
Abstract
Purpose
To investigate monoallelic CLPB variants. Pathogenic variants in many genes cause congenital neutropenia. While most patients exhibit isolated hematological involvement, biallelic CLPB variants underlie a neurological phenotype ranging from nonprogressive intellectual disability to prenatal encephalopathy with progressive brain atrophy, movement disorder, cataracts, 3-methylglutaconic aciduria, and neutropenia. CLPB was recently shown to be a mitochondrial refoldase; however, the exact function remains elusive.
Methods
We investigated six unrelated probands from four countries in three continents, with neutropenia and a phenotype dominated by epilepsy, developmental issues, and 3-methylglutaconic aciduria with next-generation sequencing.
Results
In each individual, we identified one of four different de novo monoallelic missense variants in CLPB. We show that these variants disturb refoldase and to a lesser extent ATPase activity of CLPB in a dominant-negative manner. Complexome profiling in fibroblasts showed CLPB at very high molecular mass comigrating with the prohibitins. In control fibroblasts, HAX1 migrated predominantly as monomer while in patient samples multiple HAX1 peaks were observed at higher molecular masses comigrating with CLPB thus suggesting a longer-lasting interaction between CLPB and HAX1.
Conclusion
Both biallelic as well as specific monoallelic CLPB variants result in a phenotypic spectrum centered around neurodevelopmental delay, seizures, and neutropenia presumably mediated via HAX1.
Similar content being viewed by others


INTRODUCTION
Inherited disorders of hematopoiesis disturb the differentiation of neutrophilic granulocytes and result in (severe) congenital neutropenia (CN, reviewed by1). In the bone marrow of these patients a maturation arrest of the promyelocytes is seen. These patients most often present neonatally with frequent infections. Furthermore CN bears the risk of developing myeloid malignancy. Treatment consists of granulocyte colony stimulating factor (GCSF) and significantly reduces mortality. Patients not responding to GCSF often progress to hematopoietic stem cell tsransplantation (HSCT) to avoid death by bacterial infections.
Pathogenic variants in several genes following different modes of inheritance have been linked to CN. While most of the patients with CN present with isolated hematological involvement, pathogenic variants in certain autosomal recessively inherited disease genes also lead to somatic organ involvement (SLC37A42) or neurological involvement like developmental delay (DD), intellectual disability (ID), and seizures (e.g., variants affecting both isoforms of HAX13). In these patients, the severity of neutropenia as well as of other organ involvement determine the survival.
In 2015 we described that biallelic CLPB variants cause a multisystem disease with a neurological spectrum from prenatal encephalopathy with progressive brain atrophy to isolated ID/DD as well as movement disorder. Additionally, patients show cataracts, 3-methylglutaconic aciduria (3-MGA-uria), and CN that may progress into myeloid malignancies.4 Since then more than 35 affected individuals with biallelic CLPB variants have been identified.5,6
The bacterial analog, the ClpB (caseinolytic peptidase B) chaperone of the HSP100 family, was the namesake for the human CLPB protein, as both share a significant sequence identity in the C-terminal part. The name is confusing, as ClpB lacks proteolytic activity. Bacterial ClpB is a member of the AAA + ATPase protein family. It forms hexameric ring-shaped molecules. Together with the Hsp70 protein family and its co-chaperones, ATP hydrolysis provides energy that is used to disentangle misfolded polypeptide chains (aggregates) by threading them through the hexamer’s central pore, which subsequently enables proper refolding.7 It is still unknown whether human CLPB also forms hexamers.
Human CLPB has a mitochondrial localization.4 Individuals with a CLPB defect show 3-MGA-uria, a strong biomarker for dysfunctional mitochondria. Oxidative phosphorylation in muscle and fibroblasts, however, is not affected.4 The exact function of CLPB is unknown, but it has been linked to regulation of apoptosis, maintenance of cristae formation, and modulation of antiviral innate immunity in mitochondria.8,9
Recently, human CLPB located in the mitochondrial intermembrane space was shown to interact with the prohibitin complex.9 The inner mitochondrial membrane prohibitin proteins PHB and PHB2 form a complex that regulates OPA1 proteolysis and thereby mitochondrial fusion. CLPB was further reported to interact at the outer mitochondrial membrane with the mitochondrial antiviral signaling protein.9
Human acute myeloid leukemia cells upregulate CLPB.6 The acquired resistance to the selective BCL2 inhibitor venetoclax associates to CLPB upregulation, whereas CLPB downregulation resensitizes AML cells to chemotherapy. This links CLPB to highly specific, clinically significant regulatory processes.6 An interaction of CLPB with OPA1 linked CLPB to correct cristae morphology.6 It was further shown that CLPB directly interacts with the HAX1 protein,6 another antiapoptotic factor, in line with postulations in our initial work.4
We have shown that human CLPB has both ATPase- and refoldase activity.4,10 This was recently confirmed, and it was additionally shown that CLPB activity is increased severalfold after cleavage by presenilin-associated rhomboid-like protein (PARL).11 HAX1 and PARL activate HtrA2/Omi.12,13 This might be an interesting link, as biallelic HTRA2-variants lead to a syndrome of ID/DD, seizures, and 3-MGA-uria as well as neutropenia.14
We here report six cases with de novo monoallelic missense variants in CLPB and a clinical phenotype strongly overlapping with individuals with biallelic CLPB variants.
MATERIALS AND METHODS
Genetic investigations
We performed exome (ES) or genome sequencing following local protocols.15,16 In all data sets, we prioritized variants based on an autosomal recessive filter for variants with a minor allele frequency (MAF) < 0.1% and for de novo variants with MAF < 0.01%. Sanger sequencing protocols and primers are available upon request.
Purification of CLPB, luciferase refolding activity assay, and ATPase activity assay
E. coli pMALc5X expression vectors with synthetic genes coding for human CLPB protein and its Lys404Thr, Pro427Leu, and Gly560Arg variants were obtained from GeneScript. The proteins were purified as maltose binding protein (MBP) fusions from E. coli overexpression by ammonium sulfate precipitation, dextrin-sepharose affinity chromatography, MBPcleavage by TEV protease, and Superdex200 gel filtration as described previously.10 The thermal unfolding curves were comparable with that of wild type (WT, deflection of unfolding curves for pathogenic variants were within 2.2 °C of that of WT, data not shown).
Luciferase activity was measured essentially as described,10 except that firefly luciferase (Promega) at 13 mg/ml was chemically inactivated by dilution in buffer (25 mM HEPES/KOH, 75 mM KCl, 15 mM MgCl2, pH 8.0) containing 6 M urea to 1.7 mg/ml and inactivated by incubation at 25 °C for 10 minutes and then 48 °C for 10 minutes. Subsequently it was rapidly diluted to 0.2 µM in the same buffer without urea. This preparation was used as a substrate in reactivation experiments at a final concentration of 50 nM. ATPase activity was assayed in triplicates in an enzyme-coupled assay,17 as described previously.10 Absorbance was measured using a Beckman Coulter DTX 880 Multimode Detector. Gel filtration experiments were performed as described.10
The Biolayer interferometry binding assay and the structural modeling of CLPB variants are detailed in the Supplementary methods
Complexome profiling
Mitochondrial enriched fibroblast fractions solubilized with digitonin were separated by high-resolution clear native polyacrylamide gel electrophoresis (hrCN-PAGE) for complexome profiling analysis. This method separates proteins over a very wide molecular mass range while preserving protein interactions and keeping multiprotein complexes intact (Supplementary methods, Supplementary Fig. 1AB). Entire gel lanes were fractionated and subjected to liquid chromatography tandem mass spectrometry to analyze the composition and abundance of protein complexes. Three fibroblast cell lines of patients with monoallelic variants (P2, P3, P5) and two of patients with biallelic variants (I1 = individual 6, I2 = individual 9 from4) were included. Protein abundances were determined using label-free intensity-based absolute quantification values and normalized to the maximum abundance of each protein in fractions of all samples. Migration profiles were hierarchically clustered using Pearson correlation and visualized by abundance heat maps. The migration profiles of CLPB were reconstructed from 22 of its peptides covering 38% of the total CLPB sequence (Supplementary methods, Supplementary Fig. 1).
RESULTS
Phenotypes of patients with monoallelic variants in CLPB
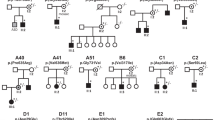
The clinical presentation and course of the six affected individuals (four females; aged 3–12 years; Table 1, Fig. 1; detailed case reports in Supplementary results) varied from a mild phenotype with isolated mild speech delay (P6) to the most severe phenotype (P2) with progressive microcephaly, absence of development, refractory seizures and infantile death (DD/ID [6/6], epilepsy [5/6], axial hypotonia with limb spasticity [4/6], secondary microcephaly [4/6] and 3-MGA-uria [6/6]). Of note none had cataracts. Five of six individuals had neutropenia, of whom two underwent HSCT.

a Individual P2 at the age of 9 months. b,c,g Individual P3 at the age of 16 months and 4 and 7 years, respectively. d,e,f Individual P5 at the age of 2 weeks, 3 and 10 years, respectively. h Individual P4 at age 7.8 years. i Pedigrees of all families including the genotype and position of variants in schematic representation of human CLPB. j Magnetic resonance image (MRI) findings in the three individuals (axial T2-weighted and sagittal T1-weighted images). At the age of 17 months, P2 shows supratentorial atrophy and also mild cerebellar atrophy. White matter signal is slightly T2-hyperintense in the central white matter. P3 also shows mild global atrophy with progressive thinning of the cortical ribbon. White matter signal is diffusely abnormal, with normal signal of the corpus callosum and the anterior limb of the internal capsule. At age 3 years, the medial part of the middle cerebellar peduncle has an elevated T2 signal. P5 has diffusely elevated white matter signal, including cerebellar white matter, indicating delayed myelination at age 15 months. At age 7 years, she has developed supratentorial atrophy. Myelination has progressed, but there is still a diffusely abnormal white matter signal. Also the signal in the basal ganglia (head of the caudate nucleus and upper part of the putamen) is too high on the T2-weighted image. Also in P5, the signal of the medial part of the middle cerebellar peduncle is elevated. k Peripheral blood smear (P6) showing relative neutropenia including forms with toxic granulation, reflecting granulocyte colony stimulating factor (GCSF) therapy. Red cell and platelet indices were within normal limits. 1,000× magnification; Wright-Giemsa. l Bone marrow aspirate (P6) shows trilineage hematopoiesis with left-shifted myelopoiesis. Mild dysmyelopoiesis is present, including neutrophils with nuclear hypolobation and megaloblastoid change (inset and solid arrow). Hematogones (B-cell progenitors) are also increased in frequency (cross-hatched arrow), confirmed by flow cytometry. 1,000× magnification; Wright-Giemsa. m The bone marrow (P6) is hypercellular for age showing trilineage hematopoiesis with expanded paratrabecular cuff of left-shifted myeloid cells and mild eosinophilia, changes reflecting GCSF therapy 400× magnification; hematoxylin and eosin (H&E). n The bone marrow aspirate smear (P4) shows a marked increase in immature myeloid and lymphoid progenitor cells. Eosinophils are also increased. Myeloid maturation is megaloblastic with several large immature cells with intermediate amounts of cytoplasm, round eccentrically placed nuclei, prominent nucleoli, perinuclear hofs, smooth chromatin, and sparse but coarse cytoplasmic granulation. o The bone marrow aspirate (P4) demonstrates trilineage hematopoiesis with a decreased myeloid to erythroid ratio. Myeloid cells include promyelocytes and very rare myelocytes but no mature forms. Erythroid maturation is complete. p The bone marrow (P4) is hypercellular for age showing trilineage hematopoiesis with expanded paratrabecular cuff of left-shifted myeloid cells, changes reflecting GCSF therapy 400× magnification; H&E. Scale bars in k,l indicate 50 μm and in M-P 40 μm.
Neuro(radiological) phenotype
One individual (P6), currently aged 5.3 years, showed mild speech delay as the only neurological manifestation. MRI at that age revealed hazy, biparietal periventricular and deep white matter signals. There was also very mild thinning of the posterior body of the corpus callosum, which was associated with periventricular white matter volume loss. All other individuals had global DD/ID, and the worst affected case (P2) did not develop further from age three months until death. Two other individuals were nonambulatory and lost the skills of standing with support by the age of three years (P3) and walking with a walking frame by the age of 5.5 years (P5), respectively. These clinical features suggesting neurodegeneration are in line with the MRI finding of (progressive) brain atrophy and myelination deficit. Brain atrophy corresponded with the clinical finding of microcephaly in four children. In two individuals with cerebellar atrophy (P3,4) broad based, ataxic gait was present. For the third individual (P2) testing for cerebellar signs was impossible. Speech development varied from absent in P2 and P3 to mildly delayed in P6, while P1, 4, 5 could communicate using 1–2 word sentences and gestures or communication aids.
Hematological/immunological phenotype
Neutropenia was severe (<0.5 × 109/l) in P4-6 and moderate (0.5 −1.0 × 109/l) in P3. P2 was found to be moderately neutropenic only during infections. P1 was the only individual not presenting neutropenia. There was no obvious correlation between the severity of the hematopoietic and neurological phenotype. P3 and P5 responded with variable effect to GCSF. In P3 ANCs remained above 1.5 × 109/l with 4.3 mcg/kg/day and in P5 the Absolute Neutrophil Count (ANC)s remain low (last visit: 0.29 × 109/l) with 5 mcg/day GCSF, however, in both without clinical signs of neutropenia. HSCT was necessary in P4 at age 15 months and P6 at age 5 years, respectively, due to recurrent infections and no response to standard doses of GCSF. The bone marrow examination of P3–P6 showed trilineage hematopoiesis with left-shifted myelopoiesis and a maturation arrest at the (pro)myelocyte stage (Fig. 1, Supplementary Fig. 2).
The clinical phenotype of individuals with biallelic CLPB variants is described in4,6
Molecular findings
In all six unrelated probands, from four countries and three continents, singleton ES did not identify causative variants in disease genes matching the mode of inheritance. Subsequent trio ES in P1, 4, 6, as well as trio genome sequencing in P 518 identified monoallelic de novo missense variants in CLPB (NM_030813.4). Sanger sequencing of the parents of P2 and P3 proved absence of the heterozygous CLPB variants found by ES (Supplementary results).
We identified the following variants (Fig. 1): c.1211A>C, p.Lys404Thr (P1); c.1280C>T, p.Pro427Leu (P2) in one patient each. The variants c.1678G>A, p.Gly560Arg (P3,4), and c.1681C>T, pArg561Trp (P5,6) independently occurred at the same position in two individuals each. All variants have not been reported previously in individuals with biallelic CLPB variants and are absent from the Genome Aggregation Database (gnomAD19) and predicted to be pathogenic by several prediction programs (https://varsome.com).
Luciferase refolding activity assay and ATPase activity assay
The in vitro luciferase refolding activity assay showed that the ability of purified WT-CLPB protein to renature chemically unfolded luciferase was abolished in pathogenic variants (Fig. 2a). While the increase of luciferase activity for p.Pro427Leu and p.Gly560Arg CLPB variants at 1 µM concentration was indistinguishable from spontaneous reactivation, only CLPB-Lys404Thr exhibited some detectable residual activity (reactivation rate: ~10.9% compared to 1 uM WT). The ATPase activity of these variants was also diminished, however to a lesser extent than the renaturation activity (Fig. 2a).

a Left: In vitro luciferase disaggregation and refolding by purified wild type CLPB (WT-CLPB) protein and its variants. Chemically denatured luciferase was added to reaction mixtures containing 0.5 µM CLPB WT protein or indicated variants in the presence of 5 mM ATP. Aliquots were taken at indicated timepoints and the luciferase activity was assayed. The CLPB disaggregase and refolding activity is shown by the gradual return of luciferase activity with time during the incubation. Note the completely absent disaggregase and refolding activity in two of the mutants and some residual activity in the Lys404Thr mutant. Inset: curves for low activity variants, rescaled for clarity. Right: ATPase activity of CLPB WT and variants. b Luciferase disaggregation and refolding activity of 1:1 mixtures of WT-CLPB with each of the pathogenic CLPB variants. The WT-CLPB reaction contains 0.25 µM CLPB and 0.5 µM bovine serum albumin while reactions with indicated pathogenic variants contain both 0.25 µM of WT-CLPB and 0.25 µM of pathogenic variant (total 0.5 µM CLPB protein). The luciferase activity was measured after a 4-hour incubation period to allow reactivation. c Molecular gel filtration of WT-CLPB and 1:1 mixtures of WT and indicated variants, separated on Superose 6 Increase 10/300. Positions of molecular weight markers indicated. d Structural analysis of the pathogenic variants using a homology model of CLPB. Overview of the model of the CLPB C-terminal domain showing two neighboring monomers. Two subunits are shown in different shades of gray. The ADP molecule is shown in ball representation (yellow). The positions of the pathogenic variants, one subunit only, are shown in red (note that Glycine does not have a sidechain). Sidechain atoms are shown in ball representation where possible. e Details of pathogenic variants G560R and R561W. Sidechain atoms are shown in stick representation to show the differences between wild type (green) and mutant (red). (Glycine sidechain not visible.) f Details of pathogenic variant K404T. g Details of pathogenic variant P427L.
To elucidate the basis for the observed clinical phenotype due to monoallelic CLPB variants, reaction mixtures containing both WT and pathogenic CLPB variants in 1:1 ratio were assayed for luciferase refolding (Fig. 2b). The WT protein and its pathogenic variants were mixed together prior to the initiation of the disaggregation reaction to simulate the simultaneous presence of both forms in the patients with monoallelic pathogenic variants. The proteins are in an oligomeric state. Either simultaneous action of both forms on the substrate or the formation of mixed forms where both kinds of protein are present, is responsible for the observed effect on luciferase refolding. If the protein variants we~~re functioning independently, their activities would be additive, and, because the residual refolding activity of pathogenic variants is very low, the total activity of the mixtures should be comparable to that of 0.25 µM WT protein present in the reaction. The lower renaturation activity that we observed is therefore a result of the negative influence of pathogenic variants on the WT protein.
Biolayer interferometry binding assay and CLPB oligomer formation
Since the mixture of WT with pathogenic variants exhibited lower luciferase renaturation activity than WT alone, it may be assumed that either they exert a negative effect on WT-CLPB itself, or rather compete with it by unproductive binding to aggregates, lessening the amount of available substrate. Supplementary Fig. 3 shows that the binding rate for pathogenic variants is much lower than that of WT protein, rendering it unlikely that the latter mechanism would be the prevailing pathogenic phenomenon.
Fig. 2c shows that purified WT-CLPB, as well as 1:1 mixture of WT and purified mutant CLPB protein, exhibited gel elution profiles showing very large complexes and no or hardly any monomeric CLPB. The WT elution profile was identical as described previously.10 WT-CLPB apparently easily forms oligomers under the in vitro conditions used. Markedly, also no significant amounts of CLPB monomers or lower mass oligomers were detected in samples where pathogenic variants were present along with WT-CLPB. This shows that the pathogenic effect of the mutant CLPB cannot be caused by defective oligomerization.
Structural modeling of monoallelic CLPB variants
Bacterial CLPB is known to act in a hexameric form (Fig. 2d–h, Supplementary Figure 4 for biallelic variants). We have no data to prove a similar hexameric state for human CLPB. Data in Fig. 2, based on a limited structural modeling, show the interface between two adjacent CLPB monomers with the ATP binding site formed by the two subunits. Two pathogenic variants, p.Gly560Arg and p.Arg561Trp, are located in the direct vicinity of these ATP sites (Fig. 2e). Both variants are expected to severely affect ATPase activity, which is in line with the ATPase activity measurement (Fig. 2b). The p.Gly560Arg pathogenic variant introduces a large hydrophilic sidechain thereby causing steric clashes and possible rearrangements of the ATP binding site. This is predicted to have severe effects on protein function, and indeed our data show severely decreased ATPase activity and consequently also very low disaggregase activity (Fig. 2). Arginine561 contains a long and positively charged sidechain that is probably interacting with ATP. The p.Arg561Trp pathogenic variant causes loss of these interactions and additionally causes steric clashes to accommodate the even larger tryptophan sidechain.
The p.Lys404Thr pathogenic variant also occurs close to the ATP binding site but this residue is not directly involved in ATP binding (Fig. 2f). Instead, the positively charged lysine sidechain is found on the surface of the domain where it can be involved in ionic interactions that could stabilize complex formation. More interactions with the N-terminal domain are also possible but we cannot confirm this using the current model. The Lys404Thr pathogenic variant possibly affects the overall stability of the CLPB conformation. The experimental results indicate that this pathogenic variant leaves some residual ATPase and disaggregase activity.
The p.Pro427Leu pathogenic variant is found in the pore area, preceding the substrate binding loop -GYVG- (residues 429–432 according to11). The pathogenic variant of the rigid and small proline into the larger leucine is expected to slightly change the local structure of this loop, thereby affecting the disaggregase activity while leaving ATP binding unaffected. The pathogenic variant results in only mildly decreased ATPase activity. Indeed, the experimental results show the mildest reduction in ATPase activity of all pathogenic variants, whereas the disaggregase activity is lost.
All pathogenic variants show a severe change in sidechain properties that is likely to affect contacts between subunits in a higher oligomeric state. The role of the p.Lys404 residue for these subunit contacts is not yet clear since the sidechain seems very flexible in the current model. The mixing experiments show that all pathogenic variants can indeed inhibit normal WT function. In general, modeling data of proteins cannot prove hypotheses. This holds especially when the 3D structure is not available as in the case of CLPB. The modeling data, however, are in line with the findings in the ATPase- and refoldase activity measurements.
Complexome profiling
CLPB was found predominantly at unexpectedly high molecular mass ranges (1.3–2.1 MDa), where prohibitins also migrate (Fig. 3a upper panel). CLPB has been shown to interact with prohibitins.9 The co-migration of CLPB and prohibitins in the 1.3–2.1 MDa range is in line with this and could indicate supercomplex formation. Further experiments will be required to prove the existence of a supercomplex formation involving CLPB. The intensity values of PHBs were orders of magnitude higher than CLPB suggesting that if the two proteins indeed interact, CLPB associates to only a small fraction of PHBs. Formation of these big complexes was observed in all patient samples, indicating that CLPB pathogenic variants did not alter its association to PHBs. In addition, low abundant CLPB peaks were found in all patient cell lines at about 360 kDa, corresponding to the mass of CLPB oligomers. The effects of the monoallelic pathogenic variants on CLPB complexes and their abundance varied for each pathogenic variant. While in individuals P2 and P5 the total abundance of CLPB seemed increased and the predominant peak was found at 1.3 MDa, a decrease in CLPB total abundance was observed in individual P3, who only showed the larger 2.1 MDa peak (Fig. 3a, upper panel). Physical interaction of CLPB with HAX1 has been reported to be critical for HAX1 solubility.11 Using protein correlation profiling we identified similarities of the migration patterns of CLPB and HAX1 (Fig. 3a, lower panel). In control fibroblasts, HAX1 migrated predominantly at 40 kDa, corresponding to the mass of its monomeric form. In contrast, in all patient samples, multiple HAX1 peaks were observed at higher molecular masses. The HAX1 peaks were present at the same mass ranges as CLPB and presented the same abundance variations along different fractions, as well as between samples (Fig. 3a, lower panel). Additionally there was a simultaneous decrease in the abundance of unbound monomeric HAX1. The shifts in migration profiles observed in CLPB patient cell lines may indicate a pattern of increased propensity to physically interact for these two proteins, which may in turn have functional effects.

a Abundance heat maps and migration profiles derived from the complexome profiling analysis of mitochondrial enriched solubilizates of control and patient fibroblasts separated by high-resolution clear native polyacrylamide gel electrophoresis. Protein migration profiles were normalized to the maximum intensity of each protein across the 360 fractions (60 fractions per sample). Comparison of normalized migration patterns of CLPB with prohibitins (upper panel) and HAX1 (lower panel). Inserts in the upper panel are plots of the intensity-based absolute quantification (iBAQ) values without normalization, showing the abundance differences between CLPB and prohibitins. b Volcano plot representation of total protein intensities ratios between controls and patients with de novo pathogenic variants (left panel) and between controls and biallelic patient fibroblasts (right panel). Total protein intensities were calculated as the sum of iBAQ values from the 60 fractions corresponding to a sample. A larger number of proteins presented changes in total abundance in de novo variants compared to biallelic variants.
We further evaluated changes in the mitochondrial proteome in response to different CLPB pathogenic variants by analyzing total protein abundances in mitochondrial enriched fractions, considering only abundance changes of more than twofold and p < 0.05 (Student’s t-test). This analysis revealed changes in abundance of 245 proteins when comparing cells with variants with controls (147 increased, 98 decreased) (Fig. 3b, left panel). In contrast, cell lines with biallelic variants showed changes in abundance in 97 proteins (54 were increased, 43 proteins decreased) (Fig. 3b, right panel). Our analysis of the mitochondrial complexome in monoallelic CLPB cell lines showed an aberrant migration profile for several mitochondrial (inter)membrane proteins like MAVS, TOMM70, CLPX, ACADVL, FKBP8, and TUFM (Supplementary Fig. 5a). These proteins migrated at lower molecular mass ranges and in that respect their profiles differed from those of the control and biallelic cell lines (Supplementary Fig. 5b).
DISCUSSION
Here we present a cohort of six individuals with de novo monoallelic CLPB variants. The phenotypic spectrum encompasses mild to severe DD/ID, epilepsy, brain atrophy, and white matter alterations. Further, the maturation is arrested at the (pro)myelocyte stage leading to chronic moderate to severe neutropenia. The clinical phenotype significantly overlaps with that seen with biallelic CLPB variants and both cohorts share the biomarker 3-MGA-uria. However, while individuals with biallelic CLPB variants regularly exhibit cataracts, none of the individuals in the monoallelic cohort presented here had ophthalmological abnormalities. And while seizures are only reported in half of individuals with biallelic CLPB defect, epilepsy is a leading clinical feature in five of the six individuals presented in this study.
The consistently observed metabolic signature supports a discrete molecular genetic disorder, and by using next-generation sequencing we identified four independent, monoallelic CLPB variants that had arisen de novo in six individuals from six unrelated families. We show that CLPB variants underlie this phenotype. The gnomAD constraint metric of CLPB for loss-of-function variation is 0.00 with a LEOUF of 0.54,20 indicating that heterozygous loss-of-function alleles are tolerated. These data suggest a dominant-negative pathogenic mechanism for the monoallelic CLPB variants presented here rather than haploinsufficiency.
Monoallelic and biallelic pathogenic variation at a single locus has been shown for other mitochondrial (e.g., OPA121, ATAD3 a22) or CN genes (CSF3R encoding GCSF receptor23,24). The severity of phenotype between the monoallelic and biallelic forms of disease can vary, but usually the same organ systems are affected. In CLPB defect, primarily the hematological and nervous system are affected in both forms, while the ophthalmological system is only involved in the biallelic form. Two of the de novo pathogenic variants reported here occurred independently in two families, which suggests that at least some of these variants confer a specific property to the protein, such as dominant-negative effects.
We have shown that the monoallelic pathogenic variants of our patients result in significantly diminished ATPase activity of CLPB and almost completely abolish the disaggregase function of the CLPB. For the homologous microbial HSP100 disaggregases, including bacterial ClpB and yeast Hsp104 proteins, the active form is a characteristic spiral ring-shaped homohexamer, as recently demonstrated by cryoelectron microscopy.25,26 This arrangement is essential for the disaggregation mechanism, where ATP hydrolysis in the NBD2 domain (homologous to ATPase domain of human CLPB) provides energy for pulling the polypeptide from an aggregate through the central channel formed by six interacting protomers.25
No monomeric CLPB is observed for the WT-CLPB preparation in the gel filtration elution profiles in Fig. 2c. The CLPB elutes in the void volume indicating a higher molecular mass form, which is in concordance with our previous work.10 The same holds for the profile when pathogenic CLPB variants are present alongside WT protein. This is in line with the data from the complexome profiling experiments where monomeric CLPB is only seen in low abundant amounts. Taken together these data suggest that in both situations oligomers are the predominant CLPB form and monomers hardly seem to occur. We propose a mechanism where mixed oligomers of WT-CLPB and the specific variant subunits of patients are formed thus breaking intersubunit conformation and communication. This could explain the observed decrease in refoldase activity and the pathogenic effect of the monoallelic variants. Such a phenomenon was already observed for hexamers of ClpB/Hsp104 microbial disaggregases.27 In the mixing experiment of WT and variant CLPB the disaggregase and refolding activity is significantly lower in all incubations containing the variant CLPB than in the incubation with the WT-CLPB only. These data suggest that the mutants “poison” the activity of the CLPB mixed oligomers, but do not formally prove this. Our experiment may mimic the situation in patient cells, where both variants are simultaneously expressed and mixed oligomers interfere with CLPB activity.
The ATP binding sites can only function properly if neighboring subunits are correctly shaped. For all four monoallelic pathogenic variants our model shows that the severe change in sidechain properties will also influence the shape of the interface between neighboring subunits. The monoallelic pathogenic variants exert a negative influence on overall CLPB activity, which may be explained by the required shape of the subunit interfaces, which is not guaranteed when mutated and WT-CLPB forms are neighboring subunits in the oligomer. In that way the monoallelic pathogenic variants may abolish the overall function of the CLPB oligomer. Dominant-negative pathogenesis is a classical genetic theory in multimeric proteins where a “poisonous” mutant peptide negatively influences the co-expressed WT protein and disturbs its cellular function. Such poisoning of the disaggregase function in mixed CLPB oligomers containing mutant subunits has already been shown for AAA + ATPases.28 We would like to stress that the available model for CLPB shows 35% sequence identity, which is close to the lower modeling threshold (30%) and small mistakes in atomic positions can be expected.29
The Lys404Thr variant proved to contain residual disaggregase activity when tested alone, and when mixed together with WT-CLPB, the 404 mutant exerted a poisoning effect to a lesser degree than the other mutants. As luciferase is not a human protein, the data on luciferase refolding and renaturation should not be overinterpreted. It may, however, be meaningful that the Lys404Thr pathogenic variant seems to have significantly more residual activity than the other pathogenic variants tested.
Our complexome profiling data have shown co-migration patterns of CLPB with prohibitins and HAX1, in line with the interaction between these protein described in literature9,11 and suggesting their association in large complexes. The interaction of CLPB with HAX1 was not evident from the control profiles since the majority of HAX1 in control samples was found in unbound form. However, HAX1 profiles in all patient fibroblasts with monoallelic CLPB pathogenic variants revealed several peaks matching those of CLPB, suggesting a stronger than normal interaction between the two proteins. It seems that HAX1 is retained longer in the mutated CLPB complexes, and that a similar interaction between HAX1 and CLPB was observed in both cell lines of patients with biallelic CLPB pathogenic variants.
In conclusion, we propose that not only biallelic but also specific monoallelic CLPB variants cause a phenotypic spectrum with CN and DD/ID as key features.
Data availability
For original data, please contact the corresponding author.
Change history
23 July 2021
A Correction to this paper has been published: https://doi.org/10.1038/s41436-021-01280-0
References
Skokowa, J., Dale, D. C., Touw, I. P., Zeidler, C. & Welte, K. Severe congenital neutropenias. Nat. Rev. Dis. Primers. 3, 17032 (2017).
Wortmann, S. B. et al. Treating neutropenia and neutrophil dysfunction in glycogen storage disease IB with an SGLT2-inhibitor. Blood. 136, 1033–1043 (2020).
Boztug, K. et al. HAX1 mutations causing severe congenital neuropenia and neurological disease lead to cerebral microstructural abnormalities documented by quantitative MRI. Am. J. Med. Genet. A. 152a, 3157–3163 (2010).
Wortmann, S. B. et al. CLPB mutations cause 3-methylglutaconic aciduria, progressive brain atrophy, intellectual disability, congenital neutropenia, cataracts, movement disorder. Am. J. Hum. Genet. 96, 245–257 (2015).
Capo-Chichi, J. M. et al. Disruption of CLPB is associated with congenital microcephaly, severe encephalopathy and 3-methylglutaconic aciduria. J. Med. Genet. 52, 303–311 (2015).
Pronicka, E. et al. A scoring system predicting the clinical course of CLPB defect based on the foetal and neonatal presentation of 31 patients. J. Inherit. Metab. Dis. 40, 853–860 (2017).
Liberek, K., Lewandowska, A. & Zietkiewicz, S. Chaperones in control of protein disaggregation. EMBO J. 27, 328–335 (2008).
Chen, X. et al. Targeting mitochondrial structure sensitizes acute myeloid leukemia to venetoclax treatment. Cancer Discov. 9, 890–909 (2019).
Yoshinaka, T. et al. Structural basis of mitochondrial scaffolds by prohibitin complexes: insight into a role of the coiled-coil region. iScience. 19, 1065–1078 (2019).
Mroz, D. et al. CLPB (caseinolytic peptidase B homolog), the first mitochondrial protein refoldase associated with human disease. Biochim. Biophys. Acta Gen. Subj. 1864, 129512 (2020).
Cupo, R. R. & Shorter, J. Skd3 (human ClpB) is a potent mitochondrial protein disaggregase that is inactivated by 3-methylglutaconic aciduria-linked mutations. eLife. 9, e55279 (2020).
Pellegrini, L. et al. PAMP and PARL, two novel putative metalloproteases interacting with the COOH-terminus of Presenilin-1 and -2. J. Alzheimers Dis. 3, 181–190 (2001).
Suzuki, Y. et al. HAX-1, a novel intracellular protein, localized on mitochondria, directly associates with HS1, a substrate of Src family tyrosine kinases. J. Immunol. 158, 2736–2744 (1997).
Kovacs-Nagy, R. et al. HTRA2 defect: a recognizable inborn error of metabolism with 3-methylglutaconic aciduria as discriminating feature characterized by neonatal movement disorder and epilepsy—report of 11 patients. Neuropediatrics. 49, 373–378 (2018).
Wagner, M. et al. Mitochondrial DNA mutation analysis from exome sequencing—a more holistic approach in diagnostics of suspected mitochondrial disease. J. Inherit. Metab. Dis. 42, 909–917 (2019).
Rius, R. et al. Biparental inheritance of mitochondrial DNA in humans is not a common phenomenon. Genet. Med. 21, 2823–2826 (2019).
Norby, J. G. Coupled assay of Na+,K+-ATPase activity. Methods Enzymol. 156, 116–119 (1988).
Riley, L. G. et al. The diagnostic utility of genome sequencing in a pediatric cohort with suspected mitochondrial disease. Genet. Med. 22, 1254–1261 (2020).
Karczewski, K. J. et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 581, 434–443 (2020).
Lek, M. et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 536, 285–291 (2016).
Spiegel, R. et al. Fatal infantile mitochondrial encephalomyopathy, hypertrophic cardiomyopathy and optic atrophy associated with a homozygous OPA1 mutation. J. Med. Genet. 53, 127–131 (2016).
Harel, T. et al. Recurrent de novo and biallelic variation of ATAD3a, encoding a mitochondrial membrane protein, results in distinct neurological syndromes. Am. J. Hum. Genet. 99, 831–845 (2016).
Sinha, S., Zhu, Q. S., Romero, G. & Corey, S. J. Deletional mutation of the external domain of the human granulocyte colony-stimulating factor receptor in a patient with severe chronic neutropenia refractory to granulocyte colony-stimulating factor. J. Pediatr. Hematol. Oncol. 25, 791–796 (2003).
Triot, A. et al. Inherited biallelic CSF3R mutations in severe congenital neutropenia. Blood. 123, 3811–3817 (2014).
Deville, C., Franke, K., Mogk, A., Bukau, B. & Saibil, H. R. Two-step activation mechanism of the ClpB disaggregase for sequential substrate threading by the main ATPase motor. Cell Rep. 27, 3433–3446.e3434 (2019).
Gates, S. N. et al. Ratchet-like polypeptide translocation mechanism of the AAA+ disaggregase Hsp104. Science. 357, 273–279 (2017).
DeSantis, M. E. et al. Operational plasticity enables hsp104 to disaggregate diverse amyloid and nonamyloid clients. Cell. 151, 778–793 (2012).
Desantis, M. E. et al. Conserved distal loop residues in the Hsp104 and ClpB middle domain contact nucleotide-binding domain 2 and enable Hsp70-dependent protein disaggregation. J. Biol. Chem. 289, 848–867 (2014).
Sander, C. & Schneider, R. Database of homology-derived protein structures and the structural meaning of sequence alignment. Proteins. 9, 56–68 (1991).
Acknowledgements
Research relating to P5 was supported by a New South Wales Office of Health and Medical Research Sydney Genomics Collaborative grant and the Victorian Government’s Operational Infrastructure Support Program. We thank the Kinghorn Centre for Clinical Genomics and Lisa Riley (Children’s Hospital at Westmead, Australia) for assistance with genome sequencing and Helen Cashman (South Eastern Sydney Local Health District, Sydney, Australia) for providing the blood film images for P5. The Chair in Genomic Medicine awarded to John Christodoulou is generously supported by The Royal Children’s Hospital Foundation
Funding
This work was funded by the ERA PerMed project PerMiM (Austrian Science Fund FWF, I4704-B) to SBW. This study was supported by a “Sonata Bis 5” grant of the National Science Center Poland (2015/18/E/NZ1/00673) to SZ and DM.
Author information
Authors and Affiliations
Contributions
S.B.W., S.Z., S.G.-C., S.G., J.A.M., R.A.W. conceived the study and wrote the manuscript. S.B.W., J.R., C.E., D.W., I.H., C.v.S., U.L.-K., J.G., R.M.W., K.C.M., R.C.D., R.L., C.E.P., managed the patients, acquired and analyzed clinical data. N.I.W. analyzed the neuroimaging data. H.V. performed and analyzed the structural protein modeling. S.B.W., M.W., J.A.M. analyzed the genetic data. S.Z., D.M., H.W., R.G.F., S.G.-C., A.C.-O., S.W.G., J.A.M., J.C., R.A.W. conceived, developed and interpreted biochemical experiments. D.M., H.W. developed relevant biochemical techniques. All authors had full access to all data, reviewed drafts of the manuscript and approved the final version.
Corresponding author
Ethics declarations
Ethics declaration
The study was performed in compliance with the Declaration of Helsinki and conforming to the laws and regulations of the respective countries and institutes. All individuals gave written informed consent for genetic investigations and functional investigations in fibroblasts and for publication of data; this part of the study was approved by the local ethics committees, either Technical University Munich, Germany (5360/12S) or Land Salzburg, Austria (415-E/2552/10-2019) as appropriate. Patients gave additional written consent for publication of their photos.
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Unfortunately the funding information was not given. Please find here the Funding: This work was funded by the ERA PerMed project PerMiM (Austrian Science Fund FWF, I4704-B) to SBW. This study was supported by a “Sonata Bis 5” grant of the National Science Center Poland (2015/18/E/NZ1/00673) to SZ and DM.
Supplementary information
Rights and permissions
About this article

Cite this article
Wortmann, S.B., Ziętkiewicz, S., Guerrero-Castillo, S. et al. Neutropenia and intellectual disability are hallmarks of biallelic and de novo CLPB deficiency. Genet Med 23, 1705–1714 (2021). https://doi.org/10.1038/s41436-021-01194-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-021-01194-x
