
Abstract
The surface electron density significantly affects the photocatalytic efficiency, especially the photocatalytic CO2 reduction reaction, which involves multi-electron participation in the conversion process. Herein, we propose a conceptually different mechanism for surface electron density modulation based on the model of Au anchored CdS. We firstly manipulate the direction of electron transfer by regulating the vacancy types of CdS. When electrons accumulate on vacancies instead of single Au atoms, the adsorption types of CO2 change from physical adsorption to chemical adsorption. More importantly, the surface electron density is manipulated by controlling the size of Au nanostructures. When Au nanoclusters downsize to single Au atoms, the strong hybridization of Au 5d and S 2p orbits accelerates the photo-electrons transfer onto the surface, resulting in more electrons available for CO2 reduction. As a result, the product generation rate of AuSA/Cd1−xS manifests a remarkable at least 113-fold enhancement compared with pristine Cd1−xS.
Similar content being viewed by others
Introduction
As a representative greenhouse gas, carbon dioxide (CO2) has brought out serious environmental problems needing to solve urgently1,2. Photocatalytic CO2 conversion technology has attracted extensive interest, since the conversion of CO2 into high value-added chemicals could potentially be achieved under relatively mild conditions, which facilitates its resource utilization3,4. Generally, photocatalytic CO2 conversion is a typical photoreduction reaction, which needs electrons to participate in5,6,7. Different from other photoreduction reactions, such as hydrogen evolution reaction (two electrons), photocatalytic CO2 conversion is an inherently multi-electron reduction process8,9,10. To this end, photocatalysts are required to offer abundant electrons for the CO2 molecule to promote its transformation in the reaction process11.
Previous studies suggest that metals, especially noble metals, have been commonly introduced to enhance the surface electron density of photocatalysts12,13. It is generally agreed that the photocatalytic performance is greatly dependent on the metal center and its local coordination. Since the local electron density will be redistributed after introducing metal and the majority of electrons are accumulated around the metal and its surrounding atoms14,15, many efforts have focused on changing the metal center and/or ligands to achieve high efficiency for CO2 photoreduction. With in-depth research, it is further found that electron transfer is tremendously dependent on the size of metal nanostructures or the type of local coordination16,17. For instance, Wu et al. reported that the electrons have transferred from single Pt atoms to sulfur-doped carbon support and the direction of electron transfer will be reversed after loading Pt nanoclusters16. Meanwhile, Dai et al. found that the types of vacancies (boron or nitrogen vacancies) could affect the electron transfer direction between Pt nanoparticles and BN nanosheets17. Hence, it is speculated that the electrons may be localized at other sites instead of metal nanostructures. Indeed, previous studies suggest that the interaction of adsorbed CO2 molecule and metal site is relatively weak since the formed metal–C or metal–O bonds are weaker than the highly stable C–O bonds in CO2 molecule8,18,19. This will lead to the easy cleaving of metal–C or metal–O bonds during the reaction process, hindering the further transformation of CO28. The above considerations suggest that controlling the size of metal nanostructures and/or the type of vacancies might realize the accumulation of electrons at other sites instead of metal nanostructures, which is likely to promote the adsorption and conversion of CO2.
For this purpose, CdS is selected to be a model material due to the easy-fabricated vacancies and favorable solar light response20,21,22. According to the pre-experiments in Supplementary Fig. 1, Au is chosen as the dopant metal23,24,25. We have designed and fabricated a series of Au-anchored CdS photocatalysts and reported a conceptually different mechanism for modulating surface electron density to promote the conversion of CO2. As displayed in Fig. 1, when the type of vacancies changes from S vacancies to Cd vacancies, the direction of electron transfer will be reversed after anchoring single Au atoms, resulting in the accumulation of electrons in neighboring Cd vacancies instead of single Au atoms. Benefiting from this, CO2 could be chemically bonded on the Cd vacancies instead of physical adsorption on single Au atoms to promote its effective activation.

Schematic illustration of single Au atoms in CdS to promote CO2 photoreduction.
Results
Catalyst characterization
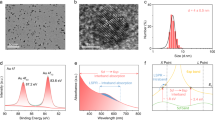
Based on the above considerations, the CdS with Cd vacancies (Cd1−xS) is first prepared by the hydrothermal method. X-ray diffraction (XRD) pattern and transmission electron microscopy (TEM) images (cf. Fig. 2a and Supplementary Fig. 2a, b) of the synthesized sample display that hexagonal CdS with the nanoparticle morphology is fabricated. Meanwhile, the electron paramagnetic resonance (EPR) signals with the g factor of 1.99 and 2.03 are observed for the synthesized sample (cf. Supplementary Fig. 3), which are ascribed to sulfur atoms surrounding Cd vacancies in hexagonal CdS26,27,28. According to the element analysis, the ratios of Cd and S are quantitated as 0.76:1 (cf. Supplementary Table 1). The above results confirm the existence of Cd vacancies in the synthesized sample. After Au loading, XRD patterns of both 1%Au/Cd1−xS and 2%Au/Cd1−xS samples have rarely changed compared with Cd1−xS (cf. Fig. 2a). For 1%Au/Cd1−xS, there are no Au nanostructures observed in TEM and HRTEM images (cf. Supplementary Fig. 2c, d). Hence, to reveal the atomic-resolution structure of 1%Au/Cd1−xS sample, aberration-corrected high-angle annular dark-field scanning TEM is shown in Fig. 2b. The bright spots corresponding to heavy Au atoms (marked by yellow circles) are homogeneously dispersed, indicating that the Au atoms are atomically dispersed on the Cd1−xS. Meanwhile, the element mapping of the 1%Au/Cd1−xS (cf. Fig. 2c) shows the uniform distribution of Au, Cd, and S elements. With the increase of loading content, some nanoparticles are observed in TEM and HRTEM images of 2%Au/Cd1−xS sample (cf. Supplementary Fig. 2e, f). The interplanar distance in these nanoclusters is ca 1.15 Å corresponding to the (222) planes of metallic Au29. This is indicated that part of single Au atoms is aggregated together to form the Au nanoclusters with the increase of loading content. According to the size distributions of Au nanoclusters and element mapping in Supplementary Fig. 4, the Au nanoclusters are homogeneously distributed on Cd1−xS and the sizes of Au nanoclusters are mainly in the range of 1–3 nm. The above results indicate that the single Au atoms (AuSA) and Au clusters (AuNC) are successfully introduced in the 1%Au/Cd1−xS and 2%Au/Cd1−xS samples, respectively. To distinguish Au nanostructures in different samples, 1%Au/Cd1−xS and 2%Au/Cd1−xS samples are referred as AuSA/Cd1−xS and AuNC/Cd1−xS for simplicity in the following study.

a XRD patterns of Cd1-xS, 1%Au/Cd1-xS, and 2%Au/Cd1-xS samples. b HAADF-STEM image of 1%Au/Cd1-xS, single atomic Au sites high-lighted by yellow circles. c Elemental mappings of 1%Au/Cd1-xS. d XAFS analysis of AuSA/Cd1-xS: Au L3 XANES spectra of the sample and the reference foil. e Corresponding k1-weighted Fourier transform (FT) EXAFS spectra.
To investigate the coordination environments of Au in the AuSA/Cd1−xS sample, Au L3-edge X-ray absorption near-edge structure (XANES) and extended X-ray absorption fine structure (EXAFS) spectra are presented in Fig. 2d, e. The white line peak of AuSA/Cd1−xS sample is different from that of the reference Au foil. The EXAFS spectrum shows that AuSA/Cd1−xS exhibits a peak near 1.7 Å without any significant Au–Au contribution between 2 and 4 Å, confirming the atomic dispersion of Au on Cd1−xS. The quantitative structural parameters of Au in the AuSA/Cd1−xS (cf. Supplementary Table 2) demonstrate that one Au atom is bonded with two or three S atoms to form the Au–S bonds. To further identify the loading position of single Au atoms, EPR spectra are carried out in Supplementary Fig. 5. The intensities of EPR signals assigned to Cd vacancies decreased after introducing single Au atoms, indicating that part of Cd vacancies are covered by single Au atoms. Furthermore, the models of Au-loaded Cd1−xS surface with different sites (surface S and Cd vacancy) are constructed by density functional theory (DFT) method (cf. Supplementary Table 3). The calculated formation energy of single Au atom anchored on Cd vacancy is −5.30 eV, which is much more negative than that on surface S atoms (Ef = −1.89 eV). It is hence suggested that single Au atoms are anchored on the Cd vacancies to form the Au–S bonds in the AuSA/Cd1−xS sample.
Similarly, the CdS with S vacancies (CdS1−x) is synthesized by the hydrothermal method (cf. Supplementary Fig. 6). There are no EPR signals assigned to S vacancies observation in the synthesized sample (cf. Supplementary Fig. 7). To confirm the existence of S vacancies, the Cd K-edge XANES and EXAFS spectra of CdS1−x and pristine CdS (CdS) samples are shown in Supplementary Fig. 8. The white line peak of the CdS1−x is similar to that of the CdS and both of them are different from that of the reference Cd foil. According to the EXAFS spectra, the CdS sample shows bulk crystalline CdS. In the case of the CdS1−x sample, intensity of the first coordination shell is lower than that of CdS, implying fewer S-neighbors than in the bulk CdS. Combined with the element analysis, the ratio of Cd and S are quantitated as 1:0.73 (cf. Supplementary Table 1). It is confirmed that there are S vacancies in the CdS1−x sample.
Furthermore, the same impregnation method is employed to synthesize the 1%Au/CdS1−x and 2%Au/CdS1−x (cf. Supplementary Fig. 6). The XANES and EXAFS spectra of CdS1−x and 1%Au/CdS1−x are shown in Supplementary Fig. 9 and 10. It is found that the intensity of the first coordination shell in CdS1−x sample increases after loading Au, suggesting that part of S vacancies are filled with Au (cf. Supplementary Fig. 9). Meanwhile, in the case of 1%Au/CdS1−x, there are no significant peaks assigned to Au–Au bonds found in the range of 2–4 Å, confirming the atomic dispersion of Au on CdS1−x (cf. Supplementary Fig. 10). The above results suggest that single Au atoms are anchored on the S vacancies. Hence, the 1%Au/CdS1−x sample is referred to AuSA/CdS1−x.
In brief, the CdS samples with different types of vacancies (Cd and S vacancies) are successfully fabricated and the introduced single Au atoms are anchored on the vacancies. In the case of the Au-anchored Cd1−xS systems, with the increase of loading content, Au has changed from single atoms into nanoclusters.
Photocatalytic CO2RR measurement
As depicted in Fig. 3 and Supplementary Fig. 11, photocatalytic CO2 reduction tests are carried out to evaluate the effect of Au on photoreduction efficiency. Cd1−xS shows low CO and CH4 generation rates of 0.2 and 0.1 μmol g−1 h−1, respectively. After anchoring Au nanoclusters, the product generation rates are enhanced. It is interesting that with the introduction of single Au atoms, the CO, CH4, and H2 generation rates are highly promoted to 32.2, 11.3, and 7.9 μmol g−1 h−1, respectively. It manifests a remarkable 161-fold and 113-fold enhancement over Cd1−xS as well as 5-fold, 4-fold, and 2-fold enhancement compared with AuNC/Cd1−xS. For CdS1−x systems (cf. Fig. 3b), the introduction of Au brings out an enhancement of CO2 photoreduction efficiency as well. Among all the samples, AuSA/CdS1−x reveals the highest CO2 photoreduction efficiency with the CO, CH4, and H2 generation rates of 3.7, 0.4, and 0.2 μmol g−1 h−1, which is a 2-fold, 4-fold, and 2-fold enhancement compared over the pristine CdS1−x. The above results indicate that, compared with S vacancies, single Au atoms bring much more significant improvement for CO2 photoreduction efficiency when they are anchored on Cd vacancies. Meanwhile, when Au changes from single atoms into nanoclusters, the CO2 photoreduction efficiency is gradually decreased. Furthermore, the photocatalytic CO2RR performance of AuSA/Cd1−xS is relatively satisfactory compared with that of other metal sulfide, CdS-based, and noble-metal based photocatalysts in the published works (cf. Supplementary Table 4)8,30,31,32,33,34,35,36,37,38. To confirm that the CO and CH4 products are generated from the CO2 conversion driven by AuSA/Cd1−xS, the isotope labeling is conducted using 13CO2 as the reactant for qualitative analysis. Supplementary Fig. 12 displays that the generated CO and CH4 originate from the CO2 conversion driven by AuSA/Cd1−xS sample instead of desorption of adsorbed species from the surface. Besides, the peak at m/z = 32 is detected, which is assigned to generated oxidation product O2. With a comparison of XRD, TEM, and HRTEM images between fresh and used AuSA/Cd1−xS samples (cf. Supplementary Fig. 13 and 14), the single Au atoms is not aggregated during the reaction process, indicating good stability.

Average yield in the photocatalytic conversion of CO2 with H2O in the presence of water vapor under UV-visible light. a Over Cd1-xS, AuSA/Cd1-xS, and AuNC/Cd1-xS samples. b Over CdS1-x, AuSA/CdS1-x, and 2%Au/CdS1-x samples. The error bar was drawn based on the calculated standard error of two parallel tests.
Discussion
CO2 adsorption during photocatalysis
To uncover the main factors of enhanced photocatalytic CO2 reduction performance, solar light absorption is first investigated by UV-vis spectroscopy (cf. Supplementary Fig. 15). The absorption edges of AuSA/Cd1−xS and AuNC/Cd1−xS have barely changed compared with Cd1−xS. For the CdS1−x system, the introduction of Au does not change the absorption edge as well. This suggests that solar light absorption is not the determining factor in the photocatalytic CO2 reduction performance. After light illumination, the excited electrons will finally transfer to the surface of the catalyst and combine with the adsorbed species to promote their conversion. The conversion of CO2 involves the following steps. The adsorption of CO2 on the surface of photocatalyst is the prerequisite step, which is relatively difficult due to the closed-shell electronic configuration of CO2. Then, adsorbed CO2 will obtain electrons from the surface to generate the carbon active species (e.g., CO2−, HCO3−, CO32−). These first two steps are aimed to activate the C–O bond of CO2 molecule. Finally, these species will be further transformed into the desired products (e.g., CO, CH4), which need abundant electrons to take part in to promote their effective transformation. Hence, the adsorption behavior of CO2 not only determines the occurrences of reaction, but significantly affects the conversion efficiency.
Based on these considerations, in situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) is performed to detect the generated intermediates during the CO2 adsorption process on the surfaces of Cd1−xS and CdS1−x systems (cf. Fig. 4 and Supplementary Fig. 16). For pristine Cd1−xS (cf. Fig. 4a), there are some absorption bands assigned to bicarbonate (HCO3−; δ(COH): 1200 and 1166 cm−1), monodentate carbonate (m-CO32−; ν(C–O): 1140–1030 cm−1) and carboxylate (CO2−; ν(O–C–O)s: 1266 cm−1) detected with the prolonged time of adsorption39. Meanwhile, the band assigned to COOH* is detected at 1634 cm−1,40,41 suggesting that the adsorbed CO2 is combined with H* and partly converted to the COOH*. After anchoring Au clusters, the intensities of absorption bands assigned to HCO3−, CO2−, and COOH* have decreased. There is a new band appearing at 1043 cm−1, which is assigned to HCO*42,43. This indicates that part of the formed COOH* is converted to HCO*. Different from it, some of the absorption bands (HCO3− and COOH*) almost disappear over AuSA/Cd1−xS. It should be noted that there is a new band detected at 1110 cm−1 assigned to H3CO*44,45. The intensity of the absorption bands assigned to HCO* is much lower than that over AuNC/Cd1−xS, revealing that the majority of formed COOH* are quickly converted into HCO* and then transform into H3CO*. The conversion pathway of CO2 over different samples in Au/Cd1−xS systems during the adsorption process could be deduced as follows:
It is indicated that the introduction of Au significantly promotes the further conversion of CO2 on Cd1−xS surface. When Au nanoclusters downsize to single Au atoms, the carbon active species can be deeply converted into H3CO*, which is beneficial for the subsequent reaction. Different from the Cd1−xS system, with the introduction of Au nanostructures, there are no absorption bands assigned to new intermediates appearing on CdS1−x surface in the process of CO2 adsorption (cf. Supplementary Fig. 16). It could be concluded that the introduction of Au has a slight effect in the CO2 adsorption and conversion on CdS1−x surface.

a In situ DRIFTS spectra of the 12CO2 adsorption process over Cd1-xS, AuNC/Cd1-xS, and AuSA/Cd1-xS. b In situ DRIFTS spectra of the 13CO2 adsorption process over AuSA/Cd1-xS. c The comparison between in situ DRIFTS spectra of 12CO2 and 13CO2 adsorption process over AuSA/Cd1-xS and AuSA/CdS1-x. d The adsorption models of CO2 on different surfaces on single Au atom in AuSA/CdS1-x and on Cd vacancy in AuSA/Cd1-xS.
The above discussions suggest that the types of vacancies and the size of Au nanostructures present a different effect on the intermediates in the CO2 adsorption process. In previous studies, reaction sites have possibly affected the generation of intermediates46. Based on it, Au nanostructures (Au site) and vacancies (vacancy site) are selected to be the different CO2 adsorption sites. The CO2 adsorption models on Au/Cd1−xS and Au/CdS1−x are constructed by the DFT method (cf. Fig. 5 and Supplementary Fig. 17 and 18). The corresponding adsorption energies are summarized in Supplementary Table 5. For the pristine CdS1−x, CO2 will spontaneously adsorb on the S vacancies due to the negative adsorption energy. After anchoring single Au atoms, the adsorption energies change slightly compared with that of pristine CdS1−x. According to the comparison of adsorption energies, CO2 prefers to adsorb on single Au atoms instead of S vacancies. Similarly, in the case of pristine Cd1−xS, CO2 will also spontaneously adsorb on the Cd vacancies (Eads = −0.43 eV). With the introduction of single Au atoms, the adsorption energy has significantly decreased, implying that single Au atoms promotes the adsorption of CO2 on Cd1−xS surface. It should be noted that CO2 is more likely to adsorb on the Cd vacancies instead of single Au atoms due to the more negative adsorption energy. To further identify the adsorption site in AuSA/Cd1−xS, the different surfaces are probed by N2 sorption (cf. Supplementary Fig. 19). The specific surface area of Cd1−xS (25.4 m2/g) has increased after anchoring single Au atoms (33.9 m2/g). It is suggested that the introduction of single Au atoms enhances the amount of adsorption site. However, the CO2 adsorption capacity of Cd1−xS is decreased from 72.9 to 66.2 μmol g−1 (cf. Supplementary Fig. 20) after introducing single Au atoms. It is hence confirmed that the real adsorption sites of CO2 in AuSA/Cd1−xS should be the vacancies instead of single Au atoms. Furthermore, combined with in situ DRIFTS spectra, CO is used as the probe to identify the reactive reaction site of CO2 reduction on AuSA/Cd1−xS surface (cf. Supplementary Fig. 21). The absorption bands at 2170 and 2109 cm−1 are assigned to gaseous CO47. Furthermore, the absorption band assigned to C=O bond and S–O are detected on the surface (v(C=O): 1621 cm−1; v(S–O): 1209 and 1162 cm−1)47,48,49,50,51. It is suggested that the CO molecule adsorbs on the Cd vacancy sites and combines with the S atoms to form C=O–S bonds. Meanwhile, there are some absorption bands detected at 1100 and 1044 cm−1, which are assigned to HCO* and H3CO*. The above results indicate that when CO adsorbs on Cd vacancy, it will combine with H* to further convert into HCO* and H3CO*, which is in accordance with the observations in DRIFTS spectra of the CO2 adsorption process.

Configurations of CO2 adsorption: a on Cd1-xS; b AuSA/Cd1-xS; d CdS1-x; e AuSA/CdS1-x. PDOS of CO2 and adsorption site: c. in AuSA/Cd1-xS; f AuSA/CdS1-x.
Furthermore, according to the partial density of states (PDOS) (cf. Fig. 5f), it is observed that the interaction between CO2 and single Au atoms in the AuSA/CdS1−x is weak. Different from it, there is a strong interaction between CO2 and Cd vacancies in the AuSA/Cd1−xS (cf. Fig. 5c). More importantly, the C and O atoms of CO2 molecule are bonded with two S atoms of the surface to form the S–C and S–O bonds (cf. Fig. 5b). As a result, the electrons transfer from the surface to the bonded CO2 molecule (0.52 e) to promote its activation, resulting in C–O–C bond angle bending and elongation of both C–O bonds. To further prove the effect of different adsorption sites in the CO2 adsorption behavior, isotope labeling is carried out on AuSA/Cd1−xS and AuSA/CdS1−x using 13CO2 instead of 12CO2 for the DRIFTS analysis (cf. Fig. 4b and Supplementary Fig. 16d). It is observed that, compared with using 12CO2 as reactants, similar absorption bands are detected on both AuSA/Cd1−xS and AuSA/CdS1−x under 13CO2 atmosphere. Meanwhile, the positions of all the absorption bands have a 13C frequency shift toward low wavenumber in varying degrees (2–16 cm−1). In the process of 12CO2 adsorption (cf. Fig. 4c), there is an absorption band detected at 1280 cm−1 on both AuSA/Cd1−xS and AuSA/CdS1−x, which is assigned to the vibration of C–O bonds in CO2− (adsorption state of CO2). In the process of 13CO2 adsorption, there is a small 13C frequency shift (6 cm−1) occurring on the surface of AuSA/CdS1−x. It should be noted that there is a large 13C frequency shift (16 cm−1) of this absorption band detected on the surface of AuSA/Cd1−xS. It is indicated that, compared with AuSA/CdS1−x surface, the adsorbed CO2 obtains more electrons from AuSA/Cd1−xS surface to weaken the C–O bond, leading to a larger 13C frequency shift. Meanwhile, according to the value of 13C frequency shift, more than one C–O bonds might be bonded to the surface11,52. Based on the above discussions, it is speculated that CO2 molecule is physically adsorbed on single atoms of AuSA/CdS1−x surface. The formation of bonds and effective electron transfer between surface and CO2 indicate that CO2 molecule is chemically adsorbed on Cd vacancies of AuSA/Cd1−xS surface. In brief, when the type of vacancies changes from S vacancies to Cd vacancies, the CO2 adsorption sites change from single Au atoms to the vacancies. Benefiting from this, CO2 could be chemically bonded on the Cd vacancies instead of physical adsorption on single Au atoms. The strong interaction between CO2 and adsorption sites (Cd vacancies) profits the formation of stable electron-transfer channels. As a result, CO2 could obtain more electrons from the adsorption sites to promote its deep transformation.
To identify the internal factors affecting adsorption sites, the charge population of Cd1−xS and CdS1−x surfaces after anchoring Au nanostructures is explored by DFT calculations (cf. Fig. 6a and 6b). For the Au anchored on Cd vacancies, the electrons (0.23 e) will transfer from single Au atoms to Cd1−xS to enhance the surface electron density. More importantly, the majority of electrons are localized at the neighboring Cd vacancy site. On the contrary, the direction of electron transfer between Au and CdS1−x is reversed, resulting in the localization of electrons at single Au atoms (0.40 e). To further identify the electron transfer between Au and Cd1−xS or CdS1−x, X-ray photoelectron spectroscopy (XPS) spectra are presented in Fig. 6. For Cd1−xS system (cf. Fig. 6c–h), the peaks indexed to the Cd–S bond in Cd 3d and S 2p spectra have a shift toward low binding energy (0.2–0.3 eV) compared with the pristine Cd1−xS, indicating that Cd1−xS receives electrons from the single Au atoms. While for CdS1−x system (cf. Fig. 6i–n), the peaks corresponding to the Cd–S bond barely change after anchoring single Au atoms. It should be noted that the peaks in Au 4f spectra of AuSA/CdS1−x system are located at 87.8 and 84.1 eV and the binding energies are much lower (c.a. 0.5 eV) than that of AuSA/Cd1−xS. This suggests that the electrons are localized at single Au atoms instead of CdS1−x in AuSA/CdS1−x system. To identify the internal factors affecting the direction of electron transfer between single Au atoms and Cd1−xS or CdS1−x, the potential energy plots are given in Supplementary Fig. 22. In the case of AuSA/CdS1−x, the potential energy of CdS1−x is much lower than that of single Au atoms. Benefiting from it, there is a driving force promoting electron transfer from CdS1−x to single Au atoms. Opposite to that, the potential energy of single Au atoms is lower than that of Cd1−xS, resulting in the reversal of electron transfer direction. Hence, it is observed that the electrons are localized at the single Au atoms when they anchor on S vacancies. After single Au atoms anchored on Cd vacancies, the electrons will conversely transfer from single Au atoms to Cd1−xS and accumulate on Cd vacancies.

Electron transfer between Au and CdS with different types of vacancies: a Cd vacancies; b S vacancies. XPS spectra: c–e Cd1-xS; f–h AuSA/Cd1-xS; i–k CdS1-x; l–n AuSA/CdS1-x samples.
Photo-induced carrier transfer during photocatalysis
According to the DFT and XPS spectra (cf. Supplementary Fig. 23–25), it is found that with the increased content of Au, the electron transfer is gradually weakened for both Cd1−xS and CdS1−x systems. Hence, to further identify the underlying roles of Au content on the surface electron density, the Au/Cd1−xS system is selected as the representative in the following discussion. In general, electron transfer might be related to the local electronic structure. According to PDOS in Supplementary Fig. 26, compared with the AuNC/Cd1−xS, the hybridization between Au 5d and S 2p orbits in Au–S bonds is much stronger for AuSA/Cd1−xS. As a result, more electrons are transferring from Au 5d orbits in single Au atoms to Cd1−xS compared with Au clusters (cf. Supplementary Fig. 27). This is identified by XPS analysis in Supplementary Fig. 25. The peaks indexed to the Cd–S bond in Cd 3d and S 2p spectra for AuSA/Cd1−xS have a shift toward low binding energy (0.2–0.3 eV) and the Au 4f spectrum shifts to high binding energy (0.4–0.6 eV) compared with AuNC/Cd1−xS. It is indicated that the interaction between single Au atoms and Cd vacancies is much stronger than that with Au clusters. Benefiting from it, under light illumination, it is observed that the lifetimes of photogenerated electrons in Cd1−xS are decreased from 2.95 to 0.44 ns (AuSA/Cd1−xS) and 2.22 (AuNC/Cd1−xS) (cf. Supplementary Fig. 28). It could be concluded that, compared with Au nanoclusters, the stronger interaction between single Au atoms and Cd vacancies significantly accelerates electron transfer to the surface to enhance the surface electron density under light illumination. Meanwhile, the intensity of transient photocurrent response is significantly increased with the introduction of Au (cf. Fig. 7a). AuSA/Cd1−xS sample reveals the highest intensity compared with AuNC/Cd1−xS and Cd1−xS. It is suggested that single Au atoms offer a significantly higher enhancement in the surface electron density of Cd1−xS compared with Au clusters. This is further identified by the surface photovoltage (SPV) in Fig. 7b. It should be noted that two peaks are appearing in the SPV spectra of Cd1−xS. The peak at c.a. 400 nm corresponds to the electrons transferring from the top of valence band to the bottom of conduction band. The other one at c.a. 500 nm is assigned to the defect level introduced by Cd vacancy on the surface which is observed in the band structure (cf. Supplementary Fig. 29). Interestingly, intensities of the two peaks increase after anchoring Au nanostructures, especially the single Au atoms. It is suggested that compared with Au nanoclusters, the introduction of single Au atoms significantly promotes the enhancement of electron density on the Cd1−xS surface as well as the Cd vacancies under light illumination. Meanwhile, a similar trend is observed in transient photocurrent response and SPV spectra of Au/CdS1−x system (cf. Supplementary Fig. 30).

a Transient photocurrent response and b SPV spectra of Cd1-xS, AuSA/Cd1-xS, and AuNC/Cd1-xS samples. c Energy barriers of CO2 reduction on AuSA/Cd1-xS and AuNC/Cd1-xS. d Reaction profiles of CO2 transition into COOH on AuSA/Cd1-xS and AuNC/Cd1-xS.
As presented in Fig. 7c and Supplementary Fig. 31, the processes of CO2 reduction on AuSA/Cd1−xS and AuNC/Cd1−xS are further simulated by DFT calculations. Most of the energy barriers on AuSA/Cd1−xS are lower than that of AuNC/Cd1−xS, indicating that single Au atoms greatly facilitate CO2 reduction compared with Au nanoclusters. More interestingly, the energy barrier of the first step (CO2+H→COOH) on AuSA/Cd1−xS is remarkably decreased by 91.1% relative to that over AuNC/Cd1−xS. This suggests that the accumulated electrons in Cd vacancies on AuSA/Cd1−xS not only promote the activation of CO2 molecule, but also facilitate its further transformations. It is identified that there is no significant absorption band assigned to COOH* observed among in situ DRIFTS test over AuSA/Cd1−xS. Furthermore, extra electron is introduced to create an electron-rich condition in the process of CO2 transition into COOH in Fig. 7d53. Interestingly, the energy barrier on AuSA/Cd1−xS presents a larger decrease (41%) compared with that on AuNC/Cd1−xS (34%). It is suggested that, compared with AuNC/Cd1−xS, the enhancement of surface electron density much more facilitates the occurrence of CO2 reduction catalyzed by AuSA/Cd1−xS.
In summary, we have designed and fabricated a series of Au/CdS photocatalysts. On the one hand, the types of vacancies highly affect the direction of electron transfer. That is the electrons will transfer from CdS1−x to Au in CdS1−x system. Quite different from it, the direction of electron transfer has been reversed in Cd1−xS system, resulting in the accumulation of electrons in Cd vacancies. Hence, the adsorption of CO2 is enabled in vacancy sites instead of Au sites. More importantly, there is a stable electron-donation channel forming between CO2 and Cd vacancies, accelerating the transfer of accumulated electrons to CO2 for its effective activation. On the other hand, under light illumination, when Au nanostructures change from Au nanoclusters to single Au atoms, the surface electron density is significantly increased and promotes much more electrons accumulating on the surface as well as the Cd vacancies, which is beneficial for the subsequent reaction. As a result, the CO and CH4 generation rates of AuSA/Cd1−xS manifest a remarkable 160-fold and 113-fold enhancement compared with Cd1−xS as well as the H2 generation rate is promoted from 0 to 7.9 μmol g−1 h−1.
Methods
Synthesis of Cd1−xS, CdS1−x and CdS
The Cd1−xS and CdS1−x were synthesized via a modified hydrothermal method54. For Cd1−xS, the synthesis process as follows: first, CdCl2·5H2O (4 mM) was dissolved in 20 mL aqueous solution (pH = 1, marked as solution A). Meanwhile, another 20 mL aqueous Na2S·9H2O (8 mM) solution was prepared, marked as solution B. Then, solution B was added dropwise into solution A under continuous stirring. The mixed solution was transferred into a Teflon-lined stainless steel autoclave (inner volume: 50 mL) and held at 140 °C for 18 h in an oven. After cooling to room temperature naturally, the product was centrifuged and washed with water and ethyl alcohol several times. Finally, the obtained product was dried overnight at 60 °C under vacuum condition. The synthesis process of CdS1−x and CdS were the same as that of Cd1−xS except that 1 and 4 mM of Na2S.9H2O were added in solution B.
Synthesis of Au/Cd1−xS and Au/CdS1−x
The Au/Cd1−xS and Au/CdS1−x were synthesized by the impregnation method. For 1%Au/Cd1−xS, 100 mg of Cd1−xS was dispersed in 30 mL water and adding 0.2 mL HAuCl4 solution (5 mg/mL). Then, the mixed solution was stirred at 70 °C for 6 h. The product was centrifuged and washed with water and ethyl alcohol several times and dried overnight at 60 °C under vacuum condition. The synthesis procedure of 2%Au/Cd1−xS was the same as that of 1%Au/Cd1−xS, except that 0.4 mL of HAuCl4 solution was used in the experiment. Meanwhile, Au/CdS1−x samples were synthesized by the same method using CdS1−x instead of Cd1−xS.
Photocatalytic performance measurement
Photocatalytic CO2 reduction performance test was carried out in a closed gas system (volume: 276 mL) at atmospheric pressure and ambient temperature. First, 30 mg of photocatalyst was dispersed with H2O in a petri dish (ø = 6 cm) and dried at 70 °C under vacuum condition. Then, petri dish was put on a stainless steel breaker with 1 mL of deionized water in the reactor, which was filled with the CO2 (concentration: 1000 ppm; carrier gas: Ar). A 300 W Xe lamp was used as the UV-vis light source (intensity: 600 mW/cm2). Every 2 h, 1 mL of gas was taken from the reactor for qualitative analysis by gas chromatography (GC7900, Tianmei Analytical Instrument Co., Ltd., China). CO and CH4 were detected by a flame ionization detector (FID) and a thermal conductivity detector (TCD) was used for detecting H2. The isotope test was carried out by using 13CO2 as the carbon source with the same reaction set as mentioned above and the gas products were analyzed by mass spectrometry (OmniStarTM, Pfeiffer Vacuum, Germany).
In situ DRIFTS investigation
In situ DRIFTS measurements were performed on a TENSOR II FT-IR spectrometer (Bruker, Germany). The spectrometer was equipped with an in situ diffuse reflectance cell (Harrick) and a high-temperature reaction chamber (HVC). A mixture of 12CO2 (20% CO2/Ar) and H2O vapors (humidity: 5%Rh) with a flow rate of 17 mL min−1 was introduced into the Harrick cell. In case of AuSA/Cd1−xS and AuSA/CdS1−x samples, 13CO2 (20% 13CO2/Ar) was used instead of 12CO2 to perform the isotope test. For CO probe test, a mixture of CO (10% CO/Ar) and H2O vapors (humidity: 5%Rh) with a flow rate of 17 mL min−1 was introduced into the Harrick cell. Before the measurement, the samples were pretreated at 100 °C for 10 min under Ar atmosphere.
Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Code availability
The code to reproduce the results is available from the corresponding author upon reasonable request.
References
Yi, Q., Li, W., Feng, J. & Xie, K. Carbon cycle in advanced coal chemical engineering. Chem. Soc. Rev. 44, 5409–5445 (2015).
Shi, R. et al. Efficient wettability-controlled electroreduction of CO2 to CO at Au/C interfaces. Nat. Commun. 11, 3028 (2020).
Zhao, Y. et al. Two-dimensional-related catalytic materials for solar-driven conversion of COx into valuable chemical feedstocks. Chem. Soc. Rev. 48, 1972–2010 (2019).
Li, Y. et al. Selective light absorber-assisted single nickel atom catalysts for ambient sunlight-driven CO2 methanation. Nat. Commun. 10, 2359 (2019).
Hao, L. et al. Surface-halogenation-induced atomic-site activation and local charge separation for superb CO2 photoreduction. Adv. Mater. 31, 1900546 (2019).
Chen, F. et al. Macroscopic spontaneous polarization and surface oxygen vacancies collaboratively boosting CO2 photoreduction on BiOIO3 single crystals. Adv. Mater. 32, 1908350 (2020).
Hu, C., et al. Coupling piezocatalysis and photocatalysis in Bi4NbO8X (X = Cl, Br) polar single crystals. Adv. Funct. Mater. 30, 1908168 (2019).
Li, X. et al. Selective visible-light-driven photocatalytic CO2 reduction to CH4 mediated by atomically thin CuIn5S8 layers. Nat. Energy 4, 690–699 (2019).
Kim, W., Seok, T. & Choi, W. Nafion layer-enhanced photosynthetic conversion of CO2 into hydrocarbons on TiO2 nanoparticles. Energ. Environ. Sci. 5, 6066–6070 (2012).
Varghese, O. K., Paulose, M., LaTempa, T. J. & Grimes, C. A. High-rate solar photocatalytic conversion of CO2 and water vapor to hydrocarbon fuels. Nano Lett. 9, 731–737 (2009).
Cao, Y. et al. B-O bonds in ultrathin boron nitride nanosheets to promote photocatalytic carbon dioxide conversion. ACS Appl. Mater. Inter. 12, 9935–9943 (2020).
Van Deelen, T. W., Hernández Mejía, C. & de Jong, K. P. Control of metal-support interactions in heterogeneous catalysts to enhance activity and selectivity. Nat. Catal. 2, 955–970 (2019).
Liam, H. F. & Andersson, G. G. Metal clusters on semiconductor surfaces and application in catalysis with a focus on Au and Ru. Adv. Mater. 32, 1904122 (2020).
Fang, S. et al. Uncovering near-free platinum single-atom dynamics during electrochemical hydrogen evolution reaction. Nat. Commun. 11, 1029 (2020).
O’Connor, N. J., Jonayat, A. S. M., Janik, M. J. & Senftle, T. P. Interaction trends between single metal atoms and oxide supports identified with density functional theory and statistical learning. Nat. Catal. 1, 531–539 (2018).
Yan, Q. et al. Reversing the charge transfer between platinum and sulfur-doped carbon support for electrocatalytic hydrogen evolution. Nat. Commun. 10, 4977 (2019).
Zhu, W. et al. Taming interfacial electronic properties of platinum nanoparticles on vacancy-abundant boron nitride nanosheets for enhanced catalysis. Nat. Commun. 8, 15291 (2017).
Ghuman, K. K. et al. Photoexcited surface frustrated lewis pairs for heterogeneous photocatalytic CO2 reduction. J. Am. Chem. Soc. 138, 1206–1214 (2016).
Wang, S., Guan, B. Y., Lu, Y. & Lou, X. W. D. Formation of hierarchical In2S3-CdIn2S4 heterostructured nanotubes for efficient and stable visible light CO2 reduction. J. Am. Chem. Soc. 139, 17305–17308 (2017).
Cheng, L., Xiang, Q., Liao, Y. & Zhang, H. CdS-based photocatalysts. Energ. Environ. Sci. 11, 1362–1391 (2018).
Zhang, K., Kim, W., Ma, M., Shi, X. & Park, J. H. Tuning the charge transfer route by p-n junction catalysts embedded with CdS nanorods for simultaneous efficient hydrogen and oxygen evolution. J. Mater. Chem. A 3, 4803–4810 (2015).
Jin, J., Yu, J., Liu, G. & Wong, P. K. Single crystal CdS nanowires with high visible-light photocatalytic H2-production performance. J. Mater. Chem. A 1, 10927–10934 (2013).
Sankar, M. et al. Role of the support in gold-containing nanoparticles as heterogeneous catalysts. Chem. Rev. 120, 3890–3938 (2020).
Wan, J. et al. Defect effects on TiO2 nanosheets: stabilizing single atomic site Au and promoting catalytic properties. Adv. Mater. 30, 1705369 (2018).
Ishida, T., Murayama, T., Taketoshi, A. & Haruta, M. Importance of size and contact structure of gold nanoparticles for the genesis of unique catalytic processes. Chem. Rev. 120, 464–525 (2020).
Taylor, A. L., Filipovich, G. & Lindeberg, G. K. Identification of Cd vacancies in neutron-irradiated CdS by electron paramagnetic resonance. Solid State Commun. 9, 945–947 (1971).
Cavenett, B. C. Optically detected magnetic resonance (O.D.M.R.) investigations of recombination processes in semiconductors. Adv. Phys. 30, 475–538 (1981).
Nakaoka, Y. & Nosaka, Y. Electron spin resonance study of radicals produced on irradiated CdS powder. J. Am. Chem. Soc. 99, 9893–9897 (1995).
O’Keefe, M. et al. Sub-angstrom high-resolution transmission electron microscopy at 300 keV. Ultramicroscopy 89, 215–241 (2001).
Wei, Y. et al. Fabrication of inverse opal TiO2-supported Au@CdS core-shell nanoparticles for efficient photocatalytic CO2 conversion. Appl. Catal. B: Environ. 179, 422–432 (2015).
Wei, Y. et al. 3D ordered macroporous TiO2-supported Pt@CdS core-shell nanoparticles: design, synthesis and efficient photocatalytic conversion of CO2 with water to methane. J. Mater. Chem. A 3, 11074–11085 (2015).
Cai, S., Zhang, M., Li, J., Chen, J. & Jia, H. Anchoring single-atom Ru on CdS with enhanced CO2 capture and charge accumulation for high selectivity of photothermocatalytic CO2 reduction to solar fuels. Sol. RRL 5, 2000313 (2021).
Jiao, X. et al. Partially oxidized SnS2 atomic layers achieving efficient visible-light-driven CO2 reduction. J. Am. Chem. Soc. 139, 18044–18051 (2017).
Dong, C. et al. Size-dependent activity and selectivity of carbon dioxide photocatalytic reduction over platinum nanoparticles. Nat. Commun. 9, 1252 (2018).
Zhu, Z. et al. A hierarchical heterostructure of CdS QDs confined on 3D ZnIn2S4 with boosted charge transfer for photocatalytic CO2 reduction. Nano Res. 14, 81–90 (2021).
Zhang, P., Wang, S., Guan, B. Y. & Lou, X. W. Fabrication of CdS hierarchical multi-cavity hollow particles for efficient visible light CO2 reduction. Energ. Environ. Sci. 12, 164–168 (2019).
Zhou, M., Wang, S., Yang, P., Huang, C. & Wang, X. Boron carbon nitride semiconductors decorated with CdS nanoparticles for photocatalytic reduction of CO2. ACS Catal. 8, 4928–4936 (2018).
Zhao, G. et al. Efficient photocatalytic CO2 reduction over Co(II) species modified CdS in aqueous solution. Appl. Catal. B: Environ. 226, 252–257 (2018).
Baltrusaitis, J., Schuttlefield, J., Zeitler, E. & Grassian, V. H. Carbon dioxide adsorption on oxide nanoparticle surfaces. Chem. Eng. J. 170, 471–481 (2011).
Ulagappan, N. & Frei, H. Mechanistic study of CO2 photoreduction in Ti silicalite molecular sieve by FT-IR Spectroscopy. J. Phys. Chem. A 104, 7834–7839 (2000).
Wang, T. et al. Photoreduction of CO2 over the well-crystallized ordered mesoporous TiO2 with the confined space effect. Nano Energy 9, 50–60 (2014).
Yates, J. T. & Cavanagh, R. R. Search for chemisorbed HCO: the interaction of formaldehyde, glyoxal, and atomic hydrogen+CO with Rh. J. Catal. 74, 97–109 (1982).
Ewing, G. E., Thompson, W. E. & Pimentel, G. C. Infrared detection of the formyl radical HCO. J. Chem. Phys. 32, 927–932 (1960).
Wu, J. & Huang, C. W. In situ DRIFTS study of photocatalytic CO2 reduction under UV irradiation. Front. Chem. Eng. China 4, 120–126 (2010).
Liu, Y., Chen, S., Quan, X. & Yu, H. Efficient electrochemical reduction of carbon dioxide to acetate on nitrogen-doped nanodiamond. J. Am. Chem. Soc. 137, 11631–11636 (2015).
Wagner, A., Sahm, C. D. & Reisner, E. Towards molecular understanding of local chemical environment effects in electro- and photocatalytic CO2 reduction. Nat. Catal. 3, 775–786 (2020).
Smith, M. L., Kumar, N. & Spivey, J. J. CO adsorption behavior of Cu/SiO2, Co/SiO2, and CuCo/SiO2 catalysts studied by in situ DRIFTS. J. Phys. Chem. C. 116, 7931–7939 (2012).
Yu, Y. et al. Promotive effect of SO2 on the activity of a deactivated commercial selective catalytic reduction catalyst: an in situ DRIFT study. Ind. Eng. Chem. Res. 53, 16229–16234 (2014).
Xu, W., He, H. & Yu, Y. Deactivation of a Ce/TiO2 catalyst by SO2 in the selective catalytic reduction of NO by NH3. J. Phys. Chem. C. 113, 4426–4432 (2009).
Wu, Q., Gao, H. & He, H. Conformational analysis of sulfate species on Ag/Al2O3 by means of theoretical and experimental vibration spectra. J. Phys. Chem. B 110, 8320–8324 (2006).
Shimizu, K.-i, Higashimata, T., Tsuzuki, M. & Satsuma, A. Effect of hydrogen addition on SO2 tolerance of silver-alumina for SCR of NO with propane. J. Catal. 239, 117–124 (2006).
Sheng, H. et al. Carbon dioxide dimer radical anion as surface intermediate of photoinduced CO2 reduction at aqueous Cu and CdSe nanoparticle catalysts by rapid-scan FT-IR spectroscopy. J. Am. Chem. Soc. 140, 4363–4371 (2018).
Cao, Y. et al. Dual functions of O-atoms in the g-C3N4/BO0.2N0.8 interface: oriented charge flow in-plane and separation within the interface to collectively promote photocatalytic molecular oxygen activation. ACS Appl. Mater. Inter. 12, 34432–34440 (2020).
Guo, L. et al. Promoting charge carriers transfer of Co-S bonds in Co/CdS for enhanced photocatalytic CO2 reduction. Chin. Sci. Bull. 65, 522-532 (2019).
Acknowledgements
We gratefully acknowledge financial support from the National Natural Science Foundation of China (U1862111), Sichuan Provincial International Cooperation Project (2019YFH0164), and International Collaboration Project of Chengdu City (2017-GH02-00014-HZ). Y. Z. thanks for the Cheung Kong Scholars Programme of China. We acknowledge the National Supercomputing Center in Shenzhen for calculation support. We thank the Synchrotron radiation source at KIT, Karlsruhe, and PETRA III at DESY, Hamburg for providing the beamtime at CAT-ACT and P65 beamlines, correspondently. We also thank Dr. Anna Zimina (KIT) and Dr. Edmund Welter (DESY) for help during measurements.
Author information
Authors and Affiliations
Contributions
Y.Z. conceived the research and designed the experiments. Y.Z. and M.D. designed the synthesis. Y.C. and L.G. performed the synthesis, characterization, and photocatalysis. Y.C. performed the DFT calculations. D.E.D. performed the XAS in terms of XANES and EXAFS test. C.H. performed the SPV tests. Y.L. and J.M. performed the TEM and EDS mapping with support from K.Z. Y.C. and Z.R. performed in situ DRIFTS test with support from Z.H. P.C. performed the EPR tests with support from F.D. Y.C. wrote the manuscript with support from Y.Z. All authors analyzed the data and commented on the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Pavel Afanasiev, Ekaterina Kozlova, and other, anonymous, reviewer(s) for their contirbutions to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Cao, Y., Guo, L., Dan, M. et al. Modulating electron density of vacancy site by single Au atom for effective CO2 photoreduction. Nat Commun 12, 1675 (2021). https://doi.org/10.1038/s41467-021-21925-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-021-21925-7
This article is cited by
-
Cobalt-doped CdS quantum dots enhanced photoelectroreduction of CO2 to formic acid with high selectivity
Environmental Chemistry Letters (2024)
-
Promoted surface charge density from interlayer Zn–N4 configuration in carbon nitride for enhanced CO2 photoreduction
Nano Research (2024)
-
Atomic Dispersed Hetero-Pairs for Enhanced Electrocatalytic CO2 Reduction
Nano-Micro Letters (2024)
-
Progress in design and preparation of multi-atom catalysts for photocatalytic CO2 reduction
Science China Materials (2024)
-
Photocatalytic CO2 reduction
Nature Reviews Methods Primers (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
