
Abstract
Objectives
To systematically analyse the NMNAT1 variant spectrum and frequency, the associated phenotypic characteristics, and potential genotype-phenotype correlations based on our data and literature review.
Methods
Biallelic potential pathogenic variants (PPV) in NMNAT1 were collected from our in-house exome sequencing data. Whole-genome sequencing was conducted subsequently for patients with only one heterozygous PPV detected in NMNAT1. The clinical data were reviewed and evaluated in detail. Furthermore, the literature was reviewed for reports of NMNAT1 variants and their associated phenotypes.
Results
Eleven NMNAT1 variants, including two novel variants, were detected in 8 families from our cohort. All of the 9 available patients showed generalized tapetoretinal dystrophy at an early age (88.9% in the first decade), and disciform macular atrophy was identified in six patients from five unrelated families. Among a total of 125 patients from 8 families of our cohort and 91 families reported by the available literature, 92.9% patients showed onset of disease in the first year after birth, and 89.0% patients showed visual acuity of 0.05 or lower. All of the 39 patients with fundus photos available presented disciform macular atrophy with generalized tapetoretinal dystrophy. Most (54/80, 67.5%) of causative NMNAT1 variants were missense. The most frequent variants in Caucasian and Asian population are p.E257K and p.R237C, respectively.
Conclusions
Early-onset age, disciform macular atrophy with generalized tapetoretinal dystrophy, and poor visual acuity are the typical features of NMNAT1-associated retinal degeneration. Different variant hot spots of NMNAT1 were observed in different populations.
Similar content being viewed by others

Introduction
Nicotinamide mononucleotide adenylyltransferase 1, encoded by the NMNAT1 gene (HGNC ID: 17877, OMIM: 608700), is a rate-limiting enzyme in the biosynthesis of nicotinamide adenine dinucleotide (NAD) [1]. NMNAT1 maps to chromosome 1p36.2 and is a 5-exon gene encoding 280 residues. NMNAT1 is a key component of the Wlds fusion protein and plays an important role in neuroprotection [2, 3].
Biallelic variants in NMNAT1 have been reported to be responsible for autosomal recessive inherited retinal degeneration (IRD), frequently for Leber congenital amaurosis (LCA), and rarely for cone and cone-rod dystrophy (CORD) [4,5,6,7,8]. Variants in NMNAT1 are an important cause of LCA, accounting for roughly 4.6~8.4% of LCA [5, 7].
In this study, sequence variants of NMNAT1 were selected from our in-house data. These samples were collected from a cohort of patients with suspected IRD who underwent whole-exome sequencing (WES) or targeted exome sequencing (TES) analysis. Eleven potential pathogenic variants (including two novel variants) in biallelic status were identified in eight families with phenotypes associated with LCA. Detailed phenotypic analyses and cosegregation analyses were conducted in these families. In addition, all variants and associated phenotypes from published literature were systemically reviewed. These results provide a brief overview of NMNAT1 variant frequencies in addition to data indicating the phenotypic features associated with NMNAT1 variants.
Materials and methods
Probands and family members
Data obtained from probands with various forms of IRD and their available family members were collected from our Pediatric and Genetic Clinic, Zhongshan Ophthalmic Center, Guangzhou, China. Genomic DNA was prepared, and clinical data were recorded in our laboratory at the State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center. Prior to the collection of clinical data and venous blood samples, written informed consent according to the tenets of the Declaration of Helsinki was obtained from participating individuals or their guardians. This study was approved by the Institutional Review Board of Zhongshan Ophthalmic Center. Genomic DNA was prepared from peripheral venous leukocytes via a previously described method [9].
Variant detection
Variants in NMNAT1 were retrieved from the exome sequencing data of patients with suspected IRD, including whole-exome sequencing (WES) data and targeted exome sequencing (TES) data. Whole-genome sequencing (WGS) was performed in one proband in whom only a single heterozygous variant in NMNAT1 was detected by WES. The procedures used to perform WES and TES were described in our previous studies [10,11,12]. In a nutshell, exons, as well as their adjacent intronic regions (at least 20 bp), were captured using the Agilent SureSelect Human All Exon Enrichment Kit (51189318 base pairs) array on the Illumina HiSequation system (Illumina, San Diego, CA, USA) with an average sequencing depth of 125-fold. The reads were mapped against the consensus sequence (UCSC hg19). Single nucleotide polymorphisms and small insertions/deletions were detected by SAMTOOLS (http://samtools.sourceforge.net/) and annotated by SnpEff (http://snpeff.sourceforge.net/) and ANNOVAR (http://annovar.openbioinf ormatics.org/en/latest/). TES was performed in our laboratory and targeted exons of 126 genes (including NMNAT1) which were mutated most frequently in genetic eye diseases. Sequencing variants in NMNAT1 were initially filtered by multi-step bioinformatics analyses, as described in our previous studies [10, 11]. Candidate variants were also filtered by removing variants with allele frequencies greater than or equal to 1% in existing databases, including the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/) and the 1000 Genomes (https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes/). The possible impact of missense changes was predicted using REVEL (https://sites.google.com/site/revelgenomics/) [13], CADD (https://cadd.gs.washington.edu/) [14], SIFT (http://sift.jcvi.org/www/SIFT_enst_submit.html) [15] and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/index.shtml) [16]. All candidate disease-associated variants were confirmed by Sanger sequencing, and cosegregation in available family members was further evaluated.
Phenotypic characterization
Routine clinical data obtained from all available patients with biallelic NMNAT1 variants were reviewed. Additional ocular examinations were carried out when necessary, including electroretinogram and fundus photographs. Probands’ parents or siblings were examined if available. Phenotypes were classified based on first symptoms, visual acuity, fundus changes, and electroretinogram data. Three LCA families with no clinical information reported by our previous study [17] were also included in the current study for phenotypic analysis.
Literature review of NMNAT1 variants
The keyword “NMNAT1” was searched on PubMed (https://www.ncbi.nlm.nih.gov/pubmed/), Web of Science (http://apps.webofknowledge.com/WOS_GeneralSearch_input.do?product=WOS&SID = 8BApKgHrMnA4WVjh45d&search_mode=GeneralSearch), and the Human Genome Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) on February, 2021. All available literature reporting NMNAT1 variants and the corresponding phenotypic data published in English were collected. The number of different variants, the frequency of each variant, and their associated phenotypes were summarized.
Results
Phenotypes of nine Chinese patients from eight families
In total, 8 families were found to harbour biallelic NMNAT1 variants. Of the 11 affected individuals in the 8 families, ophthalmological data were available for 9, including all 8 probands and one affected sibling (Table 1, Supplementary Fig. 1). Variants were detected by WES or TES in 7 families and by WGS in one family (F06). Initially, WES was done for families F02 and F06. Biallelic variants in NMNAT1 were identified in F02, while only a single heterozygous variant (c.634 G > A) was detected in F06. Therefore, WGS was conducted in F06 to determine whether there was the other NMNAT1 variant, revealing the second variant (c.713 A > G). Of the eleven variants detected, nine were missense and two were predicted to truncate the protein (Table 2). Two novel variants, c.698delG and c.−57 + 1 G > A, were identified. These variants were confirmed by Sanger sequencing and cosegregated with the disease in the families (Supplementary Fig. 1). The initial symptoms in the probands were poor vision with nystagmus in four families, no pursuit of light with nystagmus in one family, no pursuit of light with roving nystagmus and an oculo-digital sign in one family, poor vision with roving nystagmus and an oculo-digital sign in one family, and poor vision with suspected strabismus in one family. Symptoms appeared as early as three months old and no later than 1 year old. Visual acuity ranged from no pursuit of light to 0.20. Fundus observation demonstrated almost identical early generalized tapetoretinal dystrophy with disciform macular atrophy as well as attenuated retinal vessels (Figs. 1 and 2). Cone and rod responses on electroretinogram were extinguished or severely reduced.

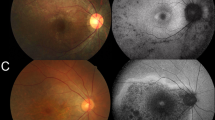
a–f A similar feature consisting of disciform coloboma-like macular atrophy was observed among patients from different families and patients within the same family. Tapetoretinal degeneration was present in the posterior pole and mid-peripheral retina. h, i OCT showed the loss of the outer retina in the region of the macular atrophic lesion with diffuse retinal atrophy of the para-lesion area. g The OCT of a macular coloboma patient served as a control. Family and individual numbers that correspond to those shown in Table 1 are shown in the bottom left corner of each plate.

a–d With age, the fundus demonstrates greater coloboma-like macular atrophy and more pigmentation around the macular region. The border of the lesion becomes unclear, forming a map-like lesion. e–f In addition to the large central macular atrophic lesion, dense intraretinal pigmentation presents in the posterior pole and mid-peripheral retina but not the periphery. Family and individual numbers are shown in the bottom left corner of each plate.
Similar macular atrophy was documented in seven of the nine available patients (Table 1, Fig. 1). The atrophic macular lesions were disciform or nummular. Optical coherent tomograghy (OCT) showed the loss of the outer retina in the region of the macular atrophy and diffuse retinal atrophy at the surrounding area (Fig. 1h, i). This type of lesion resembles pseudocoloboma while a true macular coloboma showed loss of choroid with retina preserved at the surrounding area of the coloboma (Fig. 1g). All of the nine patients developed generalized tapetoretinal degeneration no later than 12 years of age. The macular atrophy lesions enlarged with age (Fig. 2). In addition to macular atrophy, generalized retinal degeneration was also observed in the posterior and midperipheral retina, with relatively mild change in the far peripheral region (Fig. 2e).
A comprehensive review of variants and phenotypes
Profile of NMNAT1 variants
To date, 84 variants in NMNAT1 have been identified, including 80 variants in biallelic status (Supplementary Table 1) and four variants only identified as single heterozygous changes (Supplementary Table 2). The four heterozygous variants were not enrolled in this study because NMNAT1 variants were only associated with autosomal recessive IRD so far. The 80 variants identified in biallelic status were detected in 158 patients from 129 families with autosomal recessive IRD, including eight families from our cohort and 121 families reported by literature. The 80 variants can be classified as missense (54/80 [67.5%]), frameshift indel (7/80 [8.8%]), nonsense (6/80 [7.5%]), splicing defect (5/80 [6.3%]), gross indel (5/80 [6.3%]), start loss (1/80 [1.3%]), stop loss (1/80 [1.3%]), and regulatory (1/80 [1.3%]) (Fig. 3a). Of the 54 missense variants, 92.6% (50/54) were predicted to be damaging by SIFT or PolyPhen-2 (Supplementary Table 1). All variants but one were very rare, with no homozygotes detected in the ExAC database and frequencies ranging from 0 to 0.0002. The c.769 G > A (p.Glu257Lys) variant had a frequency of 0.0006 for all ethnicities and 0.0010 (66/66384) for Europeans, with no homozygotes detected in the ExAC database. The most common genotype was the combination of two missense variants, either homozygous or compound heterozygous (“missense + missense”) (89/129 [69.0%]) (Fig. 3b). The frequency of each variant (except for 5 gross indel) was summarized and is shown in Fig. 3c. Most variants are located in the adenyltransferase domain (residues 9-254). The most frequent variant, c.769 G > A (p.Glu257Lys), was present in 63 of 129 (48.8%) families. They consisted of one homozygote and 62 compound heterozygotes. The variant accounted for 24.8% (64/258) of all mutant alleles (Supplementary Table 1, Fig. 3c). Of the 64 mutant alleles, 93.8% (60/64) were identified in Caucasian families [4,5,6,7,8, 18,19,20,21,22,23,24,25,26,27,28,29] and the rest were identified in Arabian families [6, 30, 31]. The second frequent variant, c.709 C > T (p.Arg237Cys), accounted for 21 mutant alleles, most (18/21, 85.7%) of which were detected in Asian patients [5, 20, 32,33,34,35].

a Eighty variants identified in 129 families with biallelic NMNAT1 variants. Frameshift indel, frameshift deletion or frameshift insertion; gross indel, gross deletion or gross insertion; splicing, splicing defect. b Genotypes of the 129 families with biallelic NMNAT1 variants. c Distribution and frequency of variants (except for 5 gross indel) identified in the 129 families with biallelic NMNAT1 variants are shown above the mRNA structure (Ref. NM_022787.4). The position and frequency of the variants are drawn above the structure of the mRNA. Nucleotide numbering is based on the cDNA sequence of NMNAT1 (Ref. NM_022787.4), where the A of the ATG initiation codon is 1. The blue area in the middle represents the coding region. The orange acetyltransferase domain extends from codon 9 to codon 254. The white areas before and after the coding region represent the 5’ UTR and 3’ UTR, respectively. Part of the 3’ UTR sequence is omitted as shown by double-dotted slashes. The frequency of c.769 G > A (p.Glu257Lys) is 64, accounting for 24.8% of total mutant alleles.
Phenotypic characteristics of NMNAT1-associated retinal degeneration
Among the 158 patients from 129 families, the age of onset was available for 125 patients. Symptoms appeared as early as birth and no later than 11 years of age and 92.0% (115/125) presented within the first year of life. Visual acuity ranged from no pursuit of light to 0.50. Of the 114 patients with visual acuity available, 89.5% (102/114) showed visual acuity of 0.05 Snellen equivalent or lower (legal blindness). Fundus photos were available for 39 patients [4,5,6,7, 20, 22, 31, 33,34,35,36,37,38,39,40,41,42]. Despite the different diagnoses, the fundus changes of all these patients presented macular atrophy, mostly similar to those shown in Fig. 1.
Genotype-phenotype correlations
Among the 80 biallelic variants in NMNAT1, there were four missense variants predicted to be benign by both SIFT and PolyPhen-2, i.e., c.271 G > A (p. Glu91Lys), c.439 G > C (p. Ala147Pro), c.736 G > C (p.Glu246Gln), and c.769 G > A (p. Glu257Lys), and two missense variants with very low REVEL and CADD scores, i.e., c.451 G > A (p. Val151Ile) and c.500 A > G (p. Asn167Ser). The 129 families with homozygous or compound heterozygous variants were comprised of 121 families with LCA or EOSRD (early-onset severe retinal dystrophy), six families with CORD, and two families with retinitis pigmentosa. Of the two families with retinitis pigmentosa, one was reported without clinical information [43] and another was with the c.−57 + 7 T > G variant [44], which was predicted to be non-damaging by both the BDGP and HSF splice prediction tools. Of the six families with CORD, one was homozygous for the c.271 G > A (p.Glu91Lys) variant, one was homozygous for the c.500 A > G (p.Asn167Ser) variant, and four were compound heterozygotes for the c.769 G > A (p. Glu257Lys) variant [8, 29, 31, 40]. The c.439 G > C (p. Ala147Pro), c.451 G > A (p. Val151Ile) and c.736 G > C (p.Glu246Gln) variants were each identified in one compound heterozygous family with LCA [6, 7, 26]. All families with truncating variants (frameshift indel or nonsense variants) were diagnosed with LCA, and all five families with CORD had biallelic missense variants, implying a potential correlation between more severe genotypes and more severe phenotypes.
Discussion
In this study, disciform macular atrophy with early generalized retinal dystrophy was identified in seven patients from six unrelated families with NMNAT1 variants. Eleven variants, including two novel variants (c.698delG and c.−57 + 1 G > A), were detected in 8 unrelated Chinese families with LCA. The most common variant, c.769 G > A (p.Glu257Lys), was not detected in our cohort, which may be due to ethnic heterogeneity since it has previously been described only in Europeans and Arabians. A variant, c.713 A > G, was not detected by WES, but was detected by WGS. All variants were confirmed by Sanger sequencing.
Including data from our lab and the literature, we summarized the genetic spectrum of NMNAT1, phenotypic characteristics of patients with NMNAT1 variants, and the potential genotype-phenotype correlation for IRD caused by variants in this gene.
Genetically, 80 variants were detected in 129 families with biallelic NMNAT1 variants thus far. Most variants in NMNAT1 were missense (67.5%). The most common genotype was compound heterozygous (“missense + missense”). The most frequent variant, c.769 G > A (p.Glu257Lys), accounts for approximately one-fourth of mutant alleles and is enriched in the Caucasian population. The variant was predicted to be non-damaging by both SIFT and PolyPhen-2 and with low REVEL and CADD scores (Supplementary Table 1). It is outside of the adenyltranferase domain (Fig. 3c) and has a relatively high population frequency in Europeans in the ExAC database. It has been reported to have been detected in homozygous status in a patient with no ocular abnormalities, suggesting that this variant is not fully penetrant [45]. Unlike the Caucasian population, c.709 C > T (p.Arg237Cys) is the most common NMNAT1 variant in the Asian population. The variant was predicted to be damaging by both SIFT and PolyPhen-2 and with high REVEL and CADD scores (Supplementary Table 1). It is located in the adenyltranferase domain (Fig. 3c) and has a very low population frequency in East Asian in the ExAC database.
Clinically, patients with biallelic NMNAT1 variants have the following phenotypic characteristics: the onset age was early (92.0% in the first year of life); the best visual acuity was poor (89.5% were lower than 0.05 Snellen equivalent); the early generalized retinal dystrophy with disciform macular atrophy was a typical fundus finding associated with NMNAT1 variants.
Based on comparative genotypic analysis of patients with different phenotypes, a potential association between milder missense variants (predicted benign by online tools) and milder phenotypes (CORD) was suggested, which is in accordance with previous study [22]. All of the five CORD families carried one or two missense variants that were predicted to be non-damaging by both SIFT and PolyPhen-2 and with low REVEL and CADD scores. We speculate that the milder phenotype might be due to a milder reduction of NMNAT1 enzymatic activity. The correlation between phenotypic severity and variant type might be helpful for clinical gene testing.
Supplemental information is available at Eye’s website.
SUMMARY
What was known before
-
Biallelic variants in NMNAT1 can cause Leber congenital amaurosis and cone-rod dystrophy.
What this study adds
-
Early-onset age, disciform macular atrophy, and very poor visual acuity (legally blindness) are the typical features of NMNAT1-associated retinal degeneration. Two novel variants, c.698delG and c.−57 + 1 G > A, were identified. A potential association between milder missense mutations and milder phenotypes was suggested.
References
Raffaelli N, Sorci L, Amici A, Emanuelli M, Mazzola F, Magni G. Identification of a novel human nicotinamide mononucleotide adenylyltransferase. Biochem Biophys Res Commun. 2002;297:835–40.
Araki T, Sasaki Y, Milbrandt J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science. 2004;305:1010–3.
Sasaki Y, Vohra BP, Baloh RH, Milbrandt J. Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J Neurosci. 2009;29:6526–34.
Chiang PW, Wang J, Chen Y, Fu Q, Zhong J, Chen Y, et al. Exome sequencing identifies NMNAT1 mutations as a cause of Leber congenital amaurosis. Nat Genet. 2012;44:972–4.
Falk MJ, Zhang Q, Nakamaru-Ogiso E, Kannabiran C, Fonseca-Kelly Z, Chakarova C, et al. NMNAT1 mutations cause Leber congenital amaurosis. Nat Genet. 2012;44:1040–5.
Koenekoop RK, Wang H, Majewski J, Wang X, Lopez I, Ren H, et al. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat Genet. 2012;44:1035–9.
Perrault I, Hanein S, Zanlonghi X, Serre V, Nicouleau M, Defoort-Delhemmes S, et al. Mutations in NMNAT1 cause Leber congenital amaurosis with early-onset severe macular and optic atrophy. Nat Genet. 2012;44:975–7.
Nash BM, Symes R, Goel H, Dinger ME, Bennetts B, Grigg JR, et al. NMNAT1 variants cause cone and cone-rod dystrophy. Eur J Hum Genet. 2018;26:428–33.
Wang Q, Wang P, Li S, Xiao X, Jia X, Guo X, et al. Mitochondrial DNA haplogroup distribution in Chaoshanese with and without myopia. Mol Vis. 2010;16:303–9.
Jiang D, Li J, Xiao X, Li S, Jia X, Sun W, et al. Detection of mutations in LRPAP1, CTSH, LEPREL1, ZNF644, SLC39A5, and SCO2 in 298 families with early-onset high myopia by exome sequencing. Invest Ophthalmol Vis Sci. 2014;56:339–45.
Li J, Jiang D, Xiao X, Li S, Jia X, Sun W, et al. Evaluation of 12 myopia-associated genes in Chinese patients with high myopia. Invest Ophthalmol Vis Sci. 2015;56:722–9.
Wang P, Li S, Sun W, Xiao X, Jia X, Liu M, et al. An ophthalmic targeted exome sequencing panel as a powerful tool to identify causative mutations in patients suspected of hereditary eye diseases. Transl Vis Sci Technol. 2019;8:21.
Ioannidis NM, Rothstein JH, Pejaver V, Middha S, McDonnell SK, Baheti S, et al. REVEL: an ensemble method for predicting the pathogenicity of rare missense variants. Am J Hum Genet. 2016;99:877–85.
Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886–D894.
Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009;4:1073–81.
Flanagan SE, Patch AM, Ellard S. Using SIFT and PolyPhen to predict loss-of-function and gain-of-function mutations. Genet Test Mol Biomark. 2010;14:533–7.
Xu Y, Xiao X, Li S, Jia X, Xin W, Wang P, et al. Molecular genetics of Leber congenital amaurosis in Chinese: new data from 66 probands and mutation overview of 159 probands. Exp Eye Res. 2016;149:93–99.
Corton M, Nishiguchi KM, Avila-Fernandez A, Nikopoulos K, Riveiro-Alvarez R, Tatu SD, et al. Exome sequencing of index patients with retinal dystrophies as a tool for molecular diagnosis. PLoS One. 2013;8:e65574.
Siemiatkowska AM, van den Born LI, van Genderen MM, Bertelsen M, Zobor D, Rohrschneider K, et al. Novel compound heterozygous NMNAT1 variants associated with Leber congenital amaurosis. Mol Vis. 2014;20:753–9.
Coppieters F, Todeschini AL, Fujimaki T, Baert A, De Bruyne M, Van, et al. Hidden genetic variation in LCA9-associated congenital blindness explained by 5’UTR mutations and copy-number variations of NMNAT1. Hum Mutat. 2015;36:1188–96.
Thompson JA, De Roach JN, McLaren TL, Montgomery HE, Hoffmann LH, Campbell IR, et al. The genetic profile of Leber congenital amaurosis in an Australian cohort. Mol Genet Genom Med. 2017;5:652–67.
Kumaran N, Robson AG, Michaelides M. A novel case series of Nmnat1-associated early-onset retinal dystrophy: extending the phenotypic spectrum. Retin Cases Brief Rep. 2021;15:139–44.
Neveling K, Feenstra I, Gilissen C, Hoefsloot LH, Kamsteeg EJ, Mensenkamp AR, et al. A post-hoc comparison of the utility of sanger sequencing and exome sequencing for the diagnosis of heterogeneous diseases. Hum Mutat. 2013;34:1721–6.
Consugar MB, Navarro-Gomez D, Place EM, Bujakowska KM, Sousa ME, Fonseca-Kelly ZD, et al. Panel-based genetic diagnostic testing for inherited eye diseases is highly accurate and reproducible, and more sensitive for variant detection, than exome sequencing. Genet Med. 2015;17:253–61.
Bravo-Gil N, Mendez-Vidal C, Romero-Perez L, Gonzalez-del Pozo M, Rodriguez-de la Rua E, Dopazo J, et al. Improving the management of inherited retinal dystrophies by targeted sequencing of a population-specific gene panel. Sci Rep. 2016;6:23910.
Carss KJ, Arno G, Erwood M, Stephens J, Sanchis-Juan A, Hull S, et al. Comprehensive rare variant analysis via whole-genome sequencing to determine the molecular pathology of inherited retinal disease. Am J Hum Genet. 2017;100:75–90.
Stone EM, Andorf JL, Whitmore SS, DeLuca AP, Giacalone JC, Streb LM, et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology. 2017;124:1314–31.
Weisschuh N, Feldhaus B, Khan MI, Cremers FPM, Kohl S, Wissinger B, et al. Molecular and clinical analysis of 27 German patients with leber congenital amaurosis. PLoS ONE. 2018;13:e0205380.
Boulanger-Scemama E, El Shamieh S, Demontant V, Condroyer C, Antonio A, Michiels C, et al. Next-generation sequencing applied to a large French cone and cone-rod dystrophy cohort: mutation spectrum and new genotype-phenotype correlation. Orphanet J Rare Dis. 2015;10:85.
Coppieters F, Van Schil K, Bauwens M, Verdin H, De Jaegher A, Syx D, et al. Identity-by-descent-guided mutation analysis and exome sequencing in consanguineous families reveals unusual clinical and molecular findings in retinal dystrophy. Genet Med. 2014;16:671–80.
Bedoukian EC, Zhu X, Serrano LW, Scoles D, Aleman TS. Nmnat1-associated cone-rod dystrophy: evidence for a spectrum of foveal maldevelopment. Retin Cases Brief Rep. 2020. https://doi.org/10.1097/ICB.0000000000000992. Online ahead of print.
Wang H, Wang X, Zou X, Xu S, Li H, Soens ZT, et al. Comprehensive molecular diagnosis of a large chinese leber congenital amaurosis cohort. Invest Ophthalmol Vis Sci. 2015;56:3642–55.
Han J, Rim JH, Hwang IS, Kim J, Shin S, Lee ST, et al. Diagnostic application of clinical exome sequencing in Leber congenital amaurosis. Mol Vis. 2017;23:649–59.
Hosono K, Nishina S, Yokoi T, Katagiri S, Saitsu H, Kurata K, et al. Molecular diagnosis of 34 Japanese families with leber congenital amaurosis using targeted next generation sequencing. Sci Rep. 2018;8:8279.
Surl D, Shin S, Lee ST, Choi JR, Lee J, Byeon SH, et al. Copy number variations and multiallelic variants in Korean patients with Leber congenital amaurosis. Mol Vis. 2020;26:26–35.
Jin X, Qu LH, Meng XH, Xu HW, Yin ZQ. Detecting genetic variations in hereditary retinal dystrophies with next-generation sequencing technology. Mol Vis. 2014;20:553–60.
Deng Y, Huang H, Wang Y, Liu Z, Li N, Chen Y, et al. A novel missense NMNAT1 mutation identified in a consanguineous family with Leber congenital amaurosis by targeted next generation sequencing. Gene. 2015;569:104–8.
Hedergott A, Volk AE, Herkenrath P, Thiele H, Fricke J, Altmuller J, et al. Clinical and genetic findings in a family with NMNAT1-associated Leber congenital amaurosis: case report and review of the literature. Graefes Arch Clin Exp Ophthalmol. 2015;253:2239–46.
Wang S, Zhang Q, Zhang X, Wang Z, Zhao P. Clinical and genetic characteristics of Leber congenital amaurosis with novel mutations in known genes based on a Chinese eastern coast Han population. Graefes Arch Clin Exp Ophthalmol. 2016;254:2227–38.
Khan AO, Budde BS, Nurnberg P, Kawalia A, Lenzner S, Bolz HJ. Genome-wide linkage and sequence analysis challenge CCDC66 as a human retinal dystrophy candidate gene and support a distinct NMNAT1-related fundus phenotype. Clin Genet. 2018;93:149–54.
Jinda W, Taylor TD, Suzuki Y, Thongnoppakhun W, Limwongse C, Lertrit P, et al. Whole exome sequencing in eight thai patients with leber congenital amaurosis reveals mutations in the CTNNA1 and CYP4V2 genes. Invest Ophthalmol Vis Sci. 2017;58:2413–20.
Bedoni N, Quinodoz M, Pinelli M, Cappuccio G, Torella A, Nigro V, et al. An Alu-mediated duplication in NMNAT1, involved in NAD biosynthesis, causes a novel syndrome, SHILCA, affecting multiple tissues and organs. Hum Mol Genet. 2020;29:2250–60.
Wang LK, Zhang JL, Chen NN, Wang L, Zhang FS, Ma ZZ, et al. Application of whole exome and targeted panel sequencing in the clinical molecular diagnosis of 319 chinese families with inherited retinal dystrophy and comparison study. Genes-Basel. 2018;9:360.
Huang H, Chen Y, Chen H, Ma Y, Chiang PW, Zhong J, et al. Systematic evaluation of a targeted gene capture sequencing panel for molecular diagnosis of retinitis pigmentosa. PLoS One. 2018;13:e0185237.
Siemiatkowska AM, Schuurs-Hoeijmakers JH, Bosch DG, Boonstra FN, Riemslag FC, Ruiter M, et al. Nonpenetrance of the most frequent autosomal recessive leber congenital amaurosis mutation in NMNAT1. JAMA Ophthalmol. 2014;132:1002–4.
Acknowledgements
The authors are grateful to the families for their participation.
Funding
This work was supported by the Medical Scientific Research Foundation of Guangdong Province (grant number A2021313) and the National Natural Science Foundation of China (grant number 81970837).
Author information
Authors and Affiliations
Contributions
QZ designed the study. SL, SW, XX, and QZ recruited the participants. ZY, SL, SW, XX, and WS collected the clinical data. SL, XX, and QZ performed the genotyping. QZ and ZY performed the statistical analysis. ZY reviewed the data from the literature. ZY and QZ discussed the results and wrote the paper. All authors reviewed and gave final approval of this paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Yi, Z., Li, S., Wang, S. et al. Clinical features and genetic spectrum of NMNAT1-associated retinal degeneration. Eye 36, 2279–2285 (2022). https://doi.org/10.1038/s41433-021-01853-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41433-021-01853-y
This article is cited by
-
Molecular background of Leber congenital amaurosis in a Polish cohort of patients—novel variants discovered by NGS
Journal of Applied Genetics (2023)
