
Abstract
A major goal of evolutionary genetics is to understand the genetic and molecular mechanisms underlying adaptation. Previous work has established that changes in gene regulation may contribute to adaptive evolution, but most studies have focused on mRNA abundance and only a few studies have investigated the role of post-transcriptional processing. Here, we use a combination of exome sequences and short-read RNA-Seq data from wild house mice (Mus musculus domesticus) collected along a latitudinal transect in eastern North America to identify candidate genes for local adaptation through alternative splicing. First, we identified alternatively spliced transcripts that differ in frequency between mice from the northern-most and southern-most populations in this transect. We then identified the subset of these transcripts that exhibit clinal patterns of variation among all populations in the transect. Finally, we conducted association studies to identify cis-acting splicing quantitative trait loci (cis-sQTL), and we identified cis-sQTL that overlapped with previously ascertained targets of selection from genome scans. Together, these analyses identified a small set of alternatively spliced transcripts that may underlie environmental adaptation in house mice. Many of these genes have known phenotypes associated with body size, a trait that varies clinally in these populations. We observed no overlap between these genes and genes previously identified by changes in mRNA abundance, indicating that alternative splicing and changes in mRNA abundance may provide separate molecular mechanisms of adaptation.
Similar content being viewed by others


Introduction
Nearly 50 years ago, King and Wilson (1975) argued that the proteins of humans and chimpanzees were so similar that changes in gene regulation might underlie much of organismal evolution. More recent work has provided strong evidence in support of the idea that differences in gene expression underlie adaptation (e.g. Wray et al. 2003; Jones et al. 2012; Fraser 2013; Mack et al. 2018). Most of this work has focused on differences in mRNA abundance, but gene regulation involves many steps, including transcription, post-transcriptional modifications, translation, and post-translational modifications. Due to challenges in measuring post-transcriptional modifications, these have been less studied as mechanisms for evolution and local adaptation (but see Artieri and Fraser 2014; Yablonovitch et al. 2017; Steward et al. 2022). One important post-transcriptional modification is alternative splicing. During mRNA splicing, introns are typically removed and some or all of the exons in a gene are joined together in a processed mRNA, or isoform. While some exons may be constitutively spliced in, other exons may be alternatively spliced in or out to generate numerous unique mRNA isoforms from a single gene (Fig. 1A). Through alternative splicing of exons and introns, protein domains can also be included or excluded in the translated protein. Mutations in regions for cis- and trans- regulatory control of splicing can therefore alter protein structure and function and generate different phenotypes (Manning and Cooper 2017). These phenotypes, in turn, present opportunities for selection to drive divergence between populations.

A An overview of alternative splicing. Following transcription of a gene, exons are joined together in different combinations to produce different isoforms. These isoforms are related, but by including and excluding exons in the processed mRNA, protein folding can be changed and entire domains can be included or excluded in the translated product. Created with Biorender.com. B Mapping RNA-Seq short reads sequenced from mRNA isoforms. Short-read expression data are unable to reliably capture isoform diversity. Instead of defining alternative splicing by quantifying isoforms or exon usage, short-reads can be used to define alternative splicing events trough intron excisions. Reads that map to two different exons (split reads) within a gene capture splicing events between two junctions. C Leafcutter (Li et al. 2018) uses split reads counts to quantify the frequency of splicing out an intron, which in turn serves as a proxy for the frequency of splicing between two exons, as seen in this cluster diagram. For example, the blue split reads indicate that exon 1 and 2 are spliced together at a frequency of 0.32 and 0.44 in two different populations. The change in percent splice in (ΔPSI) is 0.12.
Alternative splicing has been shown to underlie adaptation over short timescales in diverse animals from vertebrates to insects (e.g. Jarosch et al. 2011; Ceinos et al. 2018; reviewed in Wright et al. 2022). For example, alternative splicing underlies differential heat sensitivity in vampire bats. Both vampire bats (Desmodus rotundus) and closely related fruit bats (Carollia brevicauda) express full length TRPV1, a heat sensitive ion channel. Full length TRPV1 is activated at temperatures higher than 38 °C to detect noxious heat. TRPV1 isoforms expressed in the specialized pit organs of D. rotundus are alternatively spliced to include an exon containing a premature stop codon that is translated into a truncated protein (Gracheva et al. 2011). Truncated TRPV1 is activated at 30 °C and improves sensitivity for detecting homeothermic prey for blood feeding. Alternative splicing has also been shown to facilitate adaptation by producing isoforms with loss-of-function phenotypes. Threespine sticklebacks (Gasterosteus aculeatus) are well known to have marine and freshwater ecotypes, with a reduction of spines in freshwater ecotypes. Marine sticklebacks produce full length isoforms of the homeodomain transcription factor MSX2A while freshwater sticklebacks produce a high proportion of non-functional MSX2A by splicing out an exon encoding a DNA binding domain (Howes et al. 2017). Transgenic expression of marine isoforms in freshwater sticklebacks increased spine length, suggesting that alternative splicing can facilitate adaptation by producing non-functional proteins.
The Western European house mouse (Mus musculus domesticus) provides a useful system for exploring the potential role of alternative splicing in local adaptation. Native to the Mediterranean region and Western Europe, house mice were spread around the world in association with humans in the last few hundred years (Morgan et al. 2022). In this short time they have adapted to a wide range of different environments through changes in morphology, physiology, and behavior. For example, mice in Eastern North America show clinal patterns of variation in body size, with larger mice in colder environments consistent with Bergmann’s Rule (Bergmann 1847), and these differences persist in a common lab environment (Lynch 1992). In eastern North America, mice from colder environments also build bigger nests, differ in metabolic traits, and show population-level differences in gene expression compared to mice from warmer environments when reared in a common laboratory environment (Phifer-Rixey et al. 2018). Moreover, patterns of genomic variation in mice sampled along a latitudinal transect from Florida to New York identified specific candidate genes underlying adaptive phenotypic differences (Phifer-Rixey et al. 2018). Most of these candidate genes did not harbor non-synonymous variants, suggesting that much of the response to selection is driven by changes in gene regulation. By identifying overlap between loci exhibiting clinally varying patterns of gene expression, genes with cis-eQTL, and loci showing signatures of selection, Mack et al. (2018) were able to identify a small number of genes responsible for adaptive differences in body size.
In this study we expand on previous work in house mice by exploring the role of splice variants in environmental adaptation. In particular, we analyzed RNA-Seq data from liver in 50 wild-caught mice sampled in five populations along a latitudinal transect (Fig. 2). First, we compared mice from the ends of this transect to identify genes showing population-level differences in alternative splicing. Next, we asked which of these genes showed clinal patterns of alternative splicing as might be expected if selection is driving differences. Third, we looked for associations between SNPs near each gene and splice variants to identify cis-acting splicing quantitative trait loci (cis-sQTL). Fourth, we explored the overlap among these loci and those showing signatures of selection from previous work (Phifer-Rixey et al. 2018) to identify a small set of candidate splice variants underlying environmental adaptation. Finally, we compared these results on alternative splicing with similar analyses conducted on patterns of mRNA abundance (Mack et al. 2018). We discovered five genes that showed differential splicing between mice sampled from the ends of this transect, clinal patterns of splicing variation across all five populations, and cis-sQTL that overlapped with previously identified targets of selection (Phifer-Rixey et al. 2018). We observed little to no overlap between genes showing alternative splicing and those showing overall differences in mRNA abundance.

Five populations were included for study: New Hampshire/Vermont, Pennsylvania, Virginia, Georgia, and Florida. Latitude was used as a proxy for bioclimatic variables, including mean winter minimum temperature from 1970 to 2000 (climate data is from Bioclim.org).
Materials and methods
Sampling and mRNA-sequencing
mRNA-sequencing (RNA-Seq) data were from Mack et al. (2018) (BioProject ID: PRJNA407812). Fifty male and female Mus musculus domesticus were collected from five populations along a latitudinal transect in the eastern United States (12 from New Hampshire/Vermont, 10 from Pennsylvania, 10 from Virginia, 10 from Georgia, and 8 from Florida; Fig. 2; Phifer-Rixey et al. 2018). Mice were sacrificed in the field, and liver tissues were extracted and stored in RNAlater at 4 °C overnight before being frozen at −80 °C. Liver was studied since it plays an important role in metabolism and growth, traits that are known to distinguish mice from this transect both in the wild and in the laboratory (Phifer-Rixey et al. 2018). Other tissues, such as brain, might be useful for studying adaptive differences in other traits such as nest-building behavior (Lynch 1992). RNA was extracted with Qiagen’s RNeasy Mini kit. RNA-Seq libraries of 100-base pair paired-end reads were generated for each population on the Illumina HiSeq 4000 platform.
mRNA-sequence data processing
We trimmed RNA-Seq reads from Mack et al. (2018) with TrimGalore! version 0.5.0 and we mapped these reads with STAR version 2.7.9a (Spliced Transcripts Alignment to a Reference) to the GRCm39 reference genome (GenBank accession GCA_000001635.9; Dobin et al. 2013). STAR performs well at identifying annotated exon junctions and also contains the twopassMode option that uses exon junctions found in the first run of mapping as annotations in a second run of mapping. This option helps to identify unannotated exon junctions. The output from STAR consists of mapped reads in BAM files which we then converted into JUNC files containing the mapped reads that span exon junctions (Regtools junction version 0.5.2; Cotto et al. 2020). We used the CollectRnaSeqMetrics tool of Picard Tools version 2.27.5 to verify that libraries did not have a 3’ transcript bias introduced from library preparation.
Intron clustering for alternative splicing and differential splicing analysis
Alternative splicing is often discussed in terms of changes in isoform abundance although in practice it is challenging to estimate isoform abundance from short-read RNA-Seq data because the same exon can be found in multiple isoforms. Instead, we utilized Leafcutter to quantify alternative splicing using split reads, with intron excision serving as a proxy for exon expression (Li et al. 2018). Split reads are reads that map to two different exons in a gene and thus directly capture a splicing event that is defined by the excision of the intervening intron (Fig. 1B). Leafcutter uses split read abundance to estimate the frequency of splicing events (Fig. 1C).
We used Leafcutter to identify clusters of alternatively spliced exons from JUNC files (Li et al. 2018). All junctions that shared either a 5’ splice site or a 3’ splice site were considered part of the same cluster, and genes typically had multiple alternatively spliced clusters (Fig. 3A). Testing for differential splicing between clusters has improved sensitivity over tests for differential splicing between introns individually (Li et al. 2018). Leafcutter measures differential splicing as the ΔPSI (change in percent-spliced in) between two groups (Fig. 3B). Here, we searched for differentially spliced intron clusters between the northernmost (New Hampshire/Vermont) and southernmost (Florida) populations from the latitudinal transect using Leafcutter’s Dirichlet-multinomial generalized linear model. The New Hampshire/Vermont population (n = 12) and the Florida population (n = 8) differed in sample size. To assess the effects of this difference in sample size, we repeated the analysis comparing these two populations using eight randomly selected individuals from the New Hampshire/Vermont population and the results were largely unchanged. Leafcutter also calculates principal components explaining variation in junction counts between populations.

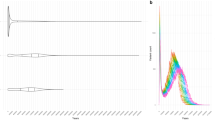
Lasp1 is a candidate for adaptive splicing among house mice in eastern North America. A Overlapping splicing 5’ and 3’ splice sites were used to identify clusters of alternatively spliced exons. The Lasp1 gene contains three differentially spliced clusters (arbitrarily named 29112, 29113, and 29114). B Differential splicing was assessed between the northernmost (New Hampshire/Vermont) and southernmost (Florida) populations on the latitudinal cline. Lasp1 had one cluster (29114) that exhibited differential splicing (FDR < 0.05). Cluster 29114 contained exons three, four, and five of Lasp1. Leafcutter measures differential splicing in terms of the change in percent-spliced in (ΔPSI) between populations. In Lasp1, exon 3 and 4 are spliced in at an average frequency of 0.34 and 0.17 between Florida and New Hampshire/Vermont populations, respectively and the ΔPSI for those exons is 0.17. C For each differentially spliced cluster between New Hampshire/Vermont and Florida, the splice-in frequency for the pair of exons with the highest ΔPSI was fitted to a linear model with latitude. The frequency of splicing between exon 3 and 4 of Lasp1 is correlated with latitude (p = 1.50 ×10−5, R2 = 0.32). D cis-sQTL for each clinally varying splicing event were identified with a linear-mixed model. Linkage disequilibrium between cis-sQTL (blue triangle) and LFMM outliers (asterisks) was calculated based on correlation.
Identification of clinally varying splicing events
For each differentially spliced cluster between New Hampshire/Vermont and Florida, the intron with the great ΔPSI was selected to test for clinal variation since it is likely that introns in the same cluster are not spliced independently of each other. We used linear regression models comparing splice-in-frequency and latitude from five populations to identify differentially spliced introns whose frequency varies clinally across latitude (p < 0.05, ΔPSI > 0.10). We chose ΔPSI > 0.10 as a minimum frequency difference to exclude changes of minor effect. A similar threshold was not used in the comparison between the New Hampshire/Vermont population and the Florida population (above) because Leafcutter’s differential splicing model tests for changes in intron usage across the entire cluster. This enables detection of differential splicing even if clusters contain introns with small ΔPSI. Linear regression models were calculated in R (version 4.2.3).
Mapping and variant calling on exome-capture data
To identify variants for mapping cis-sQTL, we mapped reads from the exome capture data of Phifer-Rixey et al. (2018) to GRCm39 with Bowtie2 version 2.4.1 (BioProject ID: PRJNA397150; Langmead and Salzberg 2012). The same data and the same mapping and variant calling approaches were used in Mack et al. (2018) to identify cis-eQTL. Of the 50 individuals with RNA-Seq data here and in Mack et al. (2018), 41 individuals had exome-capture libraries from Phifer-Rixey et al. (2018). Variant calling was performed with Genome Analysis Toolkit version 4.1 (GATK) HaplotypeCaller (McKenna et al. 2010) to match the variant calling method from Mack et al. (2018). The Base Recalibrator tool in GATK was used to identify and remove sequencing errors. Alleles missing from 30% of individuals and with a minor allele frequency less than 10% were removed from analysis. The filtered set of SNPs was then used in association studies to detect cis-sQTL.
Cis-sQTL discovery and overlap with signals of selection
We used a generalized linear mixed model implemented in the program GEMMA version 0.98.3 (Genome-wide Efficient Mixed Model Association) to identify cis-sQTL (Zhou and Stephens 2012). This model calculates a relatedness matrix among genotypes and includes the relatedness matrix as a covariate when testing for association between SNPs and splice variants. Associations were tested for each gene that contained a differentially spliced cluster with an intron whose splice-in frequency varied clinally. SNPs found within 200 kb of the 5’ splice site were tested for associations with splice variants. We applied the Benjamini–Hochberg correction to each p-value, and SNPs with adjusted p-values < 0.05 were considered significant.
To identify splice variants under selection, we looked for overlap between cis-sQTL and SNPs with signatures of selection from Phifer-Rixey et al. (2018). In that study, Latent-Factor Mixed Modeling (LFMM) was used to detect signatures of selection. LFMM accounts for covariance between environmental and genetic variation (Frichot et al. 2013). To enable comparison of these LFMM outliers to cis-sQTL, we remapped the LFMM outlier coordinates from GRCm38 to GRCm39 using the NCBI Remap tool. In Phifer-Rixey et al. (2018) and in this study, read mapping of exome-capture data was performed with Bowtie2 (versions 2.1.0 and 2.4.1, respectively). Phifer-Rixey et al. (2018) called variants with ANGSD while we called variants with GATK version 4.1, as in Mack et al. (2018). We identified SNPs that were both cis-sQTL and LFMM outliers. In cases where the same gene had a cis-sQTL and LFMM outlier at different SNPs, linkage disequilibrium (LD) was calculated between them using PLINK and Haploview (Purcell et al. 2007; Barrett et al. 2005). To determine whether the observed overlap between cis-sQTL and LFMM outliers was due to chance, we performed a permutation test. Genes were randomly sampled 24 times without replacement and then 5643 times without replacement from a set of 11,918 genes, representing the 24 cis-sQTL, the 5643 LFMM outliers, and the 11,918 genes expressed in liver. This sampling was repeated 100,000 times and for each iteration the size of the overlap was recorded to create a distribution.
Gene ontology annotation and enrichment analyses
We used DAVID (Database for Annotation, Visualization, and Integrated Discovery) for gene ontology annotation and enrichment analyses (Dennis et al. 2003; Sherman et al. 2022). DAVID compiles multiple sources of annotation but primarily draws from the Entrez Gene and Uniprot databases. This enrichment analysis uses modified Fisher Exact tests and Benjamini–Hochberg correction and provides False Discovery Rates for these tests. We provided the 11,918 genes expressed in Mus musculus liver as a background set of genes for the analyses. We defined an expressed gene as one with TPM > 1 (transcript per million) in at least one individual. With this background set of genes, enrichment tests were performed on differentially spliced genes in the comparison between New Hampshire/Vermont and Florida, genes exhibiting clinal variation in splicing, and genes associated with cis-sQTL. We found GO annotation for 144 of the 154 differentially spliced genes, 36 of the 41 genes exhibiting clinal variation, and each of the 24 genes associated with cis-sQTL.
Comparison mRNA abundance and alternative splicing
We compared the number and identity of genes that showed differences in mRNA abundance and alternative splicing (1) between populations at the ends of the transect, (2) that varied clinally with latitude, (3) that were associated with a cis-QTL, and (4) showed overlap with LFMM outliers. Data on mRNA abundance were from Mack et al. (2018) who mapped reads using Tophat (Kim and Salzberg 2011). In contrast, we used STAR (Dobin et al. 2013) to map reads since it is well suited for detecting splice junction. To assess whether these different mapping methods affected the comparison between mRNA abundance and alternative splicing, we repeated the splicing analyses using reads mapped with Tophat (as in Mack et al. 2018) and obtained similar results.
Results
Differential splicing in wild-house mice
After mapping with STAR, we identified ~1.8 billion RNA-Seq short-reads across all individuals, and ~720 million (39%) of those were split reads, averaging ~14 million split reads per sample (Supplementary Table 1). Clusters were only considered for differential splicing if each junction was supported by at least 10 reads. Principal component analysis of all five populations using junction count data showed that individuals did not cluster by population, although individuals clustered more tightly in populations from warmer climates (Supplementary Fig. 1). The first two principal components explained 3.6% and 2.9% of the variance respectively. The first 10 principal components accounted for 27.1% of the variation and none resulted in population-level clustering.
We used a Dirichlet-multinomial generalized linear model implemented in Leafcutter to test for differential splicing between the New Hampshire/Vermont and Florida populations at the cluster level. Differential splicing was measured as the difference between two populations in the proportion of excised introns (Fig. 3B). This test identified 166 differentially spliced exon clusters in 152 genes between the two populations (Supplementary Table 2; p < 0.05, Benjamini–Hochberg correction).
Clinal patterns of alternative splicing
Patterns of genetic variation among the five sampled populations do not show isolation-by-distance (Phifer-Rixey et al. 2018). Because of this, traits that co-vary with environmental gradients may be indicative of selection. We tested for clinal patterns of variation among differentially spliced clusters using latitude as a variable because latitude is strongly correlated with many climatic variables. For example, latitude shows a strong negative correlation with the first principal component summarizing climatic variables from the WorldClim database (Hijmans et al. 2005) (Pearson’s r = −0.99, p < 0.0006, Phifer-Rixey et al. 2018). We considered clusters with a maximum |ΔPSI| less than 0.10 to have a very small effect, and these were excluded from the analysis. Of the 152 genes with differentially spliced exons, we identified 44 excised introns in 41 genes that were correlated with latitude and showed |ΔPSI|>0.10 (Fig. 3C; Supplementary Table 3, p < 0.05). A gene ontology overrepresentation test for this set of 41 genes showed significant enrichment for genes involved in complement activation, a function of the immune system (Fisher’s Exact test, FDR < 0.05).
Identification of cis-sQTL and overlap with signals of selection
To identify cis-sQTL for each of the 44 clinally varying excised introns, we used GEMMA to test for associations between all variants within 200 kb of the 5’ splice site and the excision frequency of the corresponding intron. Relatedness between individuals was included as a covariate. In total, 829 variants from the exome-capture data of Phifer-Rixey et al. (2018) were tested. Out of the 44 clinally varying excised introns, 24 excisions in 24 genes had at least one cis-sQTL (Supplementary Table 4; Supplementary Fig. 2, -log10(p) > 1.30, p < 0.05, Benjamini–Hochberg correction).
Next, we looked for overlap between the 24 clusters associated with at least one cis-sQTL and signals of selection based on LFMM outliers with |Z-scores|>2 across all genes from Phifer-Rixey et al. (2018). Five of the 24 cis-sQTL overlapped with these LFMM hits at the gene level, and this amount of overlap is no more than expected by chance (Supplementary Fig. 3, permutation test, p = 0.601). To assess whether these five genes contained cis-sQTL and LFMM outliers on the same haplotype, we calculated linkage disequilibrium among all SNPs in the 400 kb window for each gene (Fig. 3D, Supplementary Fig. 4). For three of these genes (Mbl2, Pex26, Rnd2), LFMM outliers and cis-sQTL were the same SNPs. For two of the genes (Lasp1 and Atg7) the LFMM outlier SNPs were not the same as the cis-sQTL SNPs, but in both cases, the SNPs fell on the same haploblock. These five genes with cis-regulatory variants underlying clinal patterns of splicing are strong candidates for adaptive evolution mediated by alternative splicing (Table 1). Of these five genes, only four have phenotypes documented in the Mouse Genome Informatics database, and each of these four are associated with phenotypes known to distinguish mice from different latitudes (Phifer-Rixey et al. 2018). For example, Pex26, Rnd2, and Atg7 are associated with differences in growth and body size, and Lasp1 and Atg7 are associated with metabolic phenotypes (Table 1). Mice from these populations have been shown to differ in body size and growth rates and exhibit differences in aspects of blood chemistry related to metabolism (Phifer-Rixey et al. 2018).
Reflecting the probe set, nearly all overlapping cis-sQTL and LFMM outliers were found in intronic regions and gene bodies. Only one LFMM outlier overlapping a cis-sQTL was found in an intergenic region. Of the five candidate genes, Rnd2 and Pex26 had LFMM outliers on haplotypes that extended to adjacent genes (Wnk4 and Mical3, respectively). In general, LD decays over distances of 10’s of kb in mouse populations (e.g. Laurie et al. 2007) and thus haplotypes are correspondingly long. We identified cis-sQTL but cannot identify specific causative SNPs underlying splicing variation since causative SNPs may lie anywhere on the associated haplotype, including in un-surveyed regions.
Comparison between alternative splicing and mRNA abundance
We compared patterns of alternative splicing to the patterns of mRNA abundance (i.e., gene expression) from Mack et al. (2018) (Table 2). In comparisons between the two populations at the ends of this transect, 152 genes showed differential splicing and 458 genes showed differential transcription, with only 6 genes in common. Of these, 41 genes showed clinal patterns of alternative splicing and 274 genes showed clinal patterns of mRNA abundance, with no overlap between these sets of genes. Thus, there is little overlap between genes that show alternative splicing and those that show differences in mRNA abundance. Of the genes with clinal patterns of mRNA abundance, five of those with a cis-eQTL overlapped with a LFMM outlier, while for genes showing clinal patterns of splicing variation, five of those with a cis-sQTL overlapped with a LFMM outlier, suggesting that a similar proportion of cis-eQTL and cis-sQTL are targets of selection (Table 2).
Discussion
We used short-read RNA-Seq data from five populations of house mice sampled along a latitudinal transect to study the role of alternative spicing in environmental adaptation. We found genes that differed in the frequency of alternatively spliced introns in comparisons between mice sampled from the ends of this transect. A subset of those genes showed clinal patterns of splicing variation, and a further subset of five genes harbored cis-sQTL that overlapped with previously identified targets of selection. Notably, four of these five genes are associated with phenotypes that distinguish mice from the ends of the transect.
The impact of post-transcriptional processing on adaptation is a small but growing field of research. Some previous studies have focused on a particular phenotype associated with alternative splicing in a single gene (Gracheva et al. 2011; Jarosch et al. 2011; Howes et al. 2017; Ceinos et al. 2018). More recently, several studies have taken a genome-wide approach to study alternative splicing (e.g. Huang et al. 2021; Carruthers et al. 2022; Rodríguez‐Ramírez et al. 2023). Here, we looked for signatures of adaptive, alternative splicing genome-wide by studying mice along an environmental gradient. Phifer-Rixey et al. (2018) assayed the phenotypes of house mice collected from the same populations at the ends of the latitudinal transect (i.e. New Hampshire/Vermont and Florida). In addition to differences in nest-building behavior, activity, and blood chemistry, mice from these populations differ in body size. The larger body size of mice in New Hampshire/Vermont compared to mice from Florida reflects adaptation to colder environments and is consistent with Bergmann’s Rule. Four of the five candidate genes identified here are known to affect metabolism or growth and body size, suggesting potential links between alternative splicing and adaptive phenotypic change. In addition to these five genes, the other 36 genes showing clinal variation in splicing (but not harboring a cis-sQTL) might also underlie adaptive differences. Such genes may reflect downstream effects of trans-acting mutations elsewhere in the genome. They could also reflect splicing variants controlled in cis- for which we lacked power to detect the cis-sQTL. Ten of the 41 genes with clinal variation in splicing are also known to be associated with growth and body size in house mice. It is also possible that some of the genes showing clinal variation (particularly those without a cis-sQTL) reflect phenotypic plasticity in different environments (Steward et al. 2022).
Heritable variation in gene expression is a well-known source of adaptive evolution but has been primarily studied in the context of transcript abundance. Nonetheless, there is an increasing recognition of the importance of post-transcriptional regulation, and a handful of studies have used genome-wide short-read RNA-Seq to study mRNA abundance in conjunction with alternative splicing in the context of local adaptation (Huang et al. 2021; Jacobs and Elmer 2021; Carruthers et al. 2022; Singh et al. 2017; Steward et al. 2022; Huang et al. 2023; Rodríguez‐Ramírez et al. 2023). Several patterns have emerged from these studies. First, alternative splicing seems to play an important role in local adaptation in distantly related species, such as insects and vertebrates. Second, cis-sQTL have been identified that underlie splicing differences between locally adapted populations. Third, in most cases, there seem to be fewer differentially spliced genes than differentially transcribed genes (but see Carruthers et al. 2022), although this may be an artefact arising from differences in methodology as discussed below. Fourth, most studies find that the genes involved in alternative splicing and alternative transcription are not the same (but see Singh et al. 2017). Our results support and build upon the findings from these previous studies. We observed many genes showing differential splicing between populations in different environments and a substantial fraction also had a cis-sQTL. We also found little overlap between genes showing differences in transcription and those showing differences in splicing. Unlike previous studies, we explored variation along an environmental gradient, identifying 41 genes where splicing variants were correlated with latitude. Variation along environmental gradients is consistent with local adaptation as a response to spatially varying selection (Endler 1977). Moreover, by identifying cis-sQTL that overlap with targets of selection, we identified a small subset of genes that are strong candidates for adaptive alternative splicing.
By using short-read RNA-Seq data, we were able to directly compare alternative splicing and mRNA abundance. Using these same data, Mack et al. (2018) identified 458 differentially transcribed genes between New Hampshire/Vermont Florida while we identified 152 differentially spliced genes between these populations (Table 2). Differences in the methods and power of these two studies make it difficult to compare the total number of genes identified using each approach. For example, using Leafcutter, short-read data can only identify alternative splicing via split reads and these comprise only 39% of the total reads from the RNA-Seq data. However, the proportions of genes in subsequent analyses (i.e. clinally varying, harboring a cis-QTL, and overlapping with LFMM outliers) can be directly compared between these two studies (Table 2). Only six genes showed both differential transcription and differential splicing. Furthermore, there was no overlap in the genes identified for transcription and splicing in any of the other analyses. These results are consistent with previous work comparing adaptation through transcription and splicing. Jacobs and Elmer (2021) also found low overlap between these two mechanisms in the context of adaptation among benthic and pelagic ecotypes of salmonid fishes. Similarly, Rodríguez‐Ramírez et al. (2023) found low overlap between splicing and transcription as mechanisms facilitating adaptation in stickleback marine and freshwater Eda alleles on a shared marine genomic background. Of the genes that harbored a cis-QTL for either splicing or transcription, we observed the same proportion (5/24) showing overlap with LFMM hits. It is possible that splicing plays as important a role as transcription in adaptation and that the two mechanisms fulfill unique roles in adaptation.
RNA-Seq remains the most cost-effective technique for studying transcriptomes, but it is difficult to quantify whole isoforms from short-read data. In this study, we used split reads to identify alternative splicing events. Advances in long-read sequencing using PacBio and Nanopore technologies are now opening new avenues for studying isoforms. For example, tens of thousands of novel isoforms have been identified even in well-studied organisms like humans (Glinos et al. 2022; Yamaguchi et al. 2022), suggesting that these approaches will be particularly useful for studying alternative splicing in an evolutionary context.
Code availability
Bash, Python, and R scripts for data processing and subsequent analyses are available at https://github.com/dmanahan1/dmanahan2023_clinal_adaptive_splicing.
References
Artieri CG, Fraser HB (2014) Accounting for biases in riboprofiling data indicates a major role for proline in stalling translation. Genome Res 24(12):2011–2021
Barrett JC, Fry B, Maller J, Daly MJ (2005) Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21(2):263–265
Bergmann C (1847) Über die verhältnisse der wärmeökonomie der thiere zu ihrer grösse. Gottinger Stud 3:595–708
Carruthers M, Edgley DE, Saxon AD, Gabagambi NP, Shechonge A, Miska EA et al. (2022) Ecological speciation promoted by divergent regulation of functional genes within African cichlid fishes. Mol Biol Evol 39(11):msac251
Ceinos RM, Frigato E, Pagano C, Fröhlich N, Negrini P, Cavallari N et al. (2018) Mutations in blind cavefish target the light-regulated circadian clock gene, period 2. Sci Rep. 8(1):1–11
Cotto KC, Feng YY, Skidmore ZL, Ramu A, Kunisaki J, Conrad DF et al. (2020) RegTools: Integrative analysis of genomic and transcriptomic data to identify splice altering mutations across 35 cancer types. Cancer Res 80(16_Supplement):2136–2136
Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA (2003) DAVID: database for annotation, visualization, and integrated discovery. Genome Biol 4(9):1–11
Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S et al. (2013) STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29(1):15–21
Endler JA (1977) Geographic variation, speciation, and clines (No. 10). Princeton University Press, Princeton, NJ
Fraser HB (2013) Gene expression drives local adaptation in humans. Genome Res 23(7):1089–1096
Frichot E, Schoville SD, Bouchard G, François O (2013) Testing for associations between loci and environmental gradients using latent factor mixed models. Mol Biol Evol 30(7):1687–1699
Glinos DA, Garborcauskas G, Hoffman P, Ehsan N, Jiang L, Gokden A (2022) Transcriptome variation in human tissues revealed by long-read sequencing. Nature 608(7922):353–359
Gracheva EO, Cordero-Morales JF, González-Carcacía JA, Ingolia NT, Manno C, Aranguren CI et al. (2011) Ganglion-specific splicing of TRPV1 underlies infrared sensation in vampire bats. Nature 476(7358):88–91
Hijmans RJ, Cameron SE, Parra JL, Jones PG, Jarvis A (2005) Very high resolution interpolated climate surfaces for global land areas. Int J Climatol 25(15):1965–1978
Howes TR, Summers BR, Kingsley DM (2017) Dorsal spine evolution in threespine sticklebacks via a splicing change in MSX2A. BMC Biol 15:1–16
Huang Y, Lack JB, Hoppel GT, Pool JE (2021) Parallel and population-specific gene regulatory evolution in cold-adapted fly populations. Genetics 218(3):iyab077
Huang X, Li H, Shenkar N, Zhan A (2023) Multidimensional plasticity jointly contributes to rapid acclimation to environmental challenges during biological invasions. RNA 29(5):675–690
Jacobs A, Elmer KR (2021) Alternative splicing and gene expression play contrasting roles in the parallel phenotypic evolution of a salmonid fish. Mol Ecol 30(20):4955–4969
Jarosch A, Stolle E, Crewe RM, Moritz RF (2011) Alternative splicing of a single transcription factor drives selfish reproductive behavior in honeybee workers (Apis mellifera). Proc Natl Acad Sci 108(37):15282–15287
Jones FC, Grabherr MG, Chan YF, Russell P, Mauceli E, Johnson J et al. (2012) The genomic basis of adaptive evolution in threespine sticklebacks. Nature 484(7392):55–61
Kim D, Salzberg SL (2011) TopHat-Fusion: an algorithm for discovery of novel fusion transcripts. Genome biol 12(8):1–15
King MC, Wilson AC (1975) Evolution at two levels in humans and chimpanzees: Their macromolecules are so alike that regulatory mutations may account for their biological differences. Science 188(4184):107–116
Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat methods 9(4):357–359
Laurie CC, Nickerson DA, Anderson AD, Weir BS, Livingston RJ, Dean MD et al. (2007) Linkage disequilibrium in wild mice. PLoS Genet 3(8):e144
Li YI, Knowles DA, Humphrey J, Barbeira AN, Dickinson SP, Im HK et al. (2018) Annotation-free quantification of RNA splicing using LeafCutter. Nat Genet 50(1):151–158
Lynch CB (1992) Clinal variation in cold adaptation in Mus domesticus: verification of predictions from laboratory populations. Am Naturalist 139(6):1219–1236
Mack KL, Ballinger MA, Phifer-Rixey M, Nachman MW (2018) Gene regulation underlies environmental adaptation in house mice. Genome Res 28(11):1636–1645
Manning KS, Cooper TA (2017) The roles of RNA processing in translating genotype to phenotype. Nat Rev Mol cell Biol 18(2):102–114
McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A et al. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20(9):1297–1303
Morgan AP, Hughes JJ, Didion JP, Jolley WJ, Campbell KJ, Threadgill DW et al. (2022) Population structure and inbreeding in wild house mice (Mus musculus) at different geographic scales. Heredity 129(3):183–194
Phifer-Rixey M, Bi K, Ferris KG, Sheehan MJ, Lin D, Mack KL et al. (2018) The genomic basis of environmental adaptation in house mice. PLoS Genet 14(9):e1007672
Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. (2007) PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81(3):559–575
Rodríguez‐Ramírez CE, Hiltbrunner M, Saladin V, Walker S, Urrutia A, Peichel CL (2023) Molecular mechanisms of Eda‐mediated adaptation to freshwater in threespine stickleback. Mol Ecol https://doi.org/10.1111/mec.17201
Sherman BT, Hao M, Qiu J, Jiao X, Baseler MW, Lane HC et al. (2022) DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic acids Res 50(W1):W216–W221
Singh P, Börger C, More H, Sturmbauer C (2017) The role of alternative splicing and differential gene expression in cichlid adaptive radiation. Genome Biol Evol 9(10):2764–2781
Steward RA, de Jong MA, Oostra V, Wheat CW (2022) Alternative splicing in seasonal plasticity and the potential for adaptation to environmental change. Nat Commun 13(1):755
Wray GA, Hahn MW, Abouheif E, Balhoff JP, Pizer M, Rockman MV et al. (2003) The evolution of transcriptional regulation in eukaryotes. Mol Biol Evol 20(9):1377–1419
Wright CJ, Smith CW, Jiggins CD (2022) Alternative splicing as a source of phenotypic diversity. Nat Rev Genet 23(11):697–710
Yablonovitch AL, Fu J, Li K, Mahato S, Kang L, Rashkovetsky E et al. (2017) Regulation of gene expression and RNA editing in Drosophila adapting to divergent microclimates. Nat Commun 8(1):1570
Yamaguchi K, Ishigaki K, Suzuki A, Tsuchida Y, Tsuchiya H, Sumitomo S et al. (2022) Splicing QTL analysis focusing on coding sequences reveals mechanisms for disease susceptibility loci. Nat Commun 13(1):4659
Zhou X, Stephens M (2012) Genome-wide efficient mixed-model analysis for association studies. Nat Genet 44(7):821–824
Acknowledgements
The authors thank Leah Hyee Ryun Lee and Yocelyn T. Gutiérrez-Guerrero for their help with bioinformatic analyses, and Yocelyn T. Gutiérrez-Guerrero and Mallory A. Ballinger for their suggestions and comments. We thank Katya L. Mack and Megan Phifer-Rixey for providing data on adaptive gene expression and genome-wide signatures of selection, respectively. This work was possible thanks to an allocation of Advanced Cyberinfrastructure Coordination Ecosystem: Services and Support (ACCESS) to Michael W. Nachman and Megan Phifer-Rixey. This work was supported by the National Institutes of Health (R01 GM127468 and R35 GM149304).
Author information
Authors and Affiliations
Contributions
DNM and MWN designed the project. DNM conducted the analyses and produced the figures. Both authors reviewed and interpreted the data and contributed to writing the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Arne Jacobs.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Manahan, D.N., Nachman, M.W. Alternative splicing and environmental adaptation in wild house mice. Heredity 132, 133–141 (2024). https://doi.org/10.1038/s41437-023-00663-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-023-00663-0
