
Abstract
With the advent of next-generation sequencing (NGS), a blended phenotype has been shown to be caused by multilocus molecular diagnosis. Here, we present siblings of neurofibromatosis type 1 (NF1) with discordant phenotypes. Further genetic investigation revealed that the younger sister had trisomy 8 mosaicism with a low ratio and a known pathogenic mutation in the CASK gene. This is the first report of a blended phenotype caused by NF1, CASK disorder, and trisomy 8 mosaicism.
Similar content being viewed by others

NF1 is an autosomal dominant disorder characterized by multiple café-au-lait spots, growth of tumors along nerves, macrocephaly, learning difficulties and other symptoms. NF1 is also characterized by considerable inter- and intrafamilial variability in phenotypic expression1. However, its underlying mechanism is not well understood. Here, we present a good case showing the usefulness of next-generation sequencing (NGS) to uncover the cause of intrafamilial phenotypic discordance of NF1.
The proband (patient 1) was a 5-year-old boy (III-2 in Fig. 1a). He was the first child of nonconsanguineous parents. He was delivered at 36 weeks of gestation after an uneventful pregnancy. His birth weight was 2776 g (+1.1 SD). He developed multiple café-au-lait spots after birth. He had more than ten macules over 5 mm in diameter by the age of 2 years. He started controlling his head at 4 months, sitting without support at 7 months, and walking unsupported at 18 months. He spoke his first word with meaning at 36 months and two-word sentences at 48 months. He had attended a local education center for the follow-up of his developmental delay (IQ = 54) and autistic behaviors. He was referred to our hospital at the age of 5 years. His karyotype was 46,XY. Brain magnetic resonance imaging (MRI) was normal. Upon physical examination at 7 years and 10 months, his weight was 17.6 kg (−0.5 SD), height 105.9 cm (−0.9 SD), and occipital frontal circumference (OFC) 53.0 cm (+1.2 SD). He did not have specific facial features except for the relative macrocephaly (Fig. 1b).

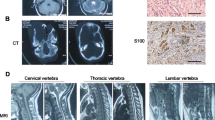
a Three-generation pedigree of the family in this study. Each symbol is annotated below. b, c Photograph of patients 1 and 2. Macrocephaly was noted in patient 1, but microcephaly and bulbous nose were noted in patient 2. d, e Brain MRI of patient 2 (d axial FLAIR image view, e sagittal T1-weighted view). MRI reveals mild hypoplasia of the cerebellum and brainstem. MRI magnetic resonance imaging
The younger sibling (patient 2) was a 5-year-old girl (III-3 in Fig. 1a). She was the second child of their parents and delivered at 40 weeks of gestation after an uneventful pregnancy. Her birth weight was 3068 g (−0.1 SD). She also had multiple café-au-lait spots after birth with seven macules over 5 mm in diameter. She began to control her head at 3 months, sit without support at 7 months, and walk alone at 16 months. She was referred to our hospital along with patient 1 at the age of 3 years for assessment of developmental delay. Upon examination, she could not speak any words with meaning. Her weight was 13.0 kg (−0.9 SD), height 94.3 cm (−0.8 SD), and OFC 46.0 cm (−1.7 SD). She was hypotonic. Her gait was ataxic with a widened base and truncal instability. Her behavior was autistic with restlessness, and she had difficulty making eye contact. She had a round face, sparse scalp hair, large ears, and downturned corners of her mouth (Fig. 1c). Her karyotype was mos 47,XX,+8[3]/46,XX[47]. Brain MRI revealed hypoplasia of the cerebellum and brainstem (Fig. 1d, e).
Written informed consent was obtained from the parents of the patients, in accordance with the Kanagawa Children’s Medical Center Review Board and Ethics Committee. Genomic DNA was extracted from peripheral blood samples of patients. Extracted DNA was captured using the TruSight One Sequencing Panel and sequenced on an MiSeq platform (Illumina, Inc., San Diego, CA, USA) with 151-bp paired-end reads. Exome data alignment, variant calling, and variant annotation were performed as previously described2,3. Copy number variations (CNVs) were assessed by analyzing the NGS data based on log-ratio analysis and the z-score of read depth (XHMM) on each exon.
We first examined the NGS data of patient 1 to elucidate any underlying genetic causes. The average coverage depth of the entire panel was 53.97 reads, and 96.1% of the targeted bases were covered with more than 10× sequence reads. We identified a novel frameshift mutation in NF1 (NM_000267:c.6060dupA:p.Asn2021Lysfs*3). Sanger sequencing confirmed that patient 2 had the same mutation and that this mutation was inherited from their mother (Fig. 2a). To elucidate the cause of the discordant phenotype in the siblings, we further analyzed patient 2 using the TruSight One Sequencing Panel. The average coverage depth of the entire panel was 86.4 reads, and 99.4% of the targeted bases were covered with more than 10× sequence reads. We found a recurrent nonsense mutation in CASK (NM_003688:c.2041 C > T:p.Arg681*). Sanger sequencing confirmed that it was a de novo mutation. Patient 1 did not have this mutation (Fig. 2b). There was no apparent pathogenic CNV in either patient.

a NF1 mutation. Electropherogram of the siblings and their parents. b CASK mutation. c Blended phenotype of patient 2 with NF1 and CASK mutation
In this study, we demonstrated the discordant phenotypes among the siblings with café-au-lait spots and developmental delay. The younger sister had a more severe developmental delay and additional phenotypes, including ataxic gait, hypotonia, microcephaly, and hypoplasia of the cerebellum and brainstem. We identified a novel frameshift mutation in NF1 of both siblings and their mother. The discordant phenotype of the siblings allowed us to consider the possibility of a blended phenotype caused by multilocus variations. We identified a de novo nonsense mutation in CASK of the younger sister. An X-linked dominant disorder of CASK is characterized by severe developmental delay, microcephaly, ataxic gait, and hypoplasia of the cerebellum and brainstem4. CASK belongs to the membrane-associated guanylate kinase (MAGUK) family and possesses a calcium/calmodulin-dependent serine protein kinase domain. It attaches to the membrane of synapses and functions in signaling pathways. CASK can also translocate to the nucleus and interact with transcription factors responsible for the expression of several genes required for normal brain formation4,5. In the context of phenotype−genotype correlation, we concluded that the NF1 mutation underlay common phenotypes of the siblings, including café-au-lait spots and autistic behavior, but the CASK mutation contributed to the sister’s distinct phenotypes, including severe developmental delay, microcephaly, ataxic gait, hypotonia, and brain abnormalities (Fig. 2c). We could not exclude the possibility of somatic mosaicism for CASK mutation on the proband’s mother. However, we concluded that the CASK mutation of the sister was de novo because her mother did not possess any phenotypes caused by CASK mutations, including intellectual disability, ataxic gait, or microcephaly. Patient 2 had another possible pathogenic background, mosaic trisomy 8. However, she did not have typical phenotypes of mosaic trisomy 8, including scaphocephaly, frontal bossing, thin and long body trunk, and aberrant creases on the sole6. Although we concluded that mosaic trisomy 8 only slightly affected her phenotype considering the very low mosaic rate (6%), this may be the case for a variation in mosaic trisomy 8.
To our knowledge, this is the first case report of blended phenotypes caused by combinatory variants: frameshift mutation of NF1 and nonsense mutation of CASK. The recent prevalence of NGS has unveiled the phenotypic variability of known genetic disorders and the presence of multilocus pathogenic variants in a single patient. Large-scale analyses of whole exome sequencing revealed that 3.5−4.9% of unselected cases have multilocus molecular diagnoses7,8. We should reevaluate the results of genetic analysis when we recognize atypical phenotypic expansion, especially in the case of disease characterized by inter- and intrafamilial phenotypic variability such as NF1.
HGV Database
The relevant data from this Data Report are hosted at the Human Genome Variation Database at https://doi.org/10.6084/m9.figshare.hgv.2564; https://doi.org/10.6084/m9.figshare.hgv.2567.
References
Sabbagh, A. et al. Unravelling the genetic basis of variable clinical expression in neurofibromatosis 1. Hum. Mol. Genet. 18, 2768–2778 (2009).
Enomoto, Y., Tsurusaki, Y., Harada, N., Aida, N. & Kurosawa, K. Novel AMER1 frameshift mutation in a girl with osteopathia striata with cranial sclerosis. Congenit. Anom. 58, 145–146 (2018).
Tsurusaki, Y. et al. A novel UBE2A mutation causes X-linked intellectual disability type Nascimento. Hum. Genome Var. 4, 17019 (2017).
Najm, J. et al. Mutations of CASK cause an X-linked brain malformation phenotype with microcephaly and hypoplasia of the brainstem and cerebellum. Nat. Genet. 40, 1065–1067 (2008).
Hsueh, Y. P., Roberts, A. M., Volta, M., Sheng, M. & Roberts, R. G. Bipartite interaction between neurofibromatosis type I protein (neurofibromin) and syndecan transmembrane heparan sulfate proteoglycans. J. Neurosci. 21, 3764–3770 (2001).
Danesino, C. et al. Constitutional trisomy 8 mosaicism: mechanism of origin, phenotype variability, and risk of malignancies. Am. J. Med. Genet. 80, 540 (1998).
Posey, J. E. et al. Resolution of disease phenotypes resulting from multilocus genomic variation. N. Engl. J. Med. 376, 21–31 (2017).
Balci, T. B. et al. Debunking Occam’s razor: diagnosing multiple genetic diseases in families by whole-exome sequencing. Clin. Genet. 92, 281–289 (2017).
Acknowledgements
We thank the patients and their family for participating in this work. We also thank Shizuka Shiiya for excellent technical assistance. This research was supported in part by the Japan Agency for Medical Research and Development (AMED) grant number 18kk02050h0003; JSPS KAKENHI 17K10069 (K.K.).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Murakami, H., Kimura, Y., Enomoto, Y. et al. Discordant phenotype caused by CASK mutation in siblings with NF1. Hum Genome Var 6, 20 (2019). https://doi.org/10.1038/s41439-019-0051-0
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41439-019-0051-0
This article is cited by
-
A de novo variant in CASK gene causing intellectual disability and brain hypoplasia: a case report and literature review
Italian Journal of Pediatrics (2022)
