
Abstract
Atrial fibrillation (AF) and hypertension (HTN) are both associated with impaired cerebrovascular carbon dioxide reactivity (CVRCO2), an indicator of cerebral vasodilatory reserve. We hypothesised that CVRCO2 would be lower in patients with both AF and HTN (AF + HTN) compared to normotensive AF patients, due to an additive effect of AF and HTN on CVRCO2. Forty AF (68 ± 9 years) and fifty-seven AF + HTN (68 ± 8 years) patients underwent transcranial Doppler ultrasound measurement of middle cerebral artery blood velocity (MCA Vm) during stepped increases and decreases in end-tidal carbon dioxide (PETCO2). A cerebrovascular conductance index (CVCi) was calculated as the ratio of MCA Vm and mean arterial pressure (MAP). CVRCO2 was determined from the linear slope for MCA Vm and MCA CVCi vs PETCO2. Baseline MAP was higher in AF + HTN than AF (107 ± 9 vs. 98 ± 9 mmHg, respectively; p < 0.001), while MCA Vm was not different (AF + HTN:49.6 [44.1–69.0]; AF:51.7 [45.2–63.3] cm.s−1; p = 0.075), and CVCi was lower in AF + HTN (0.46 [0.42–0.57] vs. 0.54 [0.44–0.63] cm.s−1.mmHg−1; p < 0.001). MCA Vm CVRCO2 was not different (AF + HTN: 1.70 [1.47–2.19]; AF 1.74 [1.54–2.52] cm/s/mmHg−2; p = 0.221), while CVCi CVRCO2 was 13% lower in AF + HTN (0.013 ± 0.004 vs 0.015 ± 0.005 cm.s−1.mmHg−1; p = 0.047). Our results demonstrate blunted cerebral vasodilatory reserve (determined as MCA CVCi CVRCO2) in AF + HTN compared to AF alone. This may implicate HTN as a driver of further cerebrovascular dysfunction in AF that may be important for the development of AF-related cerebrovascular events and downstream cognitive decline.

We demonstrated reduced cerebrovascular CO2 responsiveness in atrial fibrillation with hypertension (AF+HTN) vs. atrial fibrillation (AF). Furthermore, AF per se (as opposed to normal sinus rhythm) predicts reduced cerebrovascular CO2 responsiveness. Our findings suggest additional cerebrovascular dysfunction in AF+HTN vs. AF.
Similar content being viewed by others
Introduction
Atrial fibrillation (AF) is a supraventricular arrhythmia affecting 0.5–1% of the global population [1], making it the most prevalent sustained cardiac rhythm disorder. Alarmingly, this number is projected to rapidly increase over the next 30 years as populations age and modern healthcare improves survival outcomes for acute cardiac events [1, 2].
Hypertension (HTN) is a powerful risk factor for AF, and has previously been reported in 40–90% of AF cohorts [3]. In addition, HTN is likely responsible for up to 50% of all AF diagnoses [3]. Downstream complications of AF include thromboembolic events such as stroke, of which AF patients are five times more likely to develop [4], and neurodegenerative disorders including cognitive decline and dementia, even in the absence of overt stroke [5]. Similarly, HTN is a known risk factor for stroke, present in ~60% of stroke occurrences [6], as well as cognitive impairment disorders [7]. Possible mechanisms for this phenomena include silent cerebral infarction [8], inflammation [9] and cerebral hypoperfusion [10].
In AF, emerging evidence suggests that impaired cerebrovascular function may also contribute to this heightened risk of stroke and cognitive decline. It is currently unknown if HTN exacerbates cerebrovascular dysfunction in AF. Peripheral vascular dysfunction is well-documented in AF. Endothelium-dependent brachial artery flow-mediated dilatation is reduced in AF patients and coupled with increased levels of plasma von Willebrand factor, a known biomarker of endothelial dysfunction [11]. Furthermore, increased oxidative stress [12] and the shift to a pro-inflammatory state [13] in AF patients synergistically worsen endothelial function. Moreover, hypertension evokes similar effects on peripheral vascular function. Structural vascular remodelling evoked by increased pressures and pulsatility increases oxidative stress and inflammation [14], causing vessel stiffening and subsequent endothelial dysfunction, as evidenced by reductions in nitric oxide (NO) production in HTN [15]. Interestingly, peripheral vascular function was previously reported to not be different between a group of HTN patients and a group with both AF and HTN (AF + HTN) [16], likely due to the shared vascular pathologies between AF and HTN. Evidence of cerebrovascular dysfunction also exists in AF. Cerebrovascular CO2 reactivity (CVRCO2), defined as the change in (∆) cerebral blood flow (CBF) versus ∆ partial pressure of CO2 (PaCO2), is widely recognized as a marker of cerebral vasodilatory reserve, and has been associated with increased risk of mortality when impaired [17]. AF patients exhibit worsened CVRCO2 compared to healthy controls, indicative of cerebrovascular dysfunction [18]. Furthermore, neurovascular coupling, whereby cerebral blood flow is increased to support neuronal metabolic needs, is reduced in AF [19]. CVRCO2 responses have also been reported to be worsened in hypertension [20] although this is not a universal finding [21]. The combined effects of AF and HTN on CVRCO2 are yet to be determined; thus, it is still unknown whether their effects on CVRCO2 are additive or occlusive when concurrently present.
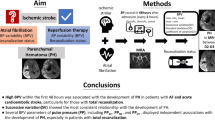
The aim of this study was to assess whether AF patients with HTN exhibit worsened cerebral vasodilatory reserve (i.e., poorer CVRCO2) compared to normotensive AF patients. Considering the current evidence indicating the effects of AF and HTN independently on CVRCO2, we hypothesized that AF patients with hypertension would exhibit lowered CVRCO2 compared with normotensive AF patients.
Methods
Ethical approval
The National Research Ethics Service Committee West Midlands (15/WM/0447) and North West (17/NW/0714) approved all study procedures, all of which conformed with the Declaration of Helsinki. Prospective participants received an information sheet which they were verbally guided through in detail. After addressing any questions, participants provided written informed consent.
Participant characteristics
Ninety-seven participants were included across two AF groups: AF (n = 40) and AF + HTN (n = 57). Of these, we included fifteen participants (which met the current inclusion criteria) from a previous study in which we used the same experimental approaches and outcome measures to cross-sectionally analyse cerebral vasodilatory reserve in three groups (AF, HTN and healthy controls) [18]. Patients were recruited from the following sites: Cardiology clinics at City Hospital, Birmingham, UK and Liverpool Heart and Chest Hospital, Liverpool, UK, along with National Institute of Health Research Clinical Research Network General Practitioner clinics in the UK.
All participants had previously been clinically diagnosed with AF and were included regardless of AF temporality (i.e. paroxysmal AF [fibrillation episodes are transient and spontaneously resolve within 48 h] or persistent AF [untreated fibrillation episodes last longer than 7 days before spontaneously resolving]). Study exclusion criteria included major illnesses such as cancer, liver or kidney disease, significant previous cardiovascular disease including myocardial infarction, left ventricular dysfunction, valvular heart disease, uncontrolled thyroid disorders, viral illness, connective tissue, inflammatory or neurological disease, significant cerebrovascular events such as stroke or transient ischaemic attack, alcohol abuse (>28 units per week), intravenous drug use or smoking, premenopausal women, use of oral nitrates or hormone replacement therapy and age <18 years. Tables 1 and 2 list participant characteristics and medication use respectively.
Experimental measures
A thorough participant history was recorded, including medication use, alcohol consumption and smoking habits. Using the recorded participant characteristics, each participant was calculated a CHA2DS2-VASc score. Anthropometric measures included height, weight, waist (umbilical level) and hip (femoral trochanter level) circumference. Non-invasive supine brachial BP was measured using an automated oscillometric device (M2, Omron, Kyoto, Japan). Beat-to-beat BP was continuously recorded by finger photoplethysmography (Finometer MIDI, Finapres Medical Systems, Amsterdam, The Netherlands), and heart rate (HR) by lead II ECG (BioAmp, ADInstruments, Dunedin, New Zealand). An oronasal mask connected to a heated pneumotach (Hans Rudolph, Kansas City, KS, USA) was used to record minute ventilation. A sampling line connected the oronasal mask to a capnograph (RespSense, Nonin Medical, Plymouth, MN, USA) for recording of end-tidal partial pressure of CO2 (PETCO2). Bilateral 2 MHz transcranial Doppler probes (Doppler BosX, DWL, Sipplingen, Germany) were used to insonate the cerebral arteries fixed at their respective temporal windows, providing mean blood flow velocity (Vm) for the left posterior cerebral artery (PCA) and the right middle cerebral artery (MCA). Insonation of the MCA was successful in all participants, but only achieved in forty-seven participants (48%) for the PCA.
Experimental protocol
Pre-study guidelines included no strenuous exercise, food and caffeine intake for 12 h prior to their study, no alcohol the day before and day of their study and to refrain from consuming medication (excluding anticoagulants) on the morning of study. Physiological assessments were undertaken with participants lying supine on a bed, with their head on a pillow. After instrumentation, a 10-minute baseline period was acquired whilst participants breathed room air. During this baseline, the mean of 3 brachial BP recordings was used to correct beat-to-beat BP obtained by finger photoplethysmography. Stepped increases and decreases in PETCO2 were then used to assess CVRCO2. Hypercapnic steps involved participants breathing through a Douglas bag circuit containing 4% CO2, followed by 7% CO2. Gas mixtures both contained 21% oxygen (N2 balanced). Each gas mixture was breathed for 4 min. Following hypercapnic steps, participants breathed room air to allow PETCO2 to return to baseline. They were then required to alter their respiratory rate and depth to lower their PETCO2 to an equal and opposite value to that achieved in the two hypercapnic steps. Each hypocapnic step was 2 minutes in duration. All participants completed the 4% hypercapnic step and its equal opposite hypocapnic step. Two participants did not complete the 7% hypercapnic step and one participant did not complete it’s equal opposite hypocapnic step. These participants were removed from our CVRCO2 analyses.
Data analysis
The ratio of height and weight squared was used to express participants BMI. Waist to hip and waist to height ratios were calculated for each participant. A multi-channel data acquisition system (Powerlab 16/35 and Labchart Pro 8.1.13, ADInstruments) was used to record and digitize analogue signals at 1 KHz. HR was calculated on a beat-to-beat basis from the ECG. Mean arterial pressure (MAP) and was obtained by integrating the arterial BP waveform over the complete cardiac cycle on a beat-to-beat basis. Similarly, Vm was obtained by integrating the MCA and PCA Vm waveforms over the complete cardiac cycle on a beat-to-beat basis. Cerebrovascular conductance index (CVCi) was calculated as the ratio of MCA Vm or PCA Vm with MAP. CVCi was not calculated if either Vm or MAP values were determined to be statistical outliers. Baseline measures were averaged over the entire 10-minute duration. Hypercapnic and hypocapnic step change measures were obtained from the final minute of the experimental period. The linear slopes of MCA Vm and PCA Vm versus PETCO2 across the range of hypo- and hypercapnic steps provided CVRCO2. Outliers were defined as ± interquartile range (IQR)*2.2 and excluded from analyses.
Statistical analysis
Shapiro-Wilk test was used to assess data normality. Levene’s test was used to assess homogeneity of variance between group data. Independent two-tailed Students t-test was implemented to analyse normally distributed continuous data and Mann–Whitney U test for non-normally distributed continuous data. Fisher’s Exact Probability Test was used to analyse normally distributed categorical data and Pearson chi-square for non-normally distributed categorical data. Effect size for significant continuous variables reported with Cohen’s d (d) and Phi (φ) for significant categorical variables. Two-way analysis of variance (ANOVA) was implemented to compare the effect of patient group and PETCO2 on MCA Vm, MCA CVCi, PCA Vm, PCA CVCi and MAP. For ANOVA, data normality was assessed by inspection of Q-Q plots for standardized residuals. Mauchly’s test of sphericity was used to assess the assumption of sphericity. If the assumption of sphericity was violated, a Greenhouse-Geisser correction was applied. Post-hoc pairwise comparisons were performed upon identification of significant main effects using t-test with Bonferroni correction. A forced entry regression model was used to determine the influence of variables of interest on baseline and exponential CVRCO2 parameters (MCA Vm, MCA CVCi, PCA Vm and PCA CVCi). Multicollinearity and autocorrelation were tested to reduce the risk of regression model violations. Tolerance, variance inflation factor, and Durbin-Watson statistic were 0.570–0.950, 1.053–1.755 and 1.754–2.520 respectively. Mean ± standard deviation (SD) are presented for normally distributed data, median [interquartile range] for non-normally distributed data and frequency (percentage) for categorical data. Statistical significance was considered as p < 0.05. Analysis was performed using SPSS, version 27 (IBM Corp., Armonk, NY, USA).
Results
Participant characteristics
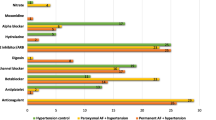
Age and the proportion of males and females was not different in the AF and AF + HTN groups (Table 1). AF + HTN exhibited greater systolic BP (p < 0.001), diastolic BP (p = 0.001), MAP (p < 0.001), HR (p = 0.047), weight (p = 0.004), BMI (p < 0.001), waist (p = 0.008) and hip (p = 0.003) measurements, waist-to-height ratio (p = 0.007), and CHA2DS2-VASc scores (p < 0.001). The proportion of participants who were fibrillating at the time of the study, had a diagnosis of persistent AF, were diabetic, had previously smoked or previously consumed excessive alcohol ( > 14 units a week) were not different between AF and AF + HTN groups (p > 0.05). AF + HTN had more frequent ACE inhibitor (p < 0.001), Ca2+ channel inhibitor (p < 0.001), ARB (p < 0.001), statin (p = 0.017) and SSR inhibitor (p = 0.018) use (Supplementary Table 1).
Baseline cerebral hemodynamics
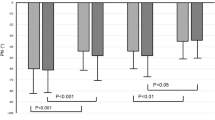
Figure 1 shows baseline cerebrovascular measures. MCA Vm was numerically lower in AF + HTN (49.6 [44.1–60.0] cm.s−1) compared to AF (51.7 [45.2–63.3] cm.s−1), but did not reach statistical significance (p = 0.075). MCA CVCi was lower in AF + HTN (0.46 [0.42–0.55] cm.s−1.mmHg−1) compared to AF (0.56 [0.46–0.71] cm.s−1.mmHg−1; p < 0.001). No differences were observed between groups for either PCA Vm (AF: 30.5 [26.4–33.6]; AF + HTN: 31.8 [24.3–38.4] cm.s−1; p = 0.731) or PCA CVCi (AF: 0.30 ± 0.05; AF + HTN: 0.29 ± 0.07 cm.s−1.mmHg−1; p = 0.548). Baseline PETCO2 was not different between groups (AF: 39.8 ± 0.7 mmHg; AF + HTN: 40.1 ± 0.7 mmHg; p = 0.813).

Baseline MCA Vm (A), MCA CVCi (B), PCA Vm (C), and PCA CVCi (D) in participants with AF or AF + HTN. Individual values plotted with bars displaying median and interquartile range for MCA Vm MCA CVCi and PCA Vm, and mean and standard deviation for PCA CVCi. AF atrial fibrillation; AF + HTN atrial fibrillation with hypertension; MCA Vm middle cerebral artery mean blood velocity; PCA Vm posterior cerebral artery mean blood velocity; MCA CVCi middle cerebral artery cerebrovascular conductance index; PCA CVCi posterior cerebral artery cerebrovascular conductance index
Cerebrovascular reactivity
MCA Vm, MCA CVCi, PCA Vm, and PCA CVCi increased progressively with each stepped increase in PETCO2 (all p < 0.001). MAP increased during the 7% hypercapnic step versus all other steps (all p < 0.001). MCA CVCi and MAP were lower in AF + HTN than AF (group effect, p < 0.001 and p = 0.002, respectively), while MCA Vm, PCA Vm and PCA CVCi were not different between AF and AF + HTN groups (all p > 0.05). No interaction was observed between group and PETCO2 for any variable examined (p > 0.05), although there was a strong tendency (p = 0.083) for an interaction between group and PETCO2 for MCA CVCi (Supplementary Fig. 1).
Indices of CVRCO2 are displayed in Table 2 and Fig. 2. MCA CVCi CVRCO2 was significantly lower in AF + HTN (0.013 ± 0.004 cm/s/mmHg−2) compared to AF (0.015 ± 0.005 cm/s/mmHg−2; p = 0.047). However, MCA Vm CVRCO2 (AF: 1.741 [1.543–2.517]; AF + HTN: 1.701 [1.472–2.186] cm/s/mmHg−2; p = 0.221), PCA Vm (AF: 1.076 ± 0.246; AF + HTN: 1.127 ± 0.352 cm/s/mmHg−2; p = 0.587) and PCA CVCi (AF: 0.008 ± 0.002; AF + HTN: 0.009 ± 0.003 cm/s/mmHg−2; p = 0.892) were not different between groups.

MCA Vm (A), MCA CVCi (B), PCA Vm (C) and PCA CVCi (D) CVRCO2 in participants with AF or AF + HTN. CO2 reactivity is expressed as the change in (∆) MCA Vm, MCA CVCi, PCA Vm and PCA CVCi versus ∆ PETCO2 in mmHg. Bars display median and interquartile range for MCA Vm CVRCO2, and mean and standard deviation for MCA CVCi, PCA Vm and PCA CVCi CVRCO2. AF atrial fibrillation; AF + HTN atrial fibrillation with hypertension; CVRCO2 cerebrovascular carbon dioxide reactivity; MCA Vm middle cerebral artery mean blood velocity; PCA Vm posterior cerebral artery mean blood velocity; MCA CVCi middle cerebral artery cerebrovascular conductance index; PCA CVCi posterior cerebral artery cerebrovascular conductance index; PETCO2 partial pressure of end-tidal carbon dioxide
Forced entry regression
Significant predictors of baseline variables and CVRCO2 as identified from forced entry linear regression are shown in Table 3. Fibrillation (vs normal sinus rhythm [NSR]) at the time of the experimental visit was associated with a lower MCA Vm CVRCO2. The presence of hypertension was associated with a lower baseline MCA CVCi, and being female was associated with higher baseline MCA Vm and baseline MCA CVCi. None of our entered independent variables were found to be associated with baseline PCA Vm, baseline PCA CVCi, MCA CVCi CVRCO2, PCA Vm CVRCO2 and PCA CVCi CVRCO2.
Discussion
This is the first study to investigate whether the presence of HTN as a comorbidity in AF worsens CVRCO2. The major novel finding of the present study is that CVCi CVRCO2 is lowered in AF + HTN compared to AF, and may implicate an additional level of cerebrovascular dysfunction in AF + HTN. This may be important for the development of cerebrovascular events and downstream cognitive decline commonly associated with AF.
Baseline MCA Vm was not different between AF and AF + HTN groups, indicating that cerebral perfusion at rest is retained in AF + HTN similar to that in AF. It is well accepted that there is a rightward shift in the cerebral autoregulatory curve in HTN, whereby CBF is maintained similarly to that in normotensives with higher MAP [22]. This may explain our conflicting baseline responses in terms of MCA Vm and CVCi. As a consequence of increased baseline MAP in AF + HTN, MCA CVCi is lowered, in order to buffer the transmission of peripheral BP to CBF. This may be explained due to HTN evoked remodelling, hypertrophy and stiffening within the cerebral arteries and arterioles, resulting in increased wall-lumen ratios and decreased vessel diameters [23]. These effects on the vasculature may be explained by a number of interconnected processes. Oxidative stress is heightened in HTN, and promotes proliferation of vascular smooth muscle cells as well as extracellular matrix remodelling [14]. Elevated reactive oxygen species (ROS) can also drive inflammation in HTN, resulting in further vascular hypertrophy. Indeed, elevated markers of inflammation, including C-reactive protein and interleukin-6, are independently associated with HTN [24]. Furthermore, the direct mechanical effects of heightened pressure on vessel walls can promote hypertrophy and remodelling through activation of extracellular matrix protein cascades, whereby noncellular material accumulates within vessel walls [25]. This can also drive ROS production, resulting in a pathological cycle of cerebrovascular remodelling.
Interestingly, in the present study we have shown a reduction in MCA CVCi CVRCO2 in AF + HTN but no difference in MCA Vm CVRCO2 between AF and AF + HTN. This offers distinct insight from our previous work, where we compared CVRCO2 in separate groups with either AF, HTN, or healthy controls (i.e. normotensive and with a normal sinus rhythm), and demonstrated lower MCA Vm CVRCO2 in AF versus HTN and healthy controls. The current study, in which all patients studied had AF, suggests that in AF + HTN, CO2-mediated increases in CBF per unit pressure are blunted. The use of a conductance index allows us to somewhat account for changes in perfusion pressure and provides more insight to vasodilator responsiveness. Lowered CVCi CVRCO2 may be indicative of additional cerebrovascular dysfunction evoked by HTN. The aforementioned HTN induced cerebrovascular remodelling will have functional consequences in terms of vascular responsiveness to CO2 that may drive differences in CVCi CVRCO2 between our AF + HTN and AF groups. In addition, endothelial damage/dysfunction associated with oxidative stress, inflammation, and reductions in NO bioavailability [26] and known to be present in both HTN and AF may also contribute and summate to worsen CVCi CVRCO2 in AF + HTN. In contrast, brachial artery flow mediated dilatation, a marker of peripheral endothelial function, is not different in HTN versus AF + HTN [16], and might suggest a stronger contribution of HTN than AF to peripheral/cerebrovascular dysfunction [22].
Forced entry regression suggested patients that were fibrillating at the time of study (as opposed to being in NSR) exhibited lower MCA Vm CVRCO2. MRI and xenon inhalation studies have previously shown improved CBF in AF patients who underwent NSR restoration procedures [27, 28], suggesting a direct effect of fibrillation on cerebral perfusion per se. However, in earlier work [18] we observed that MCA Vm was lower in AF patients that were fibrillating at the time of study compared to those in NSR, but MCA Vm CVRCO2 was not different. Differences in sample size (n = 97 in the present study vs. n = 31 in Junejo et al. [18]) and the analytical approach employed, may partly explain the discrepant findings, and further work is required to better understand how AF per se affects CVRCO2 and by what mechanisms.
The anterior and posterior cerebral circulations differ in regards to CVRCO2, with the posterior cerebral circulation exhibiting absolute CVRCO2 values ~ 50% lower than that seen in the anterior cerebral circulations [29, 30], which our results generally agree with. Additionally, differences may exist in endothelial function between the two circulations, as endothelium-derived NO has been shown to play a greater role in vasodilatory responses in the anterior cerebral circulation [31]. Notably, baseline PCA and PCA CVRCO2 measures were not different between AF and AF + HTN. This may indicate preserved endothelial function in the posterior cerebral circulation in AF + HTN vs. AF groups.
Our findings should be interpreted in light of several experimental considerations. While transcranial Doppler ultrasound offers real-time measurements of cerebrovascular function, is non-invasive and easy to implement during physiological interventions, its inability to directly measure blood flow is a limitation. Vessel diameter cannot be measured with transcranial Doppler, thus changes in cerebral blood velocity may not be directly indicative of CBF changes. However, as transcranial Doppler-derived Vm changes have been significantly correlated with gold standard measures of CBF (i.e., gadolinium tracer MRI and arterial spin labelling MRI [32]) Vm values are generally considered proportional to CBF. PETCO2 has a strong positive correlation with PaCO2 [33], and acts as a reliable non-invasive surrogate measure, though we acknowledge its possible underestimation of PaCO2 at rest [34]. There is debate surrounding the optimal method for assessing cerebrovascular function [35]. The advantages of using fixed concentrations of CO2 (4 and 7%) as in the present study, is that the experimental set-up is relatively simple, inexpensive, convenient in the clinic, does not required a complex computerised gas delivery system, and requires minimal participant cooperation. This method has also shown good between-day test-retest reliability [18]. Our primary measure of cerebrovascular responsiveness (i.e., CVCi CVRCO2) was derived from the linear slope of the relationship between PETCO2 and MCA CVCi for each patient and was significantly (p = 0.047) lower in AF + HTN versus AF. While a tendency (p = 0.083) for an interaction between Group and PETCO2 was observed for MCA CVCi (Supplementary Fig. 1B) this did not reach significance. This discrepancy is likely caused by differences in the methods of data analysis used. We did not screen for the presence of subclinical brain infarcts, alhough patient history and clinical records did not suggest any presence of these. In addition, although participants were requested to refrain from consuming medications on the morning prior to their study visit, it is unlikely that this was sufficient to cause full washout prior to data acquisition. Finally, it is a limitation of the current study that a healthy control group (i.e., normotensive and NSR) was not recruited, however we believe that this does not diminish our ability to achieve the primary aim of the study which was to determine whether the presence of HTN in AF worsens cerebrovascular responsiveness.
In conclusion, this is the first study that demonstrates reduced CVRCO2 in AF patients with concurrent HTN as compared to normotensive AF patients. This may be important for clinical management due to the possible heightened risk of cerebrovascular events and downstream cognitive decline.
Data availability
Study data are available from the corresponding author upon reasonable request.
References
Lippi G, Sanchis-Gomar F, Cervellin G. Global epidemiology of atrial fibrillation: an increasing epidemic and public health challenge. Int J Stroke. 2021;16:217–21.
Krijthe BP, Kunst A, Benjamin EJ, Lip GYH, Franco OH, Hofman A, et al. Projections on the number of individuals with atrial fibrillation in the European Union, from 2000 to 2060. Eur Heart J. 2013;34:2746–51.
Benjamin EJ, Levy D, Vaziri SM, D’Agostino RB, Belanger AJ, Wolf PA. Independent risk factors for atrial fibrillation in a population-based cohort. The Framingham heart study. JAMA. 1994;271:840–4.
Wolf PA, Abbott RD, Kannel WB. Original contributions atrial fibrillation as an independent risk factor for stroke: the Framingham study. Stroke. 1991;22:983–8.
Knecht S, Oelschläger C, Duning T, Lohmann H, Albers J, Stehling C, et al. Atrial fibrillation in stroke-free patients is associated with memory impairment and hippocampal atrophy. Eur Heart J. 2008;29;2067–9.
Wajngarten M, Sampaio Silva G. Hypertension and stroke: update on treatment. Eur Cardiol Rev. 2019;14:111–5.
Ou YN, Tan CC, Shen XN, Xu W, Hou XH, Dong Q, et al. Blood pressure and risks of cognitive impairment and dementia: a systematic review and meta-analysis of 209 prospective studies. Hypertension. 2020;76:217–25.
Petersen P, Birk Madsen E, Brun B, Pedersen F, Gyldensted C, Boysen G. Silent cerebral infarction in chronic atrial fibrillation. Stroke. 1987;18:1098–100.
Li J, Solus J, Chen Q, Rho YH, Milne G, Stein CM, et al. Role of inflammation and oxidative stress in atrial fibrillation. Heart Rhythm. 2010;7:438–44.
Gardarsdottir M, Sigurdsson S, Aspelund T, Rokita H, Launer LJ, Gudnason V, et al. Atrial fibrillation is associated with decreased total cerebral blood flow and brain perfusion. Europace. 2018;20:1252–8.
Freestone B, Chong AY, Nuttall S, Lip GYH. Impaired flow mediated dilatation as evidence of endothelial dysfunction in chronic atrial fibrillation: Relationship to plasma von Willebrand factor and soluble E-selectin levels. Thromb Res. 2008;122:85–90.
Neuman RB, Bloom HL, Shukrullah I, Darrow LA, Kleinbaum D, Jones DP, et al. Oxidative stress markers are associated with persistent atrial fibrillation. Clin Chem. 2007;53:1652–7.
Engelmann MDM, Svendsen JH. Inflammation in the genesis and perpetuation of atrial fibrillation. Eur Heart J. 2005;26:2083–92.
Xiao L, Harrison DG. Inflammation in Hypertension. Can J Cardiol. 2020;36:635–47.
Talman WT, Nitschke Dragon D. Neuronal nitric oxide mediates cerebral vasodilatation during acute hypertension. Brain Res. 2007;1139:126–32.
Khan AA, Junejo RT, Alsharari R, Thomas GN, Fisher JP, Lip GYH. A greater burden of atrial fibrillation is associated with worse endothelial dysfunction in hypertension. J Hum Hypertens. 2021;35:667–77.
Portegies ML, de Bruijin RF, Hofman A, Koudstaal PJ, Ikram AM. Cerebral vasomotor reactivity and risk of mortality. The Rotterdam study. Stroke. 2013;45:42–7.
Junejo RT, Braz ID, Lucas SJ, van Lieshout JJ, Lip GY, Fisher JP. Impaired cerebrovascular reactivity in patients with atrial fibrillation. J Am Coll Cardiol. 2019;73:1230–2.
Junejo RT, Braz ID, Lucas SJE, van Lieshout JJ, Phillips AA, Lip GYH, et al. Neurovascular coupling and cerebral autoregulation in atrial fibrillation. J Cereb Blood Flow Metab. 2020;40:1647–57.
Hajjar I, Zhao P, Alsop D, Novak V. Hypertension and cerebral vasoreactivity: a continuous arterial spin labeling magnetic resonance imaging study. Hypertension. 2010;56:859–64.
Tominaga S, Strandgaard S, Uemura K, Ito K, Kutsuzawa T, Lassen NA, et al. Cerebrovascular CO2 reactivity in normotensive and hypertensive man. Stroke. 1976;7:507–10.
Immink RV, Van Den Born BJH, Van Montfrans GA, Koopmans RP, Karemaker JM, Van Lieshout JJ. Impaired cerebral autoregulation in patients with malignant hypertension. Circulation. 2004;110:2241–5.
Rizzoni D, De Ciuceis C, Porteri E, Paiardi S, Boari GEM, Mortini P, et al. Altered structure of small cerebral arteries in patients with essential hypertension. J Hypertens. 2009;27:838–45.
Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (C-reactive protein, interleukin-6, and TNF-α) and essential hypertension. J Hum Hypertens. 2005;19:149–54.
O’callaghan CJ, Williams B. Mechanical strain-induced extracellular matrix production by human vascular smooth muscle cells role of TGF-1. Hypertension. 2000;36:319–24.
Junejo RT, Lip GYH, Fisher JP. Cerebrovascular dysfunction in atrial fibrillation. Front Physiol. 2020;11:1–7.
Petersen P, Kastrup J, Videbrek R, Boysen G. Journal of cerebral blood flow and metabolism cerebral blood flow before and after cardioversion of atrial fibrillation. J Cereb Blood Flow Metab. 1989;9:422–5.
Gardarsdottir M, Sigurdsson S, Aspelund T, Gardarsdottir VA, Forsberg L, Gudnason V, et al. Improved brain perfusion after electrical cardioversion of atrial fibrillation. Europace. 2020;22:530–7.
Skow RJ, MacKay CM, Tymko MM, Willie CK, Smith KJ, Ainslie PN, et al. Differential cerebrovascular CO2 reactivity in anterior and posterior cerebral circulations. Respir Physiol Neurobiol. 2013;189:76–86.
Bruce CD, Steinback CD, Chauhan UV, Pfoh JR, Abrosimova M, Vanden Berg ER, et al. Quantifying cerebrovascular reactivity in anterior and posterior cerebral circulations during voluntary breath holding. Exp Physiol. 2016;101:1517–27.
Perko D, Pretnar-Oblak J, Šabovič M, Žvan B, Zaletel M. Differences between cerebrovascular reactivity to L-arginine in the anterior and posterior cerebral circulation. Cerebrovasc Dis. 2011;31:358–64.
Sorond F, Hollenberg NK, Panych LP, Fisher NDL. Brain blood flow and velocity: correlations between magnetic resonance imaging and transcranial doppler. J Ultrasound Med. 2010;29:1017–22.
McSwain SD, Hamel DS, Faarc R, Brian Smith P, Gentile MA, Srinivasan S, et al. End-tidal and arterial carbon dioxide measurements correlate across all levels of physiologic dead space. Respir Care. 2010;55:288–93.
Robbins PA, Conway J, Cunningham DA, Khamnei S, Paterson DJ, Cun-Ningham DA. A comparison of indirect methods for continuous estimation of arterial PCO2 in men. J Appl Physiol. 1990;68:1727–31.
Fierstra J, Sobczyk O, Battisti-Charbonney A, Mandell DM, Poublanc J, Crawley AP, et al. Measuring cerebrovascular reactivity: What stimulus to use? J Physiol. 2013;591;5809–21.
Acknowledgements
We wish to thank all the volunteers for their enthusiastic participation in this study.
Funding
This study was supported by Bristol-Myers Squibb / Pfizer Alliance Project Grant CV185-481, British Heart Foundation Project Grant PG/15/45/31579, Royal Society Te Apārangi Marsden Fund UOA1903, and the National Institute of Health Research Clinical Research Network (NIHR CRN). Open Access funding enabled and organized by CAUL and its Member Institutions.
Author information
Authors and Affiliations
Contributions
JPF and GYHL conception, RTJ data collection and extraction, HJW data analysis, statistical analysis, figure preparation and draft of manuscript, HJW, RTJ, GYHL and JPF revision, critique and final manuscript approval.
Corresponding author
Ethics declarations
Conflict of interest
GYHL is a consultant for Bayer/Janssen, BMS/Pfizer, Medtronic, Boehringer Ingelheim, Novartis, Verseon and Daiichi-Sankyo. Speaker for Bayer, BMS/Pfizer, Medtronic, Boehringer Ingelheim, and DaiichiSankyo. No fees are directly received personally. JPF has received funding from BMS/Pfizer for an investigator-led and competitively reviewed research project. RTJ was previously employed by University of Birmingham as a research fellow on BMS/Pfizer funded, investigator-led and competitively reviewed research project which he managed. This required regular contact with funders for continued project grant support. No fees/funds were directly received personally.
Ethical approval
Ethical approval was received from the National Research Ethics Service Committee West Midlands (15/WM/0447) and North West (17/NW/0714).
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Walsh, H.J., Junejo, R.T., Lip, G.Y.H. et al. The effect of hypertension on cerebrovascular carbon dioxide reactivity in atrial fibrillation patients. Hypertens Res (2024). https://doi.org/10.1038/s41440-024-01662-2
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41440-024-01662-2
