generated, and a bromo moiety ortho to the methyl group in a1 completely inhibited the reactivity (see the SuppIementary Information for details). These findings suggest that the reaction might be controlled by electronics, necessitating electron-donating groups (EDGs) on the substrate. Several phosphonic diamides with different EDGs were tested. Tert-butyl and dimethyl groups exhibited similar inferior enantioselectivities (85%, b2 and b3). A methoxy group at the meta-position reduced the ee value to 86% because of electronic effects (b4), while halogenation could alleviate the impact and lead to improved results (b5, b6). Substrate a7, with a strong electron-donating 3,4-methylenedioxy group, yielded much poorer results, which might be attributed to a faster background reaction. Replacing the butyl moiety on the P atom with a phenyl moiety resulted in only moderate enantioselectivity, which could be explained by the similar steric hindrance between phenyl and arylamine.

Reaction conditions unless otherwise noted: a (0.10 mmol), Ph2ISbF6 (0.15 mmol), CuCl (0.01 mmol), L6 (0.015 mmol), NaOTf (0.0005 mmol), DTBP (0.15 mmol), DCM (2 mL) in a sealed tube at 60 °C for 48 h. DTBP 2,6-di-tert-butylpyridine.
Next, a series of diaryliodonium salts were synthesized and tested in this reaction (Fig. 3). Halogenation at the para and meta positions of diaryliodonium salts has a negligible influence on the results, except para-fluorination resulted in slightly lower enantioselectivity due to the electron-withdrawing effect (b9-b14). Ortho-fluoro diaryliodonium salt produced the corresponding ortho-arylated product in moderate yield because of steric repulsion but did not affect the selectivity (b15). Other functional groups, such as methyl, trifluoromethyl, ethoxycarbonyl, methoxy, trifluoromethoxy, and tert-butyl groups, exert notable electronic effects on enantioselectivity. Strong electron-donating and electron-withdrawing abilities were both detrimental to stereo-induction, but substituents in the para-position had a more remarkable negative impact than those in the meta-position (b16-b26). The absolute configuration of product b was assigned to be R according to the X-ray crystal diffraction analysis (XRD) of product b1859. Finally, heterocyclic diaryliodonium salts were also used in this reaction, and thiophenyl groups were successfully introduced at the ortho-position of a1 with moderate yields and enantioselectivities (b27-b28).

Reaction conditions unless otherwise noted: a1 (0.10 mmol), Ar2ISbF6 (0.15 mmol), CuCl (0.01 mmol), L6 (0.015 mmol), NaOTf (0.0005 mmol), DTBP (0.15 mmol), DCM (2 mL) in a sealed tube at 60 °C for 48 h. b[ArIMes]SbF6 was used. DTBP 2,6-di-tert-butylpyridine.
Axially chiral biaryl monophosphines are an important class of ligands in asymmetric catalysis, and the development of the new synthetic protocol for these compounds has attracted much attention60. As phosphine oxides are common precursors of trivalent phosphines, we envisioned the developed phosphine oxide-directed C–H arylation reaction could be extended to the construction of axially chiral biaryl monophosphine oxides61. We chose diphenylphosphine oxide indole c1 as the standard substrate under similar reaction conditions (Fig. 4, see SuppIementary Information for detailed condition optimization), and the C-3 phenylated product was obtained in 64% yield with 87% ee. XRD results of d1 indicate the configuration to be R around the axis62.

Reaction conditions unless otherwise noted: c (0.10 mmol), Ph2ISbF6 (0.2 mmol), CuI (0.01 mmol), L6 (0.015 mmol), NaOTf (0.001 mmol), DTBP (0.2 mmol), DCM (2 mL) in a sealed tube at 60 °C for 24 h. DTBP 2,6-di-tert-butylpyridine, XRD X-ray crystal diffraction analysis.
The scope of diphenylphosphine oxide indoles was also investigated, and the isopropyl on the N atom of the indole ring gave similar results to methyl (d2), while results varied with sites of methyl on the indole ring. 5- and 7-methylated substrates produced approximate results (d3 and d5), but methyl on the 6-position generates much poorer stereoselectivity (69% ee, d4). Such results could be explained as follows: EDGs para to C-3 increased the electron cloud density at the reaction site and accelerated the background reaction. Substrate c8 with stronger EDG methoxy on the 5-position also validated this deduction (d9). Conversely, electron-withdrawing groups (EWGs) like chloro and fluoro on the indole ring severely hampered the reaction rate with slight erosion on ee values of products (d6-d8). Next, different functional groups on diphenylphosphine oxide were also screened. Generally, EDGs on phenyl rings benefit the stereocontrol, giving product in 83–90% ee with moderate yields (d11-d16). Unexpectedly, chloro on para-position of phenyl rings has distinct corruption on the stereoselectivity, generating the product in 66% ee (d10). We think this could be ascribed to the weaker directing ability of the electron-deficient diphenylphosphine oxide group.
Furthermore, the scope of diaryliodonium salts for asymmetric C-3 arylation of diphenylphosphine oxide indole c1 was examined under the same conditions (Fig. 5). Groups with different electronic effects, like bromo, chloro, fluoro, methyl, ethoxycarbonyl, methoxy, and tert-butyl, were well tolerated and gave corresponding arylated products in 70–87% ee with moderate to good yields. As a whole, meta-position of substitutes on the phenyl ring of diaryliodonium salts renders low enantioselectivity than para-position, which is contrary to the tendency observed with phosphonic diamide a1 in Fig. 3. We speculated that the steric effect plays a more important role than the electronic effect during the construction of axial chirality, opposite to P-central chirality.

Reaction conditions unless otherwise noted: c (0.10 mmol), Ar2ISbF6 (0.2 mmol), CuI (0.01 mmol), L6 (0.015 mmol), NaOTf (0.001 mmol), DTBP (0.2 mmol), DCM (2 mL) in a sealed tube at 60 °C for 24 h. DTBP 2,6-di-tert-butylpyridine.
Synthetic applications
To further demonstrate the robustness of the developed method, the two arylation reactions were performed at the gram scale under adjusted conditions. With catalyst loading reduced to 1 mol%, ortho-arylated product b6 was isolated in 83% yield with 91% ee after 72 h of heating (Fig. 6a). C-3 arylation product of c1 was obtained in 62% yield and 84% ee with 5 mol% catalyst loading (Fig. 6b). Meanwhile, slurry d1 in ethyl acetate (EA) could afford the enantio-enriched product in 96% ee, which offers further possibilities for application of this axially chiral biaryl monophosphine oxides in catalysis.

a Scale up for b6. b Scale up for d1. DTBP 2,6-di-tert-butylpyridine, EA ethyl acetate.
Mechanistic studies
As mentioned above, the electronic properties of the substrates had a substantial impact on the reactivities. To understand this reaction in more depth, unsymmetric phosphonic diamide a9 was synthesized and subjected to standard conditions (Fig. 7a). As expected, arylation predominantly occurred on the meta-methyl-substituted phenyl ring, and b29 was isolated in 61% yield, while arylated product b30 with an unsubstituted phenyl ring was observed as a minor product. This differentiating behavior of the reaction system implies that the catalyst selectively interacts with the phenyl ring in an electrophilic activated manner. Additionally, this reaction represents a successful example of distinguishing between two similar phenyl rings with subtle differences in the electronic density, which is hard to achieve by other asymmetric catalytic systems.

a Intra-molecular competition experiment. b H/D Kinetic isotope effect (KIE) experiments.
The kinetic isotope effect (KIE) was evaluated under standard conditions, and a value of 1.0 indicated that C–H bond abscission is not the rate-determining step in this reaction (Fig. 7b)63. Based on the observed results and literature reports47,48, we assumed that aryl–Cu(III) intermediate II was involved in the transformation, and this highly electrophilic complex, in a stereoselective and rate-limiting manner, activates an electron-rich phenyl ring with the aid of NaOTf to abstract H atoms and generate ortho-metalized intermediate IV, as depicted in Fig. 8. The whole C–H bond activation event determined the configuration of the product; however, more meticulous investigations are needed to elucidate the exact process of C–H bond activation and the origin of stereocontrol.

DTBP 2,6-di-tert-butylpyridine, O.A. oxidative addition, R.E. reductive elimination.
In summary, we have disclosed the Cu-catalyzed asymmetric arylation for the construction of P-chiral phosphonic diamines and axially chiral diarylphosphine oxide indoles with excellent enantioselectivities. Adequate but minimal NaOTf was found to be important for achieving the high conversion of substrates. Electronic effects are fairly obvious in this reaction, and an electrophilic aryl–Cu(III) intermediate was thereby assumed. The KIE experiment shows that C–H bond abscission is not the rate-determining step in this Cu-catalyzed arylation process. The gram-scale reaction proceeded smoothly when the catalyst loading was reduced to 1 mol%, implying potential industrial applications. Further investigations to fully understand the reaction and extension of this methodology to construct other optically active structures are underway in our lab.
Methods
General procedure for copper-bisoxazoline-catalyzed ortho-arylation
A dried Schlenk tube was charged with NaOTf/MeOH solution (c: 3.4 mg in 20 mL MeOH, 0.5 mL), and the solvent was removed under vacuum. CuCl (1.0 mg, 0.01 mmol), L6 (6.9 mg, 0.015 mmol), and CH2Cl2 (2.0 mL) were added into the tube, and the mixture was stirred at room temperature for 0.5 h under N2. Substrate (0.1 mmol, 1.0 equiv.), diaryliodonium hexafluoroantimonate salt (0.15 mmol, 1.5 equiv.), and 2,6-di-tert-butylpyridine (33 μL, 0.15 mmol, 1.5 equiv.) were added into the mixture under N2. The reaction was heated to 60 °C and stirred for 48 h. After cooling to room temperature, the reaction mixture was filtrated, and the filtrate was concentrated under vacuum to remove all volatiles. The residue was purified via column chromatography on silica gel (petroleum ether/ethyl acetate = 5:1 to 2:1) to afford the product.
Data availability
The data that support the findings of this study are available within the paper and its Supplementary Information files. Crystallographic parameters for compounds b18 and d1 are available free of charge from the Cambridge Crystallographic Data Centre under CCDC 2099084 and 2189868 (www.ccdc.cam.ac.uk/data_request/cif). All data are also available from authors upon request.
References
Kakiuchi, F. & Murai, S. Catalytic C–H/olefin coupling. Acc. Chem. Res. 35, 826–834 (2002).
Giri, R., Shi, B.-F., Engle, K. M., Maugel, N. & Yu, J.-Q. Transition metal-catalyzed C–H activation reactions: diastereoselectivity and enantioselectivity. Chem. Soc. Rev. 38, 3242–3272 (2009).
Chu, J. C. K. & Rovis, T. Complementary strategies for directed C(sp3)–H functionalization: a comparison of transition-metal-catalyzed activation, hydrogen atom transfer, and carbene/nitrene transfer. Angew. Chem. Int. Ed. Engl. 57, 62–101 (2018).
Wencel-Delord, J. & Colobert, F. Asymmetric C(sp2)–H activation. Chemistry 19, 14010–14017 (2013).
Zheng, C. & You, S.-L. Recent development of direct asymmetric functionalization of inert C–H bonds. RSC Adv. 4, 6173–6214 (2014).
Newton, C. G., Wang, S.-G., Oliveira, C. C. & Cramer, N. Catalytic enantioselective transformations involving C–H bond cleavage by transition-metal complexes. Chem. Rev. 117, 8908–8976 (2017).
Saint-Denis, T. G., Zhu, R.-Y., Chen, G., Wu, Q.-F. & Yu, J.-Q. Enantioselective C(sp3)–H bond activation by chiral transition metal catalysts. Science 359, 759–770 (2018).
Woźniak, Ł. et al. Catalytic enantioselective functionalizations of C–H bonds by chiral iridium complexes. Chem. Rev. 120, 10516–10543 (2020).
Ye, B. & Cramer, N. Chiral cyclopentadienyls: enabling ligands for asymmetric Rh(III)-catalyzed C–H functionalizations. Acc. Chem. Res. 48, 1308–1318 (2015).
Thongpaen, J., Manguin, R. & Baslé, O. Chiral N-heterocyclic carbene ligands enable asymmetric C-H bond functionalization. Angew. Chem. Int. Ed. Engl. 59, 10242–10251 (2020).
Tran, V. T., Nimmagadda, S. K., Liu, M.-Y. & Engle, K. M. Recent applications of chiral phosphoric acids in palladium catalysis. Org. Biomol. Chem. 18, 618–637 (2020).
Shao, Q., Wu, K., Zhuang, Z., Qian, S.-Q. & Yu, J.-Q. From Pd(OAc)2 to chiral catalysts: the discovery and development of bifunctional mono-N-protected amino acid ligands for diverse C–H functionalization reactions. Acc. Chem. Res. 53, 833–851 (2020).
Liu, C.-X., Zhang, W.-W., Yin, S.-Y., Gu, Q. & You, S.-L. Synthesis of atropisomers by transition-metal-catalyzed asymmetric C–H functionalization reactions. J. Am. Chem. Soc. 143, 14025–14040 (2021).
Loup, J., Dhawa, U., Pesciaioli, F., Wencel-Delord, J. & Ackermann, L. Enantioselective C–H activation with earth-abundant 3d transition metals. Angew. Chem. Int. Ed. Engl. 58, 12803–12818 (2019).
Woźniak, Ł. & Cramer, N. Enantioselective C–H bond functionalizations by 3d transition-metal catalysts. Trends Chem. 1, 471–484 (2019).
Song, G.-Y., Wylie, W. N. O. & Hou, Z.-M. Enantioselective C–H bond addition of pyridines to alkenes catalyzed by chiral half-sandwich rare-earth complexes. J. Am. Chem. Soc. 136, 12209–12212 (2014).
Loup, J. et al. Asymmetric iron-catalyzed C–H alkylation enabled by remote ligand meta-substitution. Angew. Chem. Int. Ed. Engl. 56, 14197–14201 (2017).
Lou, S.-J., Mo, Z.-B., Nishiura, M. & Hou, Z.-M. Construction of all-carbon quaternary stereocenters by scandium catalyzed intramolecular C–H alkylation of imidazoles with 1,1-disubstituted alkenes. J. Am. Chem. Soc. 2020, 1200–1205 (2020).
Lou, S.-J., Zhuo, Q.-D., Nishiura, M., Luo, G. & Hou, Z.-M. Enantioselective C–H alkenylation of ferrocenes with alkynes by half-sandwich scandium catalyst. J. Am. Chem. Soc. 143, 2470–2476 (2021).
Schmiel, D. & Butenschön, H. Directed iron-catalyzed ortho-alkylation and arylation: toward the stereoselective catalytic synthesis of 1,2-disubstituted planar-chiral ferrocene derivatives. Organometallics 36, 4979–4989 (2017).
Diesel, J., Finogenova, A. M. & Cramer, N. Nickel-catalyzed enantioselective pyridone C–H functionalizations enabled by a bulky N-heterocyclic carbene ligand. J. Am. Chem. Soc. 140, 4489–4493 (2018).
Loup, J., Müller, V., Ghorai, D. & Ackermann, L. Enantioselective aluminum-free alkene hydroarylations through C–H activation by a chiral nickel/JoSPOphos manifold. Angew. Chem. Int. Ed. Engl. 58, 1749–1753 (2019).
Zhang, W.-B., Yang, X.-T., Ma, J.-B., Su, Z.-M. & Shi, S.-L. Nickel/NHC-catalyzed asymmetric C–H alkylation of fluoroarenes with alkenes: synthesis of enantioenriched fluorotetralins. J. Am. Chem. Soc. 141, 5628–5634 (2019).
Ozols, K., Jang, Y. S. & Cramer, N. Chiral cyclopentadienyl cobalt(III) complexes enable highly enantioselective 3d-metal-catalyzed C–H functionalizations. J. Am. Chem. Soc. 141, 5675–5680 (2019).
Fukagawa, S. et al. Enantioselective C(sp3)–H amidation of thioamides catalyzed by a cobaltIII/chiral carboxylic acid hybrid system. Angew. Chem. Int. Ed. Engl. 58, 1153–1157 (2019).
Jacob, N., Zaid, Y., Oliveira, J. C. A., Ackermann, L. & Wencel-Delord, J. Cobalt-catalyzed enantioselective C–H arylation of indoles. J. Am. Chem. Soc. 144, 798–806 (2022).
Yao, Q.-J., Chen, J.-H., Song, H., Huang, F.-R. & Shi, B.-F. Cobalt/salox-catalyzed enantioselective C-H functionalization of arylphosphinamides. Angew. Chem. Int. Ed. Engl. 61, e202202892 (2022).
Hirata, Y. et al. Cobalt(III)/chiral carboxylic acid-catalyzed enantioselective synthesis of benzothiadiazine-1-oxides via C–H activation. Angew. Chem. Int. Ed. Engl. 61, e202205341 (2022).
Zhang, C., Tang, C.-H. & Jiao, N. Recent advances in copper-catalyzed dehydrogenative functionalization via a single electron transfer (SET) process. Chem. Soc. Rev. 41, 3464–3484 (2012).
Wang, F., Chen, P.-H. & Liu, G.-S. Copper-catalyzed radical relay for asymmetric radical transformations. Acc. Chem. Res. 51, 2036–2046 (2018).
Zhang, W. et al. Enantioselective cyanation of benzylic C-H bonds via copper-catalyzed radical relay. Science 353, 1014–1018 (2016).
Zhang, W., Wu, L.-L., Chen, P.-H. & Liu, G.-S. Enantioselective arylation of benzylic C-H bonds by copper-catalyzed radical relay. Angew. Chem. Int. Ed. Engl. 58, 6425–6429 (2019).
Do, H.-Q. & Daugulis, O. Copper-catalyzed arylation of heterocycle C–H bonds. J. Am. Chem. Soc. 129, 12404–12405 (2007).
Do, H.-Q., Khan, R. M. K. & Daugulis, O. A general method for copper-catalyzed arylation of arene C–H bonds. J. Am. Chem. Soc. 130, 15185–15192 (2008).
Yoshizumi, T., Tsurugi, H., Satoh, T. & Miura, M. Copper-mediated direct arylation of benzoazoles with aryl iodides. Tetrahedron Lett. 49, 1598–1600 (2008).
Ackermann, L., Potukuchi, H. K., Landsberg, D. & Vicente, R. Copper-catalyzed “click” reaction/direct arylation sequence: modular syntheses of 1,2,3-triazoles. Org. Lett. 10, 3081–3084 (2008).
Guo, X.-X., Gu, D.-W., Wu, Z.-X. & Zhang, W.-B. Copper-catalyzed C–H functionalization reactions: efficient synthesis of heterocycles. Chem. Rev. 115, 1622–1651 (2015).
Rao, W.-H. & Shi, B.-F. Recent advances in copper-mediated chelation-assisted functionalization of unactivated C–H bonds. Org. Chem. Front. 3, 1028–1047 (2016).
Shang, M., Sun, S.-Z., Wang, H.-L., Wang, M.-M. & Dai, H.-X. Recent progress on copper-mediated directing-group-assisted C(sp2)–H activation. Synthesis 48, 4381–4399 (2016).
Li, Z.-K., Jia, X.-S. & Yin, L. Recent advances in copper(II)-mediated or -catalyzed C–H functionalization. Synthesis 50, 4165–4188 (2018).
Kantam, M. L., Gadipelly, C., Deshmukh, G., Reddy, K. R. & Bhargava, S. Copper catalyzed C-H activation. Chem. Rec. 19, 1302–1318 (2019).
Aneeja, T., Neetha, M., Afsina, C. M. A. & Anilkumar, G. Progress and prospects in copper-catalyzed C–H functionalization. RSC Adv. 10, 34429–34458 (2020).
Yang, Y.-H. et al. Recent advances in copper promoted inert C(sp3)–H functionalization. ACS Catal. 11, 967–984 (2021).
Shang, M., Sun, S.-Z., Dai, H.-X. & Yu, J.-Q. Cu(OAc)2-catalyzed coupling of aromatic C–H bonds with arylboron reagents. Org. Lett. 16, 5666–5669 (2014).
Ohmiya, H., Zhang, H., Shibata, S., Harada, A. & Sawamura, M. Construction of quaternary stereogenic carbon centers through copper-catalyzed enantioselective allylic alkylation of azoles. Angew. Chem. Int. Ed. Engl. 55, 4777–4780 (2016).
Kong, W.-J. et al. Copper-mediated diastereoselective C–H thiolation of ferrocenes. Organometallics 37, 2832–2836 (2018).
Phipps, R. J., Grimster, N. P. & Gaunt, M. J. Cu(II)-catalyzed direct and site-selective arylation of indoles under mild conditions. J. Am. Chem. Soc. 130, 8172–8174 (2008).
Phipps, R. J. & Gaunt, M. J. A meta-selective copper-catalyzed C–H bond arylation. Science 323, 1593–1597 (2009).
Duong, H. A., Gilligan, R. E., Cooke, M. L., Phipps, R. J. & Gaunt, M. J. Copper(II)-catalyzed meta-selective direct arylation of α-aryl carbonyl compounds. Angew. Chem. Int. Ed. Engl. 50, 463–466 (2011).
Chen, B., Hou, X.-L., Li, Y.-X. & Wu, Y.-D. Mechanistic understanding of the unexpected meta selectivity in copper-catalyzed anilide C–H bond arylation. J. Am. Chem. Soc. 133, 7668–7671 (2011).
Ciana, C.-L., Phipps, R. J., Brandt, J. R., Meyer, F.-M. & Gaunt, M. J. Highly para-selective copper(II)-catalyzed direct arylation of aniline and phenol derivatives. Angew. Chem. Int. Ed. Engl. 50, 458–462 (2011).
Yang, Y.-Q., Li, R.-R., Zhao, Y., Zhao, D.-B. & Shi, Z.-Z. Cu-catalyzed direct C6-arylation of indoles. J. Am. Chem. Soc. 138, 8734–8737 (2016).
Yang, Y.-Q., Gao, P., Zhao, Y. & Shi, Z.-Z. Regiocontrolled direct C–H arylation of indoles at the C4 and C5 positions. Angew. Chem. Int. Ed. Engl. 56, 3966–3971 (2017).
Niu, Y., Qi, Z.-C., Lou, Q.-X., Bai, P.-B. & Yang, S.-D. Copper-catalyzed arylation of polycyclic aromatic hydrocarbons by the P=O group. Chem. Commun. 56, 14721–14724 (2020).
Xu, Q. et al. Cu(I)-catalyzed asymmetric arylation of pyrroles with diaryliodonium salts toward the synthesis of N–N atropisomers. Org. Lett. 24, 3138–3143 (2022).
Johnson, J. S. & Evans, D. A. Chiral bis(oxazoline) copper(II) complexes: versatile catalysts for enantioselective cycloaddition, aldol, Michael, and carbonyl ene reactions. Acc. Chem. Res. 33, 325–335 (2000).
Yang, G.-Q. & Zhang, W.-B. Renaissance of pyridine-oxazolines as chiral ligands for asymmetric catalysis. Chem. Soc. Rev. 47, 1783–1810 (2018).
Li, Z.-L., Fang, G.-C., Gu, Q.-S. & Liu, X.-Y. Recent advances in copper-catalysed radical-involved asymmetric 1,2-difunctionalization of alkenes. Chem. Soc. Rev. 49, 32–48 (2020).
Cambridge Crystallographic Data Centre. CCDC 2099084 https://www.ccdc.cam.ac.uk/data_request/cif (2023).
Hayashi, T. Chiral monodentate phosphine ligand MOP for transition-metal-catalyzed asymmetric reactions. Acc. Chem. Res. 33, 354–362 (2000).
Huang, X.-L., Li, C., Wang, J. & Yang, S.-D. Direct copper-catalyzed C-3 arylation of diphenylphosphine oxide indoles. Synthesis 54, 4711–4720 (2022).
Cambridge Crystallographic Data Centre. CCDC 2189868 https://www.ccdc.cam.ac.uk/data_request/cif (2023).
Simmons, E. M. & Hartwig, J. F. On the interpretation of deuterium kinetic isotope effects in C–H bond functionalizations by transition-metal complexes. Angew. Chem. Int. Ed. Engl. 51, 3066–3072 (2012).
Acknowledgements
This work was financially supported by the NSFC (21871168 and 22071139), the Priority Academic Program Development of Jiangsu Higher Education Institutions, and the Top-notch Academic Programs Project of Jiangsu Higher Education Institutions.
Author information
Authors and Affiliations
Contributions
W.-L.D. conceived and designed the study. S.-B.Y., R.W., Z.-G.L., and A.-N.L. performed the experiments. S.-B.Y., C.W., and W.-L.D. discussed the data and prepared the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Zhipeng Zhang and the other anonymous reviewers for their contribution to the peer review of this work.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yan, SB., Wang, R., Li, ZG. et al. Copper-catalyzed asymmetric C(sp2)–H arylation for the synthesis of P- and axially chiral phosphorus compounds. Nat Commun 14, 2264 (2023). https://doi.org/10.1038/s41467-023-37987-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-37987-8
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
