
Abstract
Purpose
The utility of genome sequencing (GS) in the diagnosis of suspected pediatric mitochondrial disease (MD) was investigated.
Methods
An Australian cohort of 40 pediatric patients with clinical features suggestive of MD were classified using the modified Nijmegen mitochondrial disease severity scoring into definite (17), probable (17), and possible (6) MD groups. Trio GS was performed using DNA extracted from patient and parent blood. Data were analyzed for single-nucleotide variants, indels, mitochondrial DNA variants, and structural variants.
Results
A definitive MD gene molecular diagnosis was made in 15 cases and a likely MD molecular diagnosis in a further five cases. Causative mitochondrial DNA (mtDNA) variants were identified in four of these cases. Three potential novel MD genes were identified. In seven cases, causative variants were identified in known disease genes with no previous evidence of causing a primary MD. Diagnostic rates were higher in patients classified as having definite MD.
Conclusion
GS efficiently identifies variants in MD genes of both nuclear and mitochondrial origin. A likely molecular diagnosis was identified in 67% of cases and a definitive molecular diagnosis achieved in 55% of cases. This study highlights the value of GS for a phenotypically and genetically heterogeneous disorder like MD.
Similar content being viewed by others
INTRODUCTION
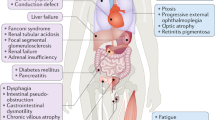
Mitochondrial disease (MD) encompasses a clinically and genetically heterogeneous group of disorders that affect mitochondrial respiratory chain function. The mitochondrial respiratory chain consists of five multimeric protein complexes that carry out oxidative phosphorylation to generate adenosine triphosphate (ATP), which supplies energy for virtually all tissues.1 Many of these disorders present during infancy, and tend to have a more severe and acute phenotype.1 Clinical presentations are highly variable and usually involve multisystem disease with combinations of the following features: skeletal muscle myopathy, cardiomyopathy, lactic acidosis, seizures, strokes, intellectual disability, ataxia, peripheral neuropathy, visual and/or hearing impairment, gastrointestinal disorders, hematological disorders, liver failure, and pancreatic exocrine and endocrine dysfunction. The majority (75–80%) of pediatric MDs are due to pathogenic variants in nuclear-encoded genes.1,2 Nuclear genes implicated encode protein subunits of the mitochondrial respiratory chain complexes and their assembly factors, as well as proteins involved in mitochondrial DNA (mtDNA) maintenance, mitochondrial protein synthesis, mitochondrial homeostasis, and metabolism.3 Nuclear gene defects commonly display autosomal recessive inheritance; however, autosomal dominant and X-linked modes of inheritance have also been reported. Pathogenic variants have been identified in over 300 of the ~1200 nuclear-encoded mitochondrial genes;3,4 however, in at least 40% of patients with presumed nuclear DNA variants, the genetic cause is not identified. MtDNA variants are responsible for around 75% of adult and 20–25% of childhood MD cases.2 Over 300 pathogenic mtDNA variants have been identified, including substitutions, deletions, and duplications.5 The variant may not be present in all of the multiple mtDNA copies per cell, a phenomenon known as heteroplasmy. Mutant load may vary between tissues within an individual, contributing to the highly variable disease phenotype. MtDNA variants are maternally inherited with unpredictable levels of heteroplasmy, or can arise spontaneously as de novo variants.
MD is difficult to diagnose and is mostly untreatable. Diagnosis has typically involved a complex combination of clinical assessment, blood and cerebrospinal fluid (CSF) metabolite profiles, brain imaging, tissue histology and enzymology, and specific nuclear gene and/or mtDNA sequencing.1,6 Histology and enzymology testing has been routine in the diagnostic workup of patients, and for those patients requiring general anesthesia to collect invasive tissue biopsies there is a potential risk of perioperative complications.7 This diagnostic method has been costly, invasive, time-consuming, and in many cases has not provided a molecular diagnosis, limiting the possibility of genetic counseling and informed reproductive decision making.
The advent of massively parallel sequencing has improved diagnostic rates and speed, and is increasingly being used as a first-line diagnostic test.8 The diagnostic success rate of massively parallel sequencing is enhanced by careful phenotypic classification of patients suspected of having a MD. Patients can be scored for mitochondrial disease severity according to one of several diagnostic criteria,9,10 based on clinical features, biochemical findings, examination of affected tissues (including morphology and enzymology), and brain imaging. Several approaches have been used for massively parallel sequencing of MD genes. The genetic heterogeneity and potential involvement of two genomes means that MD is not ideally suited to a targeted gene panel approach. To date, exome sequencing has been the favored method, with various strategies to cover the mitochondrial genome including mtDNA variant analysis using off-target reads, adding probes to the library preparation to capture mtDNA,11 or separate mtDNA sequencing. Genome sequencing (GS) has the advantage of giving good coverage of both the nuclear and mitochondrial genomes (up to 10–20,000× for the latter), and data can also be interrogated for structural variants (SVs). GS can detect heteroplasmy levels as low as 1% and mtDNA variants can be detected in blood in the majority of patients,12 reducing the need for biopsies. As costs fall, GS is likely to become the method of choice for molecular diagnosis for MD.12 Here we report on the utility of GS in diagnosis of a pediatric cohort of 40 patients with suspected MD.
MATERIALS AND METHODS
Patients
Forty pediatric patients with suspected MD were enrolled in this study. The study protocol was approved by the Human Research Ethics Committee of the Children’s Hospital at Westmead (HREC #10/CHW/113) and written informed consent was obtained for all families as approved by the local institutional human ethics committees. Patients were recruited from four Australian state based pediatric genetic metabolic service centers and were scored based on a modified Nijmegen mitochondrial disease severity scale (Supplementary Materials and Methods).9 Seventeen patients were classified as having definite MD (score 8–12), 17 probable MD (score 5–7), and 6 possible MD (score 2–4). Respiratory chain enzyme activities were determined as previously described13 in 33 of 40 patients, with 28 patients demonstrated to have a deficiency, while 5 patients had normal activities. Many of the patients had undergone previous genetic testing with array testing in 23/40, some form of mtDNA testing in 19/40, Sanger sequencing of one or more candidate nuclear genes in 15/40, gene panel testing in 5/40, and exome sequencing in 9/40. Detailed clinical histories of these cases are provided in the Supplementary Materials and Methods.
Genome sequencing
Genome sequencing was performed at the Kinghorn Centre for Clinical Genomics (Garvan Institute, Sydney) on genomic DNA extracted from blood of the patients and their parents where available. Genome sequencing libraries were prepared using Illumina TruSeq Nano HT v2.5 sample preparation kits and sequenced one lane per sample, on Illumina HiSeq X sequencers, via 2 × 150 bp reads, with >110 Gb data per lane, >75% bases with at least Q30 base quality, and >30× mean nuclear coverage. At this coverage, 95% of the nuclear genome was covered to >15× depth. Reads were aligned to the b37d5 reference genome using BWA MEM v0.7.10, sorted using novosort v1.03.01, then realigned around known indels, and base quality scores recalibrated using GATK v3.3. Variants were identified using GATK HaplotypeCaller v3.3 and GenotypeGVCFs, and variant filters established using VQSR. Variants were annotated using VEP v79, converted into a database using Gemini v0.11.0.14 Variants were filtered using Seave.15 Variants were confirmed by Sanger sequencing and classified according to the American College of Medical Genetics and Genomics guidelines.16
mtDNA variant analysis for low levels of heteroplasmy
Genome sequencing data from cases unsolved by the initial variant analysis were further analyzed for mtDNA variants with low levels of heteroplasmy using mity (Puttick et al., BioRxiv 2019; doi.org/10.1101/852210).
Structural variant analysis
Genome sequencing data from cases unsolved by the initial variant analysis were subsequently analyzed for SVs using ClinSV (Minoche et al., in preparation). SVs were filtered against population allele frequencies from 500 healthy individuals, family segregation, with all candidate variants manually inspected in Integrative Genomics Viewer (IGV).17
RESULTS
Molecular diagnosis
Mitochondrial disease genes
A definitive MD gene diagnosis was made in 15 cases and a likely MD gene diagnosis in a further five cases (Table 1; Fig. 1a). Among the definitive MD diagnoses, 11 were in nuclear genes (73%) and 4 in mtDNA (Fig. 1b), consistent with reports on the incidence of mtDNA variants in pediatric MD. Inheritance was autosomal recessive in all 11 nuclear gene cases (three homozygous and eight compound heterozygous) (Table 1). The mtDNA variants had heteroplasmy levels ranging from 40% to 100% in blood. These were identified using the standard analysis pipeline and no further low-level heteroplasmic variants were identified using mity, a sensitive analysis pipeline. No copy-number variants (CNVs) or SVs were identified in the cohort indicating CNVs/SVs only uncommonly contribute to the genetic etiology of MD. For patients with a definitive MD molecular diagnosis, 11 had been scored as definite prior to sequencing, 3 as probable, and 1 as possible MD according to a modified Nijmegen disease severity score (Fig. 1c).9

(a) The number of patients with a mitochondrial disease (MD) gene diagnosis. (b) Genomic origin of definitive MD gene variants identified. MD severity scores for patients with (c) definitive MD gene variants and (d) non-MD gene variants.
Nonmitochondrial disease genes
In seven cases, variants were identified in known disease genes with no previous evidence of causing a primary MD (Table 2, Fig. 1a). Five cases had autosomal recessive inheritance (one homozygous and four compound heterozygous), one case was X-linked recessive, and one was de novo dominant. For patients with non-MD variants, one had been scored as definite, five as probable, and one as possible MD (Fig. 1d). Three of these cases had a respiratory chain enzyme activity deficiency (ARX, NBAS, SLC39A8), three had borderline low activity (G6PC, HRAS, SKIV2L), and one was normal (EPG5) (Supplementary Appendix).
Diagnostic rate
In total, 24 novel variants were identified and 18 known variants (Tables 1 and 2). For many of the novel variants, functional assays were performed to enable classification as likely pathogenic or pathogenic (ACAD9, GFM1 data not shown; MECR,18 EPG5,19 NBAS,20 PNPT1,21 SLC39A822). Likely causal variants were identified in 27/40 cases (67.5%), with a definitive molecular diagnosis in 22 of these cases (55%).
New mitochondrial disease genes
Three potential novel disease genes were identified. Compound heterozygous variants in MECR were identified in a patient with dystonia, optic atrophy, and basal ganglia abnormalities (OMIM 617282; patient 6).18 MECR Sanger sequencing performed on a similarly affected younger sibling confirmed compound heterozygosity for the same two MECR variants identified by GS in the proband. Additional patients with similar phenotypes and variants in MECR were found using GeneMatcher,18 which is now part of Matchmaker Exchange.23 MECR is a key protein in mitochondrial fatty acid synthesis and variants result in reduced lipoylation of mitochondrial proteins. Compound heterozygous variants were identified in a respiratory chain complex assembly factor in a patient with developmental delay (patient 15) and another case identified through personal communications. In addition, compound heterozygous variants in a vitamin transporter were identified in a patient with Leigh-like disease (patient 20). Functional studies are currently underway for these two new candidate disease genes and will be published in the near future.
DISCUSSION
In this study, GS of a pediatric cohort with suspected MD resulted in identification of likely causal variants in 67.5% of cases, with a definitive diagnosis in 55% of cases. The diagnostic rate from previously reported exome sequencing of MD cohorts has ranged from 35% to 59%.24,25,26,27,28 There are no other reports of GS in MD cohorts. The diagnostic rate in GS studies is usually 40–60% compared with 25–35% in exome sequencing studies.29 Sequencing of family trios is likely to have contributed to the diagnostic success in this study as trio sequencing has been shown to increase the diagnostic rate by 8–17% in some exome sequencing studies.30
Previous studies have shown that careful phenotypic characterization of patient cohorts results in a higher diagnostic rate than poorly characterized cohorts. In this study we used a modified version of the Nijmegen mitochondrial disease severity score9 to categorize patients as having definite, probable, or possible MD. A definitive molecular diagnosis was found in 71% of definite MD cases, 47% of probable MD cases, and 33% of possible MD cases (although some of these did not have a primary MD). Similarly, in an MD exome sequencing study, 39% of the entire cohort was diagnosed; however, the diagnostic rate increased to 57% of the cohort with the highest suspicion of MD.26 Our results suggest that while the Nijmegen disease severity score captured some patients with secondary rather than primary MD, it is still a good indicator of MD and that the higher the score, the greater likelihood of reaching a genetic diagnosis. Given the phenotypic overlap between MD and some other Mendelian disorders, particularly those with secondary MD,31 and the phenotypic variability seen in MD, no scoring system is likely to exclusively capture primary MD patients. Indeed, in this study, a molecular diagnosis was found in 7 cases (17.5%) that did not have primary MD. MD exome studies have also reported up to 17% of cases diagnosed with non-MD gene causes.24,25,26 Secondary mitochondrial dysfunction has been reported in a number of other disorders including some neuromuscular, neurodegenerative, and metabolic disorders that have phenotypic overlap with MD.31 With regard to the cases diagnosed in this study, secondary mitochondrial dysfunction has previously been reported in association with variants in EPG5,32 and studies in mouse fibroblasts showed that HRAS affects mitochondrial respiration.33 SLC39A8 variants cause manganese deficiency and have previously been associated with a congenital disorder of glycosylation, type IIn (OMIM 616721); however, we have proposed that it may also cause a functional mitochondrial disorder.22 Recently G6PC knockdown mouse models were shown to have mitochondrial dysfunction.34 We could find no previous reports of mitochondrial dysfunction associated with ARX, NBAS, or SKIV2L.
The phenotypic variability of MD can contribute to the difficulty of diagnosis. A molecular diagnosis was found in four cases that had been unsolved following exome sequencing, most likely due to the phenotypic variability and/or the data analysis pipelines. The ECHS1 patient (patient 4, Supplementary Appendix), whose diagnosis was missed by previous exome sequencing, had cutis laxa, which is atypical for mitochondrial short-chain enoyl-CoA hydratase 1 deficiency (OMIM 616277).35 No additional variant was identified that could explain the cutis laxa. The EPG5 case (patient 22, Supplementary Appendix), which was also missed by exome sequencing, did not have all the cardinal features of Vici syndrome (OMIM 242840).19 The RRM2B diagnosis (patient 13) was most likely missed by exome sequencing due to the analysis pipeline failing to prioritize the synonymous splicing variant. The NBAS diagnosis (patient 25) included a deep intronic variant that was not covered by exome sequencing.20 In a consanguineous family, a PET100 variant that is a Lebanese founder variant was identified. This patient had a clear mitochondrial complex IV deficiency (OMIM 220110) but was also more severely affected than previously reported cases36 and died in the neonatal period, suffering from severe heart defects (patient 11, Supplementary Appendix). Further investigation of the GS data identified a novel homozygous variant in SCN5A, a known heart disease gene, which may have contributed to the patient phenotype. Multiple molecular diagnoses have been reported in up to 5% of positive exome sequencing results.37 Other patients in this cohort who expanded the phenotypic spectrum of their disease included the SLC39A8 patient (patient 27) who had features of both a MD and a glycosylation disorder.22
Obtaining a molecular diagnosis has impacted the management of some patients, made genetic counseling more accurate, and had a profound psychosocial impact on some families. The patient with variants in the vitamin transporter gene has been maintained on thiamine and biotin, which have dramatically reduced his titubation and tremor. Moreover, we predict that introduction of genomic sequencing earlier in the diagnostic journey will mean that invasive biopsies and their potential anesthetic risks can be avoided in a significant proportion of individuals.
Although a high diagnostic rate was achieved, a number of cases currently remain unresolved. This may be due to inability to interpret the effects of variants in noncoding regions, lack of knowledge of gene function, tissue specificity, and/or low levels of somatic or mtDNA variants. It may be that some of these cases can be solved by performing RNA-seq using an affected tissue (e.g., muscle), which can detect altered gene expression, aberrant splicing, or allelic imbalance. A recent study showed that 10% of an undiagnosed MD cohort was resolved by RNA-seq of patient fibroblast samples.38 RNA-seq becomes even more powerful when combined with GS, as it facilitates interpretation of the effects of noncoding variants.
Other challenges in improving molecular diagnostic rates include the need for functional genomic support to confirm that variants of uncertain significance in known or candidate disease genes are indeed pathogenic. A number of variants in this study were functionally evaluated including splice site variant analysis by reverse transcription polymerase chain reaction (PCR), western blotting to look at protein expression levels, and other specific functional assays (reported18,19,20,21,22 and unpublished data). While in some cases, further testing in diagnostic laboratories assisted in confirming the diagnosis, in many cases support was required from research laboratories. This remains a significant challenge moving forward, and functional genomic networks are being established in some countries to evaluate novel candidate disease genes and variants of uncertain significance39 (see also the Australian Functional Genomics Network: https://www.functionalgenomics.org.au). The identification of other cases with similar phenotypes and novel candidate disease genes through use of programs such as Matchmaker Exchange23 can also support pathogenicity and was used in this study to find other MECR patients.18
In this study, GS of a pediatric MD cohort led to identification of both nuclear and mtDNA variants, and a likely molecular diagnosis in 67.5% of cases. This is higher than reported for MD exome sequencing studies (59%). The falling cost of GS and its ability to detect both nuclear and mtDNA variants through high coverage of both genomes, intronic variants, and SVs suggests it will soon become the method of choice for the genomic diagnosis of MD.
References
Gorman S, Chinnery P, DiMauro S, et al. Mitochondrial diseases. Nat Rev Dis Primers. 2016;2:16080.
Frazier A, Thorburn D, Compton A. Mitochondrial energy generation disorders: genes, mechanisms and clues to pathology. J Biol Chem. 2017;294:5386–5395.
Stenton S, Prokisch H. Advancing genomic approached to the molecular diagnosis of mitochondrial disease. Essays Biochem. 2018;62:399–408.
Rahman J, Rahman S. Mitochondrial medicine in the omics era. Lancet. 2018;391:2560–2574.
Vafai S, Mootha V. Mitochondrial disorders as windows into an ancient organelle. Nature. 2012;491:374–383.
Menezes M, Riley L, Christodoulou J. Mitochondrial respiratory chain disorders in childhood: insights into diagnosis and management in the new era of genomic medicine. Biochim Biophys Acta. 2014;1840:1368–1379.
Parikh S, Goldstein A, Karaa A. et al. Patient care guidelines for primary mitochondrial disease: a consensus statement from the mitochondrial medicine society. Genet Med. 2017;19:1380.
McCormick E, Zolkipli-Cunningham Z, Falk M. Mitochondrial disease genetics update: recent insights into the molecular diagnosis and expanding phenotype of primary mitochondrial disease. Curr Opin Pediatr. 2018;30:714–724.
Morava E, van den Heuvel L, Hol F, et al. Mitochondrial disease criteria: diagnostic applications in children. Neurology. 2006;67:1823–1826.
Bernier F, Boneh A, Dennett X, Chow C, Cleary MT, Thorburn DR. Diagnositc criteria for respiratory chain disorders in adults and children. Neurology. 2002;59:1406–1411.
Falk MJ, Pierce EA, Consugar M, et al. Mitochondrial disease genetic diagnostics: optimized whole-exome analysis for all MitoCarta nuclear genes and the mitochondrial genome. Discov Med. 2012;14:389–399.
Raymond F, Horvath R, Chinnery P. First-line genomic diagnosis of mitochondrial disorders. Nature Rev Genet. 2018;19:399–400.
Frazier A, Thorburn D. Biochemical analyses of the electron transport chain complexes by spectrophotometry. Methods Mol Biol. 2012;837:49–62.
Paila U, Chapman B, Kirchner R, Quinlan A. GEMINI: Integrative exploration of genetic variation and genome annotations. PLoS Comput Biol. 2013;9:e1003153.
Gayevskiy V, Roscioli T, Dinger ME, Cowley MJ. Seave: a comprehensive web platform for storing and interrogating human genomic variation. Bioinformatics. 2019;35:122–125.
Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424.
Robinson J, Thorvaldsdóttir H, Winckler W, et al. Integrative Genomics Viewer. Nat Biotechnol. 2011;29:24–26.
Heimer G, Keratar JM, Riley LG, et al. MECR mutations cause childhood-onset dystonia and optic atrophy, a mitochondrial fatty acid synthesis disorder. Am J Hum Genet. 2016;99:1229–1244.
Balasubramaniam S, Riley LG, Vasudevan A, et al. EPG5-related Vici syndrome: a primary defect of autophagic regulation with an emerging phenotype overlapping with mitochondrial disorders. JIMD Rep. 2018;42:19–29.
Rius R, Riley LG, Guo Y, et al. Cryptic intronic NBAS variant reveals the genetic basis of recurrent liver failure in a child. Mol Genet Metab. 2019;126:77–82.
Rius R, Van Bergen NJ, Compton AG, et al. Clinical spectrum and functional consequences associated with bi-allelic pathogenic PNPT1 variants. J Clin Med. 2019;8:2020.
Riley LG, Cowley MJ, Gayevskiy V, et al. A SLC39A8 variant causes manganese deficiency, and glycosylation and mitochondrial disorders. J Inherit Metab Dis. 2017;40:261–269.
Philippakis AA, Azzariti DR, Beltran S, et al. The Matchmaker Exchange: a platform for rare disease gene discovery. Hum Mutat. 2015;36:915–921.
Ohtake A, Murayama K, Mori M, et al. Diagnosis and molecular basis of mitochondrial respiratory chain disorders: exome sequencing for disease gene identification. Biochim Biophys Acta. 2014;1840:1355–1359.
Pronicka E, Piekutowska-Abramczuk D, Ciara E, et al. New perspective in diagnostics of mitochondrial disorders: two years’ experience with whole-exome sequencing at a national paediatric centre. J Transl Med. 2016;14:174.
Wortmann SB, Koolen DA, Smeitink JA, van den Heuvel L, Rodenburg RJ. Whole exome sequencing of suspected mitochondrial patients in clinical practice. J Inherit Met Dis. 2015;38:437–443.
Kohda M, Tokuzawa Y, Kishita Y. et al. A comprehensive genetic analysis reveals the genetic landscape of mitochondrial respiratory chain complex deficiencies. PLoS Genet. 2016;12:e1005679.
Puusepp S, Reinson K, Pajusali S, et al. Effectiveness of whole exome sequencing in unsolved patients with a clinical suspicion of a mitochondrial disorder in Estonia. Mol Genet Metab Rep. 2018;15:80–89.
Mattick J, Dinger M, Schonrock N, Cowley M. Whole genome sequencing provides better diagnositc value than whole exome sequencing. Med J Aust. 2018;209:197–199.
Strande NT, Berg JS. Defining the clinical value of a genomic diagnosis in the era of next-generation sequencing. Annu Rev Genom Hum Genet. 2016;17:303–332.
Niyazov DM, Kahler SG, Eyre RE. Primary mitochondrial disease and secondary mitochondrial dysfunction: importance of distinction for diagnosis and treatment. Mol Syndromol. 2016;7:122–137.
Byrne S, Jansen L, U-KIng-Im J, et al. EPG-related Vici syndrome: a paradigm of neurodevelopmental disorders with defective autophagy. Brain. 2016;139:765–781.
Yang D, Wang M, Tang Y, et al. Impairment of mitochondrial respiration in mouse fibroblasts by oncogenic H-RASQ61L. Cancer Biol Ther. 2010;9:122–133.
Farah BL, Sinha RA, Wu Y. et al. Hepatic mitochondrial dysfunction is a feature of glycogen storage disease type Ia (GSDIa). Sci Rep. 2017;7:44408.
Balasubramaniam S, Riley LG, Bratkovic D, et al. Unique presentation of cutis laxa with Leigh-like syndrome due to ECHS1 deficiency. J Inherit Metab Dis. 2017;40:745–747.
Lim SC, Smith KR, Stroud DA, et al. A founder mutation in PET100 causes isolated complex IV deficiency in Lebanese individuals with Leigh syndrome. Am J Hum Genet. 2014;94:209–222.
Tayoun AA, Krock B, Spinner NB. Sequencing-based diagnostics for pediatric genetic diseases: progress and potential. Expert Rev Mol Diagn. 2016;16:987–999.
Kremer LS, Bader DM, Mertes C, et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat Commun. 2017;8:15824.
Beaulieu C, Majewski J, Schwartzentruber J, et al. FORGE Canada Consortium: outcomes of a 2-year national rare-disease gene-discovery project. Am J Hum Genet. 2014;94:809–817.
Acknowledgements
This research was supported by a New South Wales Office of Health and Medical Research Council Sydney Genomics Collaborative grant (C.M.S., J.C.), National Health and Medical Research Council (NHMRC) project grant 1026891 (J.C.), NHMRC research fellowship 1102896 (D.R.T.) and a NSW Health Early-Mid Career Fellowship (M.J.C.). We are grateful to the Crane and Perkins families for their generous financial support. The research conducted at the Murdoch Children’s Research Institute was supported by the Victorian Government’s Operational Infrastructure Support Program. We thank the Kinghorn Centre for Clinical Genomics for assistance with production and processing of genome sequencing data, and Kate Wilson, Genetic Metabolic Disorders Service, The Children’s Hospital at Westmead, for assistance in preparing some of the clinical summaries.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Disclosure
The authors declare no conflicts of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Riley, L.G., Cowley, M.J., Gayevskiy, V. et al. The diagnostic utility of genome sequencing in a pediatric cohort with suspected mitochondrial disease. Genet Med 22, 1254–1261 (2020). https://doi.org/10.1038/s41436-020-0793-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41436-020-0793-6
Keywords
This article is cited by
-
Wide diagnostic and genotypic spectrum in patients with suspected mitochondrial disease
Orphanet Journal of Rare Diseases (2023)
-
Use of dual genomic sequencing to screen mitochondrial diseases in pediatrics: a retrospective analysis
Scientific Reports (2023)
-
TEFM variants impair mitochondrial transcription causing childhood-onset neurological disease
Nature Communications (2023)
-
Implications of mitochondrial DNA variants in pediatric B-cell acute lymphoblastic leukemia
Egyptian Journal of Medical Human Genetics (2022)
-
Recommendations for whole genome sequencing in diagnostics for rare diseases
European Journal of Human Genetics (2022)
