
Abstract
Phylogeography often focuses on the spatial dimension of genetic diversity, rarely including the temporal dynamics occurring interannually among local populations, which can provide insight into past variations in reproductive success. Currently, there is an intense aquaculture industry of Mytilus spp. on the Southeast Pacific Coast which depends entirely on the spat released by natural populations forming a relevant and sensitive social-ecological system. Temporal and spatial spat variability from natural mussel beds could be related to interannual reproductive dynamics with variable reproductive success and recruitment, which leave genetic signatures. Temporal and spatial genetic structure was evaluated in six natural beds in the Southeast Pacific (from 39°25’S to 43°07’S) on the most abundant and widespread Mytilus lineage detected, Mytilus cf. chilensis, in 4 consecutive years. Analyses included data from >180 individuals per year, with a total of 751 (mitochondrial COI) and 747 (nuclear H1) individuals, respectively. Overall, both markers showed high haplotype diversity and low spatial and temporal genetic differentiation. Likely, the high dispersal capacity of Mytilus cf. chilensis maintains population homogeneity and prevents diversity erosion. The slight differences in genetic variance of COI were better explained by differences among sites (space), and conversely, the H1 genetic variance was better explained by interannual (temporal) comparisons, which could explain temporal variability in spat availability. This study highlights the important insights achieved with the evaluation of both temporal and spatial population genetic structures in marine species with high reproductive output, which can condition the success and sustainability of the relevant social-ecological system.
Similar content being viewed by others

Introduction
Phylogeography considers the temporal and spatial genetic structure within a species. However, most studies focus solely on the spatial dimension not including the dynamics of the reproductive output which could be revealed by signatures in the temporal variation of the genetic structure of populations (Berger et al. 2006; Barshis et al. 2011; Haye et al. 2021). Because populations with variable reproductive output show genetic structure over time (Flowers et al. 2002; Siegel et al. 2008; Marshall et al. 2010), the temporal genetic structure of populations could inform about past population reproductive dynamics modulated by reproductive output and provide an indirect assessment of the variability in reproductive success of populations. This is particularly relevant in marine species inhabiting variable environments.
Many marine benthic species broadcast-spawn their gametes to the water column and have a planktonic larval developmental phase of variable duration. Long reproductive larval development has been associated with high variance in reproductive output (Li and Hedgecock 1998; Pineda et al. 2010; Hedgecock and Pudovkin 2011), as a response to environmental variations linked to differential survival, and patchy and stochastic larval recruitment (Jackson et al. 2017; McKeown et al. 2017; Villacorta-Rath et al. 2018). These species are prone to displaying large variance in reproductive success and stochasticity in reproductive events (Li and Hedgecock 1998; Hedgecock and Pudovkin 2011; Pérez-Portela et al. 2018; Vendrami et al. 2021), which can generate greater population genetic differentiation between year classes and body size than between spatially discrete populations (Hilbish et al. 2002). This dispersal and connectivity pattern (Lee and Boulding 2009), known as the chaotic genetic patchiness paradigm (Johnson and Black 1984; David et al. 1997; Eldon et al. 2016), is associated with patchy and stochastic larval and recruitment survival, collective dispersal, and sweepstakes reproductive success (Hedgecock 1994; Flowers et al. 2002; Bierne et al. 2003; Vadopalas et al. 2012; Owen and Rawson 2013; Kesäniemi et al. 2014; Eldon et al. 2016; Plough et al. 2016; Jackson et al. 2017; Villacorta-Rath et al. 2018). Consistently, studies have shown that species with high dispersal potential (planktonic larval dispersal >2 weeks) usually display high temporal genetic instability of populations, because of variations in the reproductive output of local populations temporarily changing the local gene pool, according to the chaotic genetic patchiness paradigm (e.g., Ng et al. 2010; Jackson et al. 2017; Villacorta-Rath et al. 2018; Salloum et al. 2019; Vendrami et al. 2021).
Mytilus chilensis Hupé 1854 is a Chilean endemic marine blue mussel found in protected areas in the southeast Pacific on the Chilean coast (Barria et al. 2012; Molinet et al. 2015; Larraín et al. 2017) with a geographical distribution from the Tirúa River (38°S) to Punta Arenas (53°S) in the Magellan province (Toro et al. 2004, 2006; Astorga et al. 2020; Gardner et al. 2021), in which individuals form discrete dense beds of sessile adults at 10 to 25 meters of depth (Brattström and Johanssen 1983). The species’ natural populations have been extensively exploited, leading to the collapse of natural populations, as has been suggested for other commercial resources in the area (Gelcich et al. 2010).
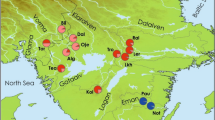
Although there are other species of the genus in the geographic area (i.e., M. edulis and M. galloprovincialis), and other mussels in sympatry (Choromytilus chorus, Aulacomya atra, Semimytilus algosus, Perumytilus purpuratus), M. chilensis is the preferred species for aquaculture in the southeast Pacific because it provides better growth outcomes (Díaz et al. 2019). Currently, natural populations of M. chilensis contribute with less than ~1% of the harvested biomass, the 99.66% remaining being provided by the mussel aquaculture (390,000 tons on average) (Sernapesca 2020, 2021). In consequence, a significant socio-ecological system has been structured around the mussel aquaculture in the inner sea of Chiloé and Reloncaví Sound in northern Patagonia (Molinet et al. 2017; Fernández et al. 2018; Gonzalez-Poblete et al. 2018; San Martin et al. 2020). This system strongly depends on natural beds for the collection of spat required for aquaculture. Understanding temporal variability is particularly relevant, given that a few sites have been used for the collection of spat, such as southern Chiloé and different areas of the Reloncaví Sound (Fig. 1). The geographic area where more than 99% of the mussel aquaculture facilities are placed coincides with the occurrence of most natural beds of M. chilensis in northern Patagonia, between 41°30’S and 43°10°S in the Reloncaví Sound and the Inner Sea of Chiloé (Sernapesca 2020, 2021). Areas of spat collection vary in time according to spat availability (Winter et al. 1984; Uriarte 2008; Molinet et al. 2017, 2021); from ~15 years ago, larval abundance and recruitment of M. chilensis have shown a strong temporal and spatial variability (Avendaño et al. 2011; Barria et al. 2012; Campos and Landaeta 2016; Lara et al. 2016), becoming a main concern for the sustainability of the mytiliculture social-ecological system.

Sampling in these natural beds was done by local fishers and were replicated during four consecutive Austral summers (2014-2017).
The reproductive cycle of M. chilensis includes broadcast spawning of gametes and external fertilization, followed by several free-living larval stages with a total duration of 30–45 days in the water column (Toro and Sastre 1995; Toro et al. 2004), providing the species with a high dispersal potential by larval connectivity (Bohonak 1999). Most of the population genetic structure studies of M. chilensis have described moderate to low genetic differentiation among mussel beds in northern Patagonia, including the Reloncaví Sound and the Inner Sea of Chiloé (Toro et al. 2004, 2006; Larraín et al. 2015; Araneda et al. 2016; Astorga et al. 2018, 2020). When considering the range of distribution including southern Patagonia (south of 51°S), significant genetic differentiation has been broadly reported between northern and southern Patagonia (e.g., Araneda et al. 2016; Astorga et al. 2020; Díaz-Puente et al. 2020), showing also that northern Patagonia is likely a single gene pool.
Taxonomical identification in the genus Mytilus can be challenging, particularly using traditional morphological and molecular DNA barcoding tools. For example, the mitochondrial marker (mtDNA) Cytochrome Oxidase I (COI), a standard and highly validated tool for DNA barcoding (Valentini et al. 2009), could not be a proper tool for taxonomical validation in the Mytilus species complex due to a lack of resolution among close species of Mytilus (Tinacci et al. 2018), the double uniparental inheritance (Garrido-Ramos et al. 1998) and the hybridization and introgression among Mytilus species (Beaumont et al. 2008; Toro et al. 2012). Altogether evidence suggests that a single mtDNA marker could not be enough information for taxonomical identification and validation of Mytilus spp. (Larraín et al. 2019). Furthermore, Guisti et al. (2020) questioned the validation of the endemic species in the Southeast pacific coast (Mytilus chilensis), suggesting that this is a lineage of M. galloprovincialis not significantly distinct from the northern hemisphere Mediterranean mussel. However, recent studies have provided robust evidence through single and multi-locus approaches of a distinct native Chilean mussel, M. chilensis, as a valid taxonomical unit, which is likely to dominate in the southeast Pacific coast (Larraín et al. 2019; Oyarzún et al. 2021; Araneda et al. 2021).
The goal of this study was to evaluate the interannual and spatial genetic structure of Mytilus spp. in northern Patagonia from six natural beds between 39°25’S, 73°13’W and 43°07’S, 73°42’W (Fig. 1), as an indirect measure of past variations in reproductive success, for 4 consecutive years. Considering the presence of syntopic species, hybridization, and introgression among species, we used COI as a phylogeographic tool to evaluate temporal and spatial genetic diversity and structure in the most abundant, widespread, and monophyletic haplogroup (lineage), putatively assigned as M. chilensis (M. cf. chilensis). Given that in northern Patagonia the spat availability is variable in space and time (Barria et al. 2012), temporal genetic population structure serves as a proxy of past variations in reproductive success and as a complementary approach to studies of variation in larval availability, reproductive output, and recruitment (Avendaño et al. 2011; Díaz et al. 2011; Barria et al. 2012), for understanding temporal reproductive variations.
Methods
Samples of Mytilus spp. were collected during four consecutive austral summers (March) from 2014 to 2017. To avoid overlapping generations, only juvenile-size individuals of ~2–3 cm of shell length were collected in each of the sites and years. Samples were obtained in situ by local fishers in six natural mussel beds located between Mehuín (ME; 39°25’S, 73°13’W) and Yaldad (YA; 43°07’S, 73°42’W) (Fig. 1 and Table 1).
Mytilus spp. is considered as a species complex, with hybridization processes among sympatric and parapatric species (Beaumont et al. 2008; Toro et al. 2012). The overlapping morphological traits among species/lineages present in north Patagonia (M. edulis and M. galloprovincialis), particularly among juveniles, impedes robust morphological species identification.
Due to the biparental inheritance of mitochondrial DNA in Mytilus spp. (Rawson et al. 1996; Garrido-Ramos et al. 1998; Burzyński et al. 2006), although supposedly low in M. chilensis (Larraín et al. 2019) and given that the M-type (Male type) mitochondrial lineage can theoretically only be detected in gonadal and mantle tissues in adult Mytilus individuals (Gérard et al. 2008), only abductor muscle tissue from juveniles was collected to ensure obtaining only matrilineal mitochondrial lineages. Moreover, using small spat drastically reduces the probability of amplifying M-type mtDNA in males, as the latter is overexpressed in ripe adult male gonadal tissue. In juvenile somatic tissue, F-type is the predominantly overrepresented mtDNA type. In addition, the M-type and F-type mitochondrial lineages have an average divergence of at least 20% (Garrido-Ramos et al. 1998), which can be detected through simple visual inspection of DNA sequence alignment afterward.
Small pieces of 2–3 mm3 of fresh posterior abductor muscle were dissected from each individual and preserved in absolute ethanol at −20 °C until DNA extractions. Genomic DNA was extracted from 25 mg of tissue using the EZNA DNA extraction kit (Omega, USA) following the manufacturer’s protocol. To amplify a portion of the mtDNA COI, PCRs were performed using the universal primers HCO and LCO (Folmer et al. 1994).
We focused our phylogeographic analyses on the population genetic structure of the single and most widespread lineage or haplogroup in the study area. For this, the presence of different lineages was determined using genetic distance among COI sequence data for all sampled Mytilus spp. individuals (Fig. 2). For further population genetic analyses, only the single haplogroup that was frequent and widespread was included, which was putatively assigned to the Chilean endemic lineage, M. cf. chilensis. This approach was not designed as a taxonomical validation method but rather as a phylogeographic criterion for the analysis of populations of a monophyletic group. With this phylogeographic approach, issues with mislabeling in public databases such as GenBank (Tinacci et al. 2018; Giusti et al. 2020), lack of resolution of COI gene sequences to distinguish in certain cases between M. chilensis and M. galloprovincialis (Tinacci et al. 2018), and the potential introgressions among Mytilus species are avoided.

Complex, displaying three haplogroups putatively assigned to Mytilus cf. chilensis, M. cf. galloprovincialis and M. cf. edulis.
To complement the mtDNA sequence data in subsequent analyses of temporal genetic diversity and spatial structure and to incorporate the information of another cellular compartment, sequence data of the nuclear gene H1 was included for individuals of the frequent and widespread COI haplogroup. To choose a nuclear sequence with variation at the population level, PCRs for several nuclear genes were tested with primers from the literature (data not shown) and with newly designed primers for H1 (H1-238F 5’-AAG GGT GTC TTC GTC GAG GT-3’ and H1-998R 5’-AGG GCT TTT GGT GGC TGA AT-3’) from aligned H1 sequences available from several species of the genus Mytilus in GenBank. Even though H1 sequences are not a commonly used marker for phylogeographic studies, we chose H1 because, in our assessment, it showed greater polymorphism than other nuclear sequence markers and great PCR and sequencing success.
PCRs for COI and H1 were carried out with the following conditions: 1× Buffer, 1.3 mM MgCl2, 0.2 μM of each primer, 0.8 mM dNTPs, 0.03 U/μL of Invitrogen Platinum Taq polymerase enzyme, 0.075 mg/mL BSA and 1 μL of DNA. The cycling conditions consisted of an initial denaturation at 94 °C for 10 min, 35 cycles of denaturation at 94 °C for 1 min, annealing for 1 min, and extension at 72 °C for 2 min, ending with a final extension at 72 °C for 13 min. PCR annealing temperatures were 40 and 57 °C, for COI and H1, respectively. Sequencing of purified amplicons was performed on an automated ABI 3730XL sequencer (Applied Biosystems). Obtained sequences were aligned using Geneious R10 (www.geneious.com).
To apply phylogeographic analytical methods designed for haplotypes to the H1 data (diploid), the nuclear H1 sequences were transformed into haplotypes using the Bayesian framework implemented PHASE 2.1.1 (Stephens et al. 2001) in the software DnaSP 6.0 (Rozas et al. 2017). The settings used for running PHASE were determined after several run tests with a different number of iterations and chains consisting of five chains of 10,000 iterations, a thinning interval of 10, and 10% Burn-in. Only haplotypes with posterior probability greater than 0.9 were used for analyses (e.g., Haye and Muñoz-Herrera 2013).
The following analyses were carried out separately for each marker (COI and H1). DnaSP 6.0 was used to obtain the genetic diversity measures for sites and years, including number of haplotypes, number of segregating sites, haplotype diversity, mean number of pairwise nucleotide differences, and nucleotide diversity). Sample sizes were evaluated to determine if they were enough to recover most of the genetic diversity within the sampled populations and years. For this, haplotype accumulation curves were constructed for each year and marker using the function haploAccum with a total of 10,000 permutations in the R 4.2.0 (R Development Core Team 2022) package SPIDER (Brown et al. 2012).
Genetic differentiation by sampling site and year was assessed with FST (Weir and Cockerham 1984), and the significance was assessed after false discovery rate correction (Q value < 0.05) (Benjamini et al. 2005). Pairwise FST among years was estimated both using haplotype frequencies and unique haplotypes in Arlequin 3.5 (Excoffier and Lischer 2010). Hierarchical evaluations of FST based on haplotype frequencies were used to compare and evaluate the effect of sampling site and sampling year in the population genetic structure, considering variation among sites within years and among years within sites, using analysis of molecular variance (AMOVA) in Arlequin 3.5.
Permutation-based multivariate analyses (PERMANOVA) were performed using the adonis function in the Vegan R package (Oksanen et al. 2020) to evaluate the decomposed variance of genetic distance matrices, with sampling site and years as crossed factors (instead of a hierarchical model such as AMOVA). For this, Bray–Curtis distance matrices based on haplotype frequencies were calculated in Vegan with the vegdist function for each marker. Obtained matrices were corrected using Hellinger’s Method with the decostand function, and significant values of PERMANOVA were assessed using 10,000 permutations.
Tests of congruence among distance matrices (CADM) were explored to evaluate the temporal stability of genetic differentiation (Legendre and Lapointe 2004) using site pairwise FST matrices for each year with the CADM.global and CADM.post functions implemented in the APE package (Paradis and Schliep 2019) in R. CADM was implemented using the pairwise FST matrices between sites for each year for both markers, with 10,000 permutations, to evaluate the congruence of the genetic distance matrix of each year against all the matrices of the other years through a Kendall coefficient of matrix concordance (W). A high and significant Kendall coefficient suggests a high concordance between year matrices (therefore temporal stability). In addition, extended Mantel tests were performed using a post hoc Holm’s correction using the CADM.post function. Considering that the null hypothesis was complete incongruence among matrices, higher Mantel values indicate greater concordance/correlation between matrices and, thus, temporal stability.
Results
We sequenced a total of 803 individuals of Mytilus spp. from 4 consecutive years in six natural beads along the Southeast Pacific coast. COI data revealed three haplogroups (Fig. 2 and Supplementary Table S1). One is highly frequent and widespread, putatively Mytilus cf. chilensis, that included 751 of the sampled individuals. The rest of the individuals corresponded to highly divergent and less frequent lineages assigned according to BLAST search to M. edulis (46 individuals, 106 mutational steps) and M. galloprovincialis (6, 21 mutational steps) (Fig. 2 and Supplementary Table S1), which displayed only haplotypes of a single frequency (with only one exception) and not randomly distributed among years and sites, being most frequent in the northern sites of Metri and Los Molinos, and particularly in years 2015 and 2016. All natural beds displayed more than one species of Mytilus in syntopy. Due to the low frequency of the two divergent haplogroups, phylogeographic analyses focused on the most frequent, widespread, and monophyletic linage, putatively M. cf. chilensis. Albeit this, all the posterior analyses based on COI data, were performed with all the sampled individuals, showing that the general patterns described in this study remain congruent when considering all Mytilus spp. collected (Supplementary Tables S1 and S5–S7).
The 751 individuals of the COI haplogroup identified as M. cf. chilensis consisted of an average of 30 individuals per sampling site and year, with 180–200 individuals per year, and were used to obtain partial sequences of the nuclear gene H1. The final COI and H1 datasets for M. cf. chilensis haplogroup contained 751 and 747 sequences of 610 and 691 base pairs of length, respectively (GenBank Accession numbers, COI MZ773651–MZ773880, and H1 MZ901916–MZ902024) (Supplementary Table S1). The COI sequences included 230 haplotypes, with a total of 121 polymorphic sites, of which 74 were informative for parsimony and 47 singletons. For H1, the 747 diploid sequences resulted in 1012 haploid sequences that corresponded to 111 haplotypes, with 77 polymorphic sites, 43 sites informative for parsimony and 34 singletons. The number of shared haplotypes between sites was 42 and 47 for the COI and H1 data, respectively, while the number of unique haplotypes for a single sampling site was 188 and 64, respectively. Overall, genetic diversity was high (Table 1); haplotype diversity values were, in all the cases, in the upper 15% (>0.85) of possible values. Simulations using haplotype accumulation curves suggest that the number of individuals sampled by year was not enough to account for the observed diversity, being this pattern is more pronounced in COI than H1 (Supplementary Figs. S1–S4). Extrapolated plateau of COI was reached with a sample size approximately three times greater than the one we used, and two times greater for H1. Number of segregating nucleotide sites was higher for COI as well as the nucleotide diversity that indicates less genetic distance within COI than H1 haplotypes. In general, diversity indices were higher in sites from the Inner Sea of Chiloé (i.e., PI, PU and YA).
The COI haplotype network had a star-like pattern with the most frequent haplotypes homogenously distributed among years and sites (Fig. 3). The seven most frequent haplotypes were present in all sites and years, and the four most common haplotypes always represented more than 50% of the individuals (Fig. 3). There was some variability of the haplotype frequencies among sites/years (Figs. 3 and 4). The year 2015, for example, showed a higher relative frequency of the less common haplotypes (still within the 14 most common ones) (Fig. 4). COI genetic differentiation between pairs of sites for each year was low (Supplementary Table S2), with FST values ranging from 0 to 0.176 between ME and MT in 2017. In 2014, the only significant FST values were those that included comparisons with the site LM; in 2015, the two significant values were restricted to population pairwise comparisons including the site PI; in 2016, most pairwise comparisons including ME and all the ones including YA were significant. Finally, the year 2017 showed the highest number of significant values across sites (Supplementary Table S2).

Median-joining networks of 230 COI (A, B) and 111 H1 (C, D) haplotypes of Mytilus cf. chilensis. In A and C, each color represents a sampling year (time) according, and in B and D, colors represent sampling sites (space). Each circle represents a haplotype, and the size is proportional to the number of individuals that display each haplotype. Each link between haplotypes corresponds to a mutational step and crossed lines in the links represent additional mutational steps. Acronyms for sample sites according to Fig. 1.

Relative frequencies per sampling site and year of the 14 most frequent haplotypes of A COI and B H1 of Mytilus cf. chilensis. Acronyms for sample sites according to Fig. 1.
The H1 haplotype network was characterized by one most frequent haplotype with the highest representation in all sites and years, and several additional shared haplotypes among sites and years which emerged subsequently one to each other from the most common one (Fig. 3). Excluding the most common haplotype, the next 13 most frequent haplotypes varied in presence and frequency between sites and years (Fig. 4). The pairwise FST values of H1 were mostly non-significant, the two significant values were from the year 2017 in pairwise values that included ME (Supplementary Table S2).
In general, there was a low genetic differentiation between years, with FST values from 0 to 0.047 (Supplementary Table S3). For COI, the sampling sites PU, and YA, showed significant temporal FST differentiation values in comparisons in LM between the years 2014 and 2015 and between 2015 and 2017, and in PU between 2016 and 2017. Differentiation using unique haplotypes UST showed significant values in ME between 2014 and 2015, in three comparisons in LM (2014–2015, 2015–2016 and 2015–2017), in PU between 2014 and 2017 and in YA between 2016 and 2017. For H1 no FST value was significant, and four values of UST were significant: in PI between the years 2014–2016 and 2014–2017, and in PU between 2014–2015 and 2015–2017.
AMOVA using year and site as a priori groupings, respectively, showed contrasting results among markers (Table 2). Most of the COI genetic variance was explained by differences within populations in both sites and years and genetic differentiation among years was non-significant (FCT = −0.004, p = 0.99), i.e., no significant temporal variation of the COI genetic variance. For H1, although only 0.33% of the genetic variance was explained by differences among years, the descriptive statistic FCT was significant (FCT = 0.003, p = 0.022), indicating that variation among years had a significant contribution to the H1 genetic variance. On the contrary, AMOVA grouping by sites instead of years in H1 led to non-significant values (FCT = 0.002, p = 0.240), suggesting that sampling sites did not contribute to the distribution of the genetic variance in H1.
PERMANOVA analyses using site and year as crossed factors were consistent with AMOVA results. For COI, there was significance when sites were considered as the fixed factor (Site F value = 1.299; p = 0.01; Year F value = 0.05, p = 0.820), while for H1 this result was inverted, i.e., the year was the most important factor to explain genetic variability (Year F value = 2.313; p < 0.001; Site F value = 0.128; p = 0.99) (Table 3). Similarly, CADM results for COI showed marginally significant Kendall coefficients of concordance (W) among distance matrices for analyses performed with the full data set of M. cf. chilensis (W = 0.39; χ2 = 22.150; p = 0.049) and using only shared haplotypes (W = 0.410; χ2 = 22.950; p = 0.042) (Table 4). The congruence of the genetic distance matrices among years suggests temporal stability for the COI data. In contrast, for H1, Kendall coefficient values of concordance were low and non-significant for the full data set (W = 0.266, χ2 = 14.888, p = 0.394) and shared haplotypes (W = 0.250, χ2 = 15.000, p = 0.452), suggesting temporal genetic variation of the H1 data (Table 4). A posteriori Mantel correlation values (CADM.post) for H1 were also incongruent, suggesting temporal genetic differentiation. Values ranged from −0.600 to 0.434 and were significant only for the comparison of 2015–2017 using shared haplotypes (Fig. 5). The year 2014 showed the lowest Mantel correlation values, ranging from −0.600 to −0.029, suggesting that in this particular year, haplotype frequencies were consistently incongruent with respect to all other years (Fig. 5).

Values and colors indicate the Mantel test statistic. A CADM with all haplotypes. B CADM with shared haplotypes. Significant p value with an asterisk.
Discussion
With more than 800 individuals sampled from six natural beds through four consecutive years in the southeast Pacific, there was one most abundant lineage of Mytilus spp., putatively Mytilus cf. chilensis, which had high genetic diversity in comparison to natural populations of Mytilus worldwide (Zardi et al. 2007; Riginos and Henzler 2008; Han et al. 2016; Pickett and David 2018). Reduced genetic diversity can be expected with overfishing and leads to increased population vulnerability (Hauser et al. 2002; Pinsky and Palumbi 2014). The absence of signals of reduced diversity in M. cf. chilensis stands out in that a highly exploited species has a high degree of genetic diversity, a main requirement for the adaptation and resilience of populations (Bitter et al. 2019).
M. cf. chilensis showed to have homogeneous and well-connected natural beds and slight but significantly contrasting differentiation in both spatial and temporal genetic structure. The mtDNA COI and the nuclear H1 markers displayed different spatial-temporal genetic structure signals. Overall, COI data had a greater spatial genetic structure than the nuclear H1, and vice-versa, H1 showed a greater temporal genetic structure than COI.
The low but significant temporal genetic structure of M. cf. chilensis based on H1 could explain the described temporal variations in the availability of spat from natural beds (Avendaño et al. 2011; Barria et al. 2012; Molinet et al. 2021). Different temporal genetic structure signals between markers could be due to their different effective population size and degree of genetic diversity. Possibly, as shown by the haplotype accumulation curves, the COI haplotype diversity was not well-captured with the studied sample size. The effects of under-sampling such a high diversity, as the one detected for COI, albeit using a relatively high sample size, could also explain detected differences in the temporal genetic structure among studied genetic markers. Mito-nuclear discordance, i.e., different phylogenetic or phylogeographic signals among mitochondrial and nuclear markers, is a widespread and relevant phenomenon (Larmuseau et al. 2010; Toews and Brelsford 2012), normally attributed to differences in the linage sorting process between markers with different effective population sizes (e.g., Debiasse et al. 2014; Lee et al. 2021), and to life history traits such as dispersal, mating, and reproductive success (Toews and Brelsford 2012). However, it is improbable that linage sorting can explain the temporal population genetic variation of M. cf. chilensis. It is more likely a consequence of the high genetic diversity of COI when compared with H1 and enhanced by the different patterns of heredity of nuclear and mitochondrial genomes.
Another explanation for the slight temporal structure detected with the H1 nuclear marker could be habitat dynamics that affect variability in post-settlement processes, causing temporal reproductive segregation (Bierne et al. 2003). Analyses of the population abundance of M. chilensis indicate temporal variation and rapid changes in local abundance in the Reloncaví fjord (Molinet et al. 2015). In addition, local environmental conditions in the study area are highly variable in time, with seasonal fluctuations and intromission of warm sub-superficial waters (>10 °C) (González et al. 2011). The Northern Patagonia area, for example (from ~41.5°S to 43.9°S), has been described mainly as an estuary zone, with the influence of the interaction of freshwater from rivers and ice melting with oceanic water masses from the south Pacific (Sievers and Silva 2006), causing significant dynamics in salinity, chlorophyll (González et al. 2011; Narvaez et al. 2019), temperature, pH and oxygen (Sievers and Silva 2006; Narvaez et al. 2019). These temporal fluctuations may impose physiological challenges to populations of benthic marine organisms. In mollusks, variation in salinity and pH (acidification) affects respiration, growth, calcification, and reproduction rates (Navarro 1988; Gazeau et al. 2007; Duarte et al. 2014, 2018). In specific, for M. chilensis the capability and magnitude of trait responses depend on the environmental history of their native habitats to which individuals and ancestors have been exposed (Duarte et al. 2014).
Early studies and several following ones that aimed at detecting temporal genetic structure in marine species were performed with conservative allozyme loci and often were able to detect temporal genetic structure (David et al. 1997; Moberg and Burton 2000; Robainas-Barcia et al. 2008), and studies performed with sequence markers such as COI have also evaluated temporal genetic structure (Flowers et al. 2002; Haye et al. 2021). Given the temporal genetic population structure detected with H1, using an interannual scale evaluating persisting natural beds of Mytilus chilensis, this study prompts further analyses involving higher resolution methodologies that could more accurately depict population structure at a fine spatial scale (e.g., Ng et al. 2010; Villacorta-Rath et al. 2018; Quintero-Galvis et al. 2020). Although studies of temporal population genetic structure are often not comparable because they use different methodological approaches (i.e., focus species, geographic location, years of sampling, and molecular markers), available studies of marine species on the southeast Pacific provide somewhat contrasting results (Rojas-Hernandez et al. 2016; Quintero-Galvis et al. 2020; Haye et al. 2021). On the one hand, the brachyuran crab Metacarcinus edwardsii showed a pattern of temporal stability attributed to high dispersal coupled with high and temporally stable reproductive output (Rojas-Hernandez et al. 2016). On the other hand, the muricid gastropod Concholepas concholepas, also with high larval dispersal potential (>3 months), displayed high temporal genetic variation, likely linked to variations in reproductive success in response to the environment (Quintero-Galvis et al. 2020). Finally, the ascidian Pyura chilensis, which has low intrinsic dispersal potential and high potential for anthropogenic transport in ships and boat hulls, harbors three well-differentiated intraspecific lineages that have disparate patterns of COI temporal genetic structure. For this species, one of the lineages had a stronger temporal than spatial genetic structure, indicating reproductive and connectivity differences among species’ lineages (Haye et al. 2021).
In the genus Mytilus, temporal population genetic variation (e.g., among year classes) has been reported in coastal areas for M. edulis (Koehn et al. 1976; Pedersen et al. 2000; Hilbish et al. 2002; Gilg and Hilbish 2003; Simon et al. 2020), M. galloprovincialis (Gardner and Palmer 1998; Gilg and Hilbish 2003; Gilg et al. 2016), and M. trossulus (Pedersen et al. 2000). In general, like for M. cf. chilensis, Mytilus species from different geographic areas display low to moderate levels of temporal genetic structure, suggesting that the presence of a moderate reproductive success variation could be a widespread pattern for mytilids. Similarly, at a broader taxonomic scope, changes in the temporal genetic structure of populations have been widely reported, to different degrees, in different species of marine invertebrates with high dispersal potential, high fecundity, and in populations without geographic differentiation, including species of crustaceans (Robainas-Barcia et al. 2008; Jackson et al. 2017), gastropod mollusks (Salloum et al. 2019), and echinoids (Flowers et al. 2002; Calderón et al. 2012; Couvray and Coupé 2018).
The human-mediated spat movement likely potentiates the autonomous intrinsic high dispersal potential of M. chilensis given by its long-lived larvae, contributing to population connectivity, and maintaining highly diverse natural beds, as has been described for M. edulis in offshore installations in the North Sea (Coolen et al. 2020). A similar pattern was suggested by Norrie et al. (2020) for the mussel Perna canaliculus in New Zealand, in which larval spillover from farmed populations provided larval subsidy for the restoration of natural beds. Simon et al. (2020) also showed that for both M. edulis and M. galloprovincialis, there was a significant influence of human-mediated transport in population genetic connectivity. Mussel aquaculture could also promote temporal genetic structure by transporting spat from different source populations in time to an extensive geographic area where mariculture is developed. The enhancement of population connectivity driven by the mussel aquaculture similes the model of spillover subsidy of aquaculture farms and provides an opportunity for a network management approach of Mytilus spp. (Ludford et al. 2012; Norrie et al. 2020).
Geographic distribution of the genetic diversity has been widely studied in M. chilensis using different markers. Overall, studies show that the study area on the southeast Pacific coast is composed of connected and diverse populations (Toro et al. 2004, 2006; Araneda et al. 2016; Astorga et al. 2020; Díaz-Puente et al. 2020), although there is variation in the degree of structure detected among studies. Differences can be attributed to the different markers used (e.g., Araneda et al. 2016; Astorga et al. 2020), to the large temporal variation in reproductive success and environmental and anthropogenic local processes (e.g., Astorga et al. 2020), and to the aquaculture industry enhancing connectivity by translocating spat (Díaz-Puente et al. 2020). Temporal population genetic variation could contribute to the differences among phylogeographic and population genetic studies performed with samples obtained in different time periods and locations.
The low yet significant temporal genetic variation among years detected between distance matrices shows a greater differentiation of the year 2014 with respect to the other years. From 2015 on, haplotype frequencies of M. cf. chilensis using both markers significantly changed in the study area with respect to 2014. This change could be attributed to the strong El Niño Southern Oscillation (ENSO) event of 2015 (Blunden and Arndt 2016; Santoso et al. 2017; Molina et al. 2021); the pre- and post-ENSO change in haplotype frequencies could be linked to the environmental variability associated with ENSO events that can impact the reproductive success and connectivity dynamics of marine populations. A similar pattern of changes in the temporal genetic structure associated with ENSO was reported in a broad geographic area along the southeast Pacific for the ascidian Pyura chilensis, which also showed greater temporal genetic differentiation in the years 2015–2016, likely associated with the ENSO event of 2015 (Haye et al. 2021). Environmental variables, such as temperature and nutrients, display great variations during ENSO events (e.g., LaVigne et al. 2013; Santoso et al. 2017), affecting species’ reproductive traits (e.g., Cantillánez et al. 2005; Manríquez et al. 2018; Avila-Poveda et al. 2021). Particularly, and in agreement with interannual genetic structure, Narváez et al. (2019) showed that temperature anomalies of 2014 differed from 2015, 2016, and 2017 in the western and the eastern coasts of Chiloé Island (41–43°S). During ENSO events, the genetic pool of broadcasting species with reproductive output may get shuffled due to changes in reproductive success, larval development (e.g., Ruiz et al. 2008), and larval settlement (e.g., Fuentes-Santos and Labarta 2015). These results highlight the importance of interannual comparisons in genetic structure to reveal signatures in genetic diversity and population structure of strong stochastic events such as ENSO.
Despite the limitations of the markers used in terms of spatial resolution (in comparison with hypervariable markers), and taxonomical validation, our phylogeographic approach appears to be relevant for the study of the temporal and spatial genetic structure of a species highly demanded for human consumption, which represents a relevant social-ecological system, particularly for the northern Patagonian area in the southeast Pacific Coast.
Conclusion and final remarks
In phylogeography, studies often focus on the spatial structure using only one temporal scale, rarely incorporating the temporal dynamics of population genetic variation in nature, which is a proxy of past variations in reproductive success. Herein, temporal interannual comparisons of natural populations of M. cf. chilensis in northern Patagonia showed that the main haplogroup conforms to a connected gene pool with high genetic diversity and with some interannual variations in haplotype frequencies, which could explain fluctuations in natural spat availability. Results also highlight the need to analyze the spatial and temporal genetic variation of populations to shed light on population dynamics resulting from reproductive success, particularly on marine broadcasting species inhabiting highly variable environments prone to variable reproductive output and reproductive success. Special attention to population structure should be given to species, such as M. chilensis, that provide relevant social-ecological services. For adequate management of persistent mussel beds and sustainability of the mussel socio-ecological system, it is necessary to include both temporal and spatial monitoring of local populations of M. chilensis and their genetic diversity and differentiation, considering environmental heterogeneity and temporal variations, such as ENSO that could impact on reproductive success.
Data availability
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession numbers are as follows: NCBI [accession: MZ773651–MZ773880 (COI) and MZ901916–MZ902024 (H1)].
References
Araneda C, Angel-Pardo M, Jimenez E, Longa A, Lee RS, Segura C et al. (2021) A comment on Giusti et al. (2020) Mussels (Mytilus spp.) products authentication: a case study on the Italian market confirms issues in species identification and arises concern on commercial names attribution. Food Control 107:379
Araneda C, Larraín MA, Hecht B, Narum S (2016) Adaptive genetic variation distinguishes Chilean blue mussels (Mytilus chilensis) from different marine environments. Ecol Evol 6:3632–3644
Astorga MP, Cárdenas L, Pérez M, Toro JE, Martínez V, Farías A et al. (2020) Complex spatial genetic connectivity of mussels Mytilus chilensis along the southeastern Pacific coast and its importance for resource management. J Shellfish Res 39:77–86
Astorga MP, Vargas J, Valenzuela A, Molinet C, Marín SL (2018) Population genetic structure and differential selection in mussel Mytilus chilensis. Aquac Res 49:919–927
Avendaño M, Cantillánez M, Le Pennec M, Varela C, Garcias C (2011) Temporal distribution of larvae of Mytilus chilensis (Hupé, 1854) (Mollusca: Mytilidae), in the interior sea of Chiloé, southern Chile. Lat Am J Aquat Res 39:416–426
Avila-Poveda OH, Abadia-Chanona QY, Alvarez-Garcia IL, Arellano M (2021) Plasticity in reproductive traits of an intertidal rocky shore chiton (Polyplacophora: Chitonida) under pre-ENSO and ENSO events. J Mollus Stud 97:eyaa033
Barria A, Gebauer P, Molinet C (2012) Spatial and temporal variability of mytilid larval supply in the Seno de Reloncaví, southern Chile. Rev Biol Mar Oceanogr 47:461–473
Barshis D, Sotka E, Kelly R, Sivasundar A, Menge B, Barth J et al. (2011) Coastal upwelling is linked to temporal genetic variability in the acorn barnacle Balanus glandula. Mar Ecol Prog Ser 439:139–150
Beaumont AR, Hawkins MP, Doig FL, Davies IM, Snow M (2008) Three species of Mytilus and their hybrids identified in a Scottish Loch: natives, relicts and invaders? J Exp Mar Biol Ecol 367:100–110
Benjamini Y, Yekutieli D, Edwards D, Shaffer JP (2005) False discovery rate-adjusted multiple confidence. J Am Stat Assoc 100:469
Berger MS, Darrah AJ, Emlet RB (2006) Spatial and temporal variability of the early post-settlement survivorship and growth in the barnacle Balanus glandula along an estuarine gradient. J Exp Mar Biol Ecol 336:74–87
Bierne N, Bonhomme F, David P (2003) Habitat preference and the marine-speciation paradox. Proc R Soc Lond 270:1399–1406
Bitter MC, Kapsenberg L, Gattuso J-P, Pfister CA (2019) Standing genetic variation fuels rapid adaptation to ocean acidification. Nat Commun 10:5821
Blunden J, Arndt DS (2016) State of the climate in 2015. B Am Meteorol Soc 97:S1–S275
Bohonak AJ (1999) Dispersal, gene flow, and population structure. Q Rev Biol 74:21–45
Brattström H, Johanssen A (1983) Ecological and regional zoogeography of the marine benthic fauna of Chile: Report N°. 49 of the Lund University Chile Expedition 1948–49. Sarsia 68:289–339
Brown SDJ, Collins RA, Boyer S, Lefort M, Malumbres-Olarte J, Vink CJ et al. (2012) SPIDER: an R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Mol Ecol Res 12:562–565
Burzyński A, Zbawicka M, Skibinski DOF, Wenne R (2006) Doubly uniparental inheritance is associated with high polymorphism for rearranged and recombinant control region haplotypes in Baltic Mytilus trossulus. Genetic 174:1081–1094
Calderón I, Pita L, Brusciotti S, Palacín C, Turon X (2012) Time and space: genetic structure of the cohorts of the common sea urchin Paracentrotus lividus in Western Mediterranean. Mar Biol 159:187–197
Campos B, Landaeta MF (2016) Moluscos planctónicos entre el fiordo Reloncaví y el golfo Corcovado, sur de Chile: ocurrencia, distribución y abundancia en invierno. R Biol Mar Oceanogr 51:527–539
Cantillánez M, Avendaño M, Thouzeau G, Le Pennec M (2005) Reproductive cycle of Argopecten purpuratus (Bivalvia: Pectinidae) in La Rinconada marine reserve (Antofagasta, Chile): response to environmental effects of El Niño and La Niña. Aquaculture 246:181–195
Coolen JWP, Boon AR, Crooijmans R, van Pelt H, Kleissen F, Gerla D et al. (2020) Marine stepping-stones: connectivity of Mytilus edulis populations between offshore energy installations. Mol Ecol 29:686–703
Couvray S, Coupé S (2018) Three-year monitoring of genetic diversity reveals a micro-connectivity pattern and local recruitment in the broadcast marine species Paracentrotus lividus. Heredity 120:110–124
David P, Perdieu MA, Pernot AF, Jarne P (1997) Finegrained spatial and temporal population genetic structure in the marine bivalve Spisula ovalis. Evolution 51:1318–1322
Debiasse MB, Nelson BJ, Hellberg ME (2014) Evaluating summary statistics used to test for incomplete lineage sorting: mito-nuclear discordance in the reef sponge Callyspongia vaginalis. Mol Ecol 24:225–238
Díaz C, Figueroa Y, Sobenes C (2011) Effect of different longline farming designs over the growth of Mytilus chilensis (Hupé, 1854) at Llico Bay, VIII Región of Bio-Bio, Chile. Aquac Eng 45:137–145
Díaz C, Sobenes C, Machino S (2019) Comparative growth of Mytilus chilensis (Hupé 1854) and Mytilus galloprovincialis (Lamarck 1819) in aquaculture longline system in Chile. Aquaculture 507:21–27
Díaz-Puente B, Pita A, Uribe J, Cuéllar-Pinzón J, Guiñez R, Presa P (2020) A biogeography-based management for Mytilus chilensis: the genetic hodgepodge of Los Lagos versus the pristine hybrid zone of the Magellanic ecotone. Aquat Conserv 30:412–425
Duarte C, Navarro JM, Acuña K, Torres R, Manríquez PH, Lardies MA et al. (2014) Combined effects of temperature and ocean acidification on the juvenile individuals of the mussel Mytilus chilensis. J Sea Res 85:308–314
Duarte C, Navarro JM, Quijón PA, Loncon D, Torres R, Manríquez PH et al. (2018) The energetic physiology of juvenile mussels, Mytilus chilensis (Hupe): the prevalent role of salinity under current and predicted pCO2 scenarios. Environ Pollut 242:156–163
Eldon B, Riquet F, Yearsley J, Jollivet D, Broquet T (2016) Current hypotheses to explain genetic chaos under the sea. Curr Zool 62:551–566
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Res 10:564–567
Fernández JF, Ponce RD, Vásquez-Lavin F, Figueroa Y, Gelcich S, Desdner J (2018) Exploring typologies of artisanal mussel seed producers in southern Chile. Ocean Coast Manag 158:24–31
Flowers JM, Schroeter SC, Burton RS (2002) The recruitment sweepstakes has many winners: genetic evidence from the sea urchin Strogylocentrotus purpuratus. Evolution 56:1445–1453
Folmer O, Black M, Hoeh W, Lutz R, Vrijenhoek R (1994) DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol Mar Biol Biotech 3:294–299
Fuentes-Santos I, Labarta U (2015) Spatial patterns of larval settlement and early post-settlement survivorship of Mytilus galloprovincialis in Galician Ría (NW Spain). Effect on recruitment success. Reg Stud Mar Sci 2:1–10
Gardner JP, Oyarzún PA, Toro JE, Wenne R, Zbawicka M (2021) Phylogeography of Southern Hemisphere blue mussels of the genus Mytilus: evolution, biosecurity, aquaculture and food labelling. Oceanogr Mar Biol 59:139–228
Gardner JPA, Palmer NL (1998) Size-dependent, spatial and temporal genetic variation at a leucine aminopeptidase (LAP) locus among blue mussel (Mytilus galloprovincialis) populations along a salinity gradient. Mar Biol 132:275–281
Garrido-Ramos MA, Stewart DT, Sutherland BW, Zouros E (1998) The distribution of male-transmitted and female-transmitted mitochondrial DNA types in somatic tissues of blue mussels: implications for the operation of doubly uniparental inheritance of mitochondrial DNA. Genome 41:818–824
Gazeau F, Quiblier C, Jansen JM, Gattuso JP, Middelburg JJ, Heip CH (2007). Impact of elevated CO2 on shellfish calcification. Geophys Res Lett 34:L07603
Gelcich S, Hughes TP, Olsson P, Folke C, Defeo O, Fernández M et al. (2010) Navigating transformations in governance of Chilean marine coastal resources. Proc Natl Acad Sci USA 107:16794–16799
Gérard K, Bierne N, Borsa P, Chenuil A, Féral JP (2008) Pleistocene separation of mitochondrial lineages of Mytilus spp. mussels from Northern and Southern Hemispheres and strong genetic differentiation among southern populations. Mol Phylogenet Evol 49:84–91
Gilg MR, Hilbish TJ (2003) Spatio-temporal patterns in the genetic structure of recently settled blue mussels (Mytilus spp.) across a hybrid zone. Mar Biol 143:679–690
Gilg MR, Martin L, Fernandez N, Murphy C, Walsh C, Rognstad RL et al. (2016) Temporal patterns in allele frequencies of the gamete-recognition locus M7 lysin within a population of Mytilus galloprovincialis in southwestern England. J Mollusca Stud 82:542–549
Giusti A, Tosi F, Tinacci L, Guardone L, Corti I, Arcangeli G et al. (2020) Mussels (Mytilus spp.) products authentication: a case study on the Italian market confirms issues in species identification and arises concern on commercial names attribution. Food Control 118:107379
González HE, Castro L, Daneri G, Iriarte JL, Silva N, Vargas CA et al. (2011) Seasonal plankton variability in Chilean Patagonia fjords: carbon flow through the pelagic food web of Aysen Fjord and plankton dynamics in the Moraleda Channel basin. Cont Shelf Res 31:225–243
Gonzalez-Poblete E, Hurtado CF, Rojo C, Normabuena R (2018) Blue mussel aquaculture in Chile: small- or large-scale industry? Aquaculture 493:113–122
Han Z, Mao Y, Shui B, Yanagimoto T, Gao T (2016) Genetic structure and unique origin of the introduced blue mussel Mytilus galloprovincialis in the north-western Pacific: clues from mitochondrial cytochrome c oxidase I (COI) sequences. Mar Freshw Res 68:263–269
Hauser L, Adcock GJ, Smith PJ, Bernal-Ramírez JH, Carvalho GR (2002) Loss of microsatellite diversity and low effective population size in an overexploited population of New Zealand snapper (Pagurus auratus). Proc Natl Acad Sci USA 99:11742–11747
Haye PA, Muñoz-Herrera NC (2013) Isolation with differentiation followed by expansion with admixture in the tunicate Pyura chilensis. BMC Evol Biol 13:1–15
Haye PA, Turon X, Segovia NI (2021) Time or Space? Relative importance of geographic distribution and interannual variation in three lineages of the ascidian Pyura chilensis in the Southeast Pacific coast. Front Mar Sci 8:657411
Hedgecock D (1994) Does variance in reproductive success limit effective population sizes of marine organisms? In: Beaumont A (ed). Genetics and evolution of aquatic organisms. Chapman and Hall, London, p 1222–1344
Hedgecock D, Pudovkin AI (2011) Sweepstakes reproduction success in highly fecund marine fish and shellfish: a review and commentary. Bull Mar Sci 87:971–1002
Hilbish T, Carson E, Plante J, Weaver L, Gilg M (2002) Distribution of Mytilus edulis, M. galloprovincialis, and their hybrids in open-coast populations of mussels in southwestern England. Mar Biol 140:137–142
Jackson TM, Roegner GC, O’Malley KG (2017) Evidence for interannual variation in genetic structure of Dungeness crab (Cancer magister) along the California Current System. Mol Ecol 27:352–368
Johnson MS, Black R (1984) Pattern beneath the chaos: the effect of recruitment on genetic patchiness in an intertidal limpet. Evolution 38:1371–1383
Kesäniemi JE, Mustonen M, Biström C, Hansen B, Knott KE (2014) Temporal genetic structure in a poecilogonous polychaete: the interplay of developmental mode and environmental stochasticity. BMC Evol Biol 14:12
Koehn RK, Milkman R, Mitton JB (1976) Population genetics of marine pelecypods. IV. Selection, migration and genetic differentiation in the blue mussel Mytilus edulis. Evolution 30:2–32
Lara C, Saldías GS, Tapia FJ, Iriarte JL, Broitman BR (2016) Interannual variability in temporal patterns of Chlorophyll–a and their potential influence on the supply of mussel larvae to inner waters in northern Patagonia (41–44°S). J Mar Syst 155:11–18
Larmuseau MHD, Raeymaekers JAM, Hellemans B, Van Houdt JKJ, Volckaert FAM (2010) Mito-nuclear discordance in the degree of population differentiation in a marine goby. Heredity 105:532–542
Larraín MA, Díaz NF, Lamas C, Uribe C, Jilberto F, Araneda C (2015) Heterologous microsatellite-based genetic diversity in blue mussel (Mytilus chilensis) and differentiation among localities in southern Chile. Lat Am J Aquat Res 5:998–1010
Larraín MA, González P, Pérez C, Araneda C (2019) Comparison between single and multi-locus approaches for specimen identification in Mytilus mussels. Sci Rep 9:1–13
Larraín MA, Zbawicka M, Araneda C, Gardner JPA, Wenne R (2017) Native and invasive taxa on the Pacific coast of South America: impacts on aquaculture, traceability and biodiversity of blue mussels (Mytilus spp.). Evol Appl 11:298–311
LaVigne M, Nurhati IS, Cobb KM, McGeregor HV, Sinclair D, Sherrell RM (2013) Systematic ENSO-driven nutrient variability recorded by central equatorial Pacific corals. Geophys Res Lett 40:3956–3961
Lee HJ, Boulding EG (2009) Spatial and temporal population genetic structure of four northeastern Pacific littorinid gastropods: the effects of mode of larval development on variation at one mitochondrial and two nuclear DNA markers. Mol Ecol 18:2165–2184
Lee Y, Ni G, Shin J, Kim T, Kern EMA, Kim Y et al. (2021) Phylogeography of Mytilisepta virgate (Mytilidae: Bivalvia) in the northwestern Pacific: cryptic mitochondrial linages and mito-nuclear discordance. Mol Phylogenet Evol 157:107067
Legendre P, Lapointe FJ (2004) Assessing congruence among distance matrices: single‐malt scotch whiskies revisited. Aust N Z J Stat 46:615–629
Li G, Hedgecock D (1998) Genetic heterogeneity detected by PCR-SSCP, among samples of Pacific oysters (Crassostrea gigas Thunberg), supports the hypothesis of a large variance in reproductive success. Can J Fish Aquat Sci 55:1025–1033
Ludford A, Cole VJ, Porri F, McQuaid CD, Nakin MDV, Erlandsson J (2012) Testing source-sink theory: the spill-over of mussel recruits beyond marine protected areas. Lands Ecol 27:859–868
Manríquez PH, Guiñez R, Olivares A, Clarke M, Castilla JC (2018) Effects of inter-annual temperature variability, including ENSO and post-ENSO events, on reproductive traits in the tunicate Pyura praeputialis. Mar Biol Res 14:462–477
Marshall DJ, Monro K, Bode M, Keough ME, Swearer S (2010) Phenotype-environment mismatches reduce connectivity in the sea. Ecol Lett 13:128–140
McKeown NJ, Hauser L, Shaw PW (2017) Microsatellite genotyping of brown crab Cancer pagurus reveals fine scale selection and ‘non-chaotic’ genetic patchiness within a high gene flow system. Mar Ecol Progr Ser 566:91–103
Moberg PE, Burton RS (2000) Genetic heterogeneity among adult and recruit red sea urchins, Strongylocentrotus franciscanus. Mar Biol 136:773–784
Molina V, Cornejo-D’Ottone M, Soto EH, Quiroga E, Alarcón G, Silva D et al. (2021) Biogeochemical responses and seasonal dynamics of the benthic boundary layer microbial communities during the El Niño 2015 in an eastern boundary upwelling system. Water 12:180
Molinet C, Astorga M, Cares L, Diaz M, Hueicha K, Marín S et al. (2021) Vertical distribution patterns of larval supply and spatfall of three species of Mytilidae in Chilean fjord used for mussel farming: insights for mussel spatfall efficiency. Aquaculture 535:736341
Molinet C, Díaz M, Marín SL, Astorga MP, Ojeda M, Cares L et al. (2017) Relation of mussel spatfall on natural and artificial substrates: analysis of ecological implications ensuring long-term success and sustainability for mussel farming. Aquaculture 467:211–218
Molinet CA, Díaz MA, Arriagada CB, Cares LE, Marín SL, Astroga MP et al. (2015) Spatial distribution pattern of Mytilus chilensis beds in the Reloncaví fjord: hypothesis on associated processes. Rev Chil Hist Nat 88:1–12
Narváez DA, Vargas CA, Cuevas LA, García-Loyola SA, Lara C, Segura C et al. (2019) Dominant scales of subtidal variability in coastal hydrography of the Northern Chilean Patagonia. J Mar Syst 193:59–73
Navarro JM (1988) The effects of salinity on the physiological ecology of Choromytilus chorus (Molina, 1782)(Bivalvia: Mytilidae). J Exp Mar Biol Ecol 122:19–33
Ng W, Leung FCC, Chak STC, Slingsby G, Williams GA (2010) Temporal genetic variation in populations of the limpet Cellana grata from Hong Kong shores. Mar Biol 157:325–337
Norrie C, Dunphy B, Roughan M, Weppe S, Lundquist C (2020) The spill-over from aquaculture may provide larval subsidy for the restoration of mussel reefs. Aquac Env Interact 12:231–249
Oksanen J, Blanchet FG, Friendly M, Kindt R, Legendre P, McGlinn D et al. (2020) vegan: Community Ecology Package. R package version 2.5-7. https://CRAN.R-project.org/package=vegan
Owen EF, Rawson PD (2013) Small-scale spatial and temporal genetic structure of the Atlantic sea scallop (Placopecten magellanicus) in the inshore Gulf of Maine revealed sing AFLPs. Mar Biol 160:3015–3025
Oyarzún PA, Toro JE, Nuñez JJ, Suárez-Villota EY, Gardner JPA (2021) Blue mussels of the Mytilus edulis species complex from South America: the application of species delimitation models to DNA sequence variation. PLoS ONE 16:e0256961
Paradis E, Schliep K (2019) Ape 5.0: an environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics 35:526–528
Pedersen EM, Hunt HL, Scheibling RE (2000) Temporal genetic heterogeneity within a developing mussel (Mytilus trossulus and M. edulis) assemblage. J Mar Biol Assoc UK 80:843–854
Pérez-Portela R, Wangensteen OS, Garcia-Cisneros A, Valero-Jiménez C, Palacín C, Turón X (2018) Spatio-temporal patterns of genetic variation in Arbacia lixula, a thermophilous sea urchin in expansion in the Mediterranean. Heredity 122:244–259
Pickett T, David AA (2018) Global connectivity patterns of the notoriously invasive mussel, Mytilus galloprovincialis Lmk using archived CO1 sequence data. BMC Res Notes 11:1–7
Pineda J, Porri F, Starczak V, Blythe J (2010) Causes of decoupling between larval supply and settlement and consequences for understanding recruitment and population connectivity. J Exp Mar Biol Ecol 392:9–21
Pinsky ML, Palumbi SR (2014) Meta‐analysis reveals lower genetic diversity in overfished populations. Mol Ecol 23:29–39
Plough LV, Shin G, Hedgecock D (2016) Genetic inviability is a major driver of type III survivorship in experimental families of a highly fecund marine bivalve. Mol Ecol 25:895–910
Quintero-Galvis JF, Bruning P, Paleo-López R, Gomez D, Sánchez R, Cárdenas L (2020) Temporal variation in the genetic diversity of a marine invertebrate with long larval phase, the muricid gastropod Concholepas. J Exp Mar Biol Ecol 530-531:151432
R Development Core Team R (2022) A language and environment for statistical computing. R Foundation for Statistical Computing. www.r-project.org
Rawson PD, Secor C, Hilbish TJ (1996) The effects of natural hybridization on the regulation of doubly uniparental mtDNA Inheritance in blue mussels (Mytilus spp.). Genetics 144:241–248
Riginos C, Henzler CM (2008) Patterns of mtDNA diversity in North Atlantic populations of the mussel Mytilus edulis. Mar Biol 155:399–412
Robainas-Barcia A, Espinosa López G, Hernández D, García-Machado E (2008) Temporal variation of the population structure and genetic diversity of Farfantepenaeus notialis assessed by allozyme loci. Mol Ecol 14:2933–2942
Rojas-Hernandez N, Veliz D, Riveros MP, Fuentes JP, Pardo L (2016) Highly connected populations and temporal stability in allelic frequencies of a harvested crab from the southern Pacific coast. PLoS ONE 11:e0166029
Rozas J, Ferrer-Mata A, Sánchez-DelBarrio JC, Guirao-Rico S, Librado P, Ramos-Onsins SE et al. (2017) DnaSP 6: DNA Sequence Polymorphism analysis of large datasets. Mol Biol Evol 34:3299–3302
Ruiz M, Tarifeño E, Llanos-Rivera A, Padget C, Campos B (2008) Temperature effect in the embryonic and larval development of the mussel, Mytilus galloprovincialis (Lamarck, 1819). Rev Biol Mar Oceanogr 42:51–61
Salloum PM, Silva MJ, Solferini VN (2019) Fine-scale genetic structure of the periwinkle Echinolittorina lineolata (Gastropoda: Littorinidae): the interplay between space and time. J Moll Stud 85:73–78
San Martin VA, Vásquez-Lavin F, Ponce Oliva RD, Lerdón XP, Rivera A, Serramalera L et al. (2020) Exploring the adaptive capacity of the mussel mariculture industry in Chile. Aquaculture 519:734856
Santoso A, Mcphaden MJ, Cai W (2017) The defining characteristics of ENSO extremes and the strong 2015/2016 El Niño. Rev Geophys 55:1079–1129
Sernapesca (2020) Anuario estadístico de pesca. http://www.sernapesca.cl/. Accessed Oct 2022
Sernapesca (2021) Anuario estadístico de pesca. http://www.sernapesca.cl/. Accessed Oct 2022
Siegel DA, Mitarai S, Costello CJ, Gaines SD, Kendall BE, Warner RR et al. (2008) The stochastic nature of larval connectivity among nearshore marine populations. Proc Natl Acad Sci USA 105:8974–8979
Sievers HA, Silva N (2006) Comité Oceanográfico Nacional—Pontificia. Universidad Católica de Valparaíso, Valparaíso, p 53–58
Simon A, Arbiol C, Nielsen EE, Couteau J, Sussarellu R, Burgeot T et al. (2020) Replicated anthropogenic hybridisations reveal parallel patterns of admixture in marine mussels. Evol Appl 13:575–599
Stephens M, Smith NJ, Donnelly P (2001) A new statistical method for haplotype reconstruction from population data. Am J Hum Genet 68:978–989
Tinacci L, Guidi A, Toto A, Guardone L, Giusti A, D’Amico P et al. (2018) DNA barcoding for the verification of supplier’s compliance in the seafood chain: how the lab can support companies in ensuring traceability. Ital J Food Saf 7:6894
Toews DPL, Brelsford A (2012) The biogeography of mitochondrial and nuclear discordance in animals. Mol Ecol 21:3907–3930
Toro JE, Castro GC, Ojeda JA, Vergara AM (2006) Allozymic variation and differentiation in the Chilean blue mussel, Mytilus chilensis, along its natural distribution. Genet Mol Biol 29:174–179
Toro JE, Ojeda JA, Vergara AM (2004) The genetic structure of Mytilus chilensis (Hupé, 1854) populations along the Chilean coast based on RAPDs analysis. Aquac Res 35:1466–1471
Toro JE, Oyarzún PA, Peñaloza C, Alcapán A, Videla V, Tillería et al. (2012) Production and performance of larvae and spat of pure and hybrid species of Mytilus chilensis and M. galloprovincialis from laboratory crosses. Lat Am J Aquat 40:243–247
Toro JE, Sastre D (1995) Induced triploidy in the Chilean blue mussel, Mytilus chilensis (Hupe 1854), and performance of triploid larvae. J Shellfish Res 14:161–164
Uriarte I (2008) Estado actual del cultivo de moluscos bivalvos en Chile. In: Lovatelli A, Uriarte I, Farias A (eds). Estado actual del cultivo y manejo de moluscos bivalvos y su proyección futura. FAO, Rome, Italy, p 61–76
Vadopalas B, Leclair LL, Bentzen P (2012) Temporal genetic similarity among year-classes of the pacific geoduck clan (Panopea generosa Gould 1850): a species exhibiting spatial genetic patchiness. J Shellfish Res 31:697–709
Valentini A, Pompanon F, Taberlet P (2009) DNA barcoding for ecologists. Trends Ecol Evol 24:110–117
Vendrami DLJ, Peck LS, Clark MS, Eldon B, Meredith M, Hoffman JI (2021) Sweepstakes reproductive success and collective dispersal produce chaotic genetic patchiness in a broadcast spawner. Sci Adv 7:eabj4713
Villacorta-Rath C, Souza CA, Murphy NP, Green BS, Gardner C, Strugnell JM (2018) Temporal genetic patterns of diversity and structure evidence chaotic genetic patchiness in a spiny lobster. Mol Ecol 27:54–65
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Winter JE, Toro JE, Navarro JM, Valenzuela GS, Chaparro OR (1984) Recent developments, status, and prospects of molluscan aquaculture on the Pacific coast of South America. Aquaculture 39:95–134
Zardi GI, McQuaid CD, Teske PR, Barker NP (2007) Unexpected genetic structure of mussel populations in South Africa: indigenous Perna perna and invasive Mytilus galloprovincialis. Mar Ecol Progr Ser 337:135–144
Acknowledgements
We thank Natalia Muñoz, Francisca Gálvez, Daniel Aliste, and especially Raúl Vera for their assistance with fieldwork and sample processing. We also extend our appreciation to Carolina Oliva, Raúl Vera, and in particular, Paulina Gyorgy for their help with DNA extractions and PCR. We would like to acknowledge the editor, Bastiaan Star, for their valuable suggestions, as well as four anonymous reviewers whose comments helped improve the final version of this manuscript. This study was funded by the Chilean National Fund for Scientific and Technological Development through Grants FONDECYT 1140862 and FONDECYT Iniciación 11220913, Universidad Católica del Norte, and the Millennium Science Initiative Program (Code ICN2019_015), Chile.
Author information
Authors and Affiliations
Contributions
PAH conceived the idea and designed the study. PAH and NIS contributed to sample collection, laboratory work, and acquisition of data, conceived and designed the data analyses, and wrote the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Associate editor: Bastiaan Star.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Haye, P.A., Segovia, N.I. Shedding light on variation in reproductive success through studies of population genetic structure in a Southeast Pacific Coast mussel. Heredity 130, 402–413 (2023). https://doi.org/10.1038/s41437-023-00615-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41437-023-00615-8
