
Abstract
It is a long-standing dream to fabricate micron-sized inorganic spheres that have nanocompartments enclosed by a permeable shell and thus resemble the shape of natural cells. Here, we demonstrate a novel synthesis protocol to attain this goal for the first time. This protocol unprecedentedly harnesses interfacial sol–gel growth around Pickering droplets coupled with a surfactant assembly-directed sol–gel process within droplet-confined spaces. This protocol enables us to fabricate novel mesoporous silica microspheres (MSMs) with tunable interior architectures, such as hollow MSMs, hollow nanosphere-containing MSMs (hn@MSMs), nanosphere-containing MSMs (n@MSMs), yolk-shell-structured MSMs (y@MSMs) and “solid” MSMs. The obtained multi-compartmentalized MSMs exhibit good permeability to external molecules and good mechanical strength against stress. Moreover, their structural features benefit practical applications of CO2 capture and enzymatic reactions. Due to the high dispersion of tetraethylenepentamine and enzyme in the spatially separated nanocompartments, the developed CO2 sorbents and catalysts exhibit significantly enhanced CO2 capture efficiency and catalysis efficiency. Meanwhile, owing to the encapsulation of these nanocompartments inside the hollow micron-sized spheres, these CO2 sorbents and catalysts can be packed directly in fixed-bed reactors, which is unattainable for nanoparticle materials.
Similar content being viewed by others


Introduction
Engineering micron-sized multi-compartmentalized systems has attracted tremendous interest from diverse fields such as catalysis1,2, microreactors3,4, and microencapsulation5, not only due to their ability to spatially control local physical and chemical processes occurring in their interiors but also due to their merits in practical applications. More challenging is the synthesis of artificial cells that possess some properties of natural cells, such as spatial compartmentalization and selective permeability. In the past two decades, polymersomes6,7, liposomes8, and multiple emulsions9 have been extensively explored to mimic the structures and functions of natural cells. Despite intriguing results achieved from these compartmentalized structures, they are soft matter in nature and inevitably suffer from poor mechanical strength and chemical robustness, thereby severely hampering their practical applications.
These drawbacks associated with these soft materials may be overcome if multi-compartmentalized inorganic microspheres can be fabricated because their frameworks consist of covalent bonds instead of molecular aggregation. In recent years, mesoporous silica particles with core-shell and yolk-shell structures have been fabricated. However, their particle sizes are limited to the nanometer to submicron range1,4. Many methods exist for the fabrication of hollow micron-sized spheres, including hard templating10,11, spray drying12,13, droplet microfluidics14,15, and multiple emulsions16,17,18,19,20,21,22,23,24. Unfortunately, hard templating and spray drying are barely able to produce inorganic microspheres with complex interior structures. The surfactant-based multiple emulsion (oil-in-water-in-oil) method seems promising to construct cell-like structures, but this method also has intrinsic limitations: (i) two surfactants with a special formulation are required to stabilize the droplet and inner droplet surfaces, thus causing difficulty in tuning the outer shell and sub-compartment structures19, and (ii) the system is not sufficiently stable to prevent coalescence induced by thermal and extraneous reagents19,25. Owing to the inherent drawbacks of these methods, existing micron-sized spheres are still rudimentary compared to natural cells, especially in terms of the multi-compartmentalization in the interior architecture.
In comparison to the surfactant-stabilized emulsions, Pickering emulsions, namely, nanoparticle-stabilized emulsions, are highly stable because the removal of nanoparticles from oil/water interfaces requires large energy, leading to the “permanent” adsorption of nanoparticles at the droplet surface26. More interestingly, the droplet size can be easily adjusted by simply varying the amount of nanoparticle emulsifier26,27, which is difficult to realize for surfactants. These features have been harnessed to synthesize polymer capsules6,28 and colloidosomes16,17,24,29. However, the obtained microspheres lack well-defined inner compartments and uniform mesopores, thus also being far different from “cell-like” structures. Therefore, developing efficient approaches to fabricate inorganic mesoporous microspheres that have multi-compartments within their interior remains an important yet unfilled challenge.
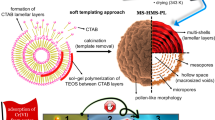
Herein, we report a novel protocol to realize this goal for the first time, exemplified by the fabrication of multi-compartmentalized mesoporous silica microspheres (MSMs). As shown in Fig. 1, our protocol combines Pickering emulsions with molecular surfactant assembly inside the droplets. This protocol is expected to break through the limitations of using conventional surfactant-based emulsions or Pickering emulsions alone. Each nanoparticle-stabilized water droplet serves as a geometrical template for growing an outer shell through an interfacial sol–gel process. The surfactants located in the water droplets are utilized to construct interior compartments through a surfactant assembly-directed sol–gel process. Such a protocol is shown to enable tuning of the sol–gel process occurring at the oil/water interface and within the droplet-confined spaces independently, yielding unprecedented MSMs ranging from hollow MSMs to hollow nanosphere-containing mesoporous silica microspheres (hn@MSMs), nanosphere-containing mesoporous silica microspheres (n@MSMs), yolk-shell-structured mesoporous silica microspheres (y@MSMs), and “solid” mesoporous silica microspheres (“solid” MSMs). Interestingly, the obtained microspheres exhibit significantly enhanced performances in CO2 capture and enzymatic catalysis due to the large void spaces and spatially separated nanocompartments within the microsphere.

Schematic illustration of the preparation of mesoporous silica microspheres with different interior architectures
Materials and methods
Synthesis of multi-compartmentalized MSMs under basic conditions
Typically, 0.1 g of SiO2 emulsifier was dispersed into 50 mL of toluene, followed by the addition of a solution of cetyltrimethylammonium chloride (CTAC, 0.25 g) and aqueous ammonia (0.5 g, 25–28 wt%) in deionized water (1.75 g). Subsequently, the mixture was homogenized at 20,000 rpm for 5 min, yielding a Pickering emulsion. Then, a given amount of tetramethoxysilane (TMOS, 0.375, 0.75, 1.5, or 3.0 g) was added to this Pickering emulsion system. After rotating at 40 °C for 24 h, the resulting particles were isolated through centrifugation, washed with ethanol and deionized water, and dried in air for 24 h. Finally, the resultant solid powder was calcined in air at 550 °C for 5 h (ramping rate, 2 °C min−1). Other structured MSMs were synthesized by varying the amounts of CTAC, ammonia, and SiO2 emulsifier.
Synthesis of multi-compartmentalized MSMs under acidic conditions
Typically, 0.12 g of SiO2 emulsifier was dispersed into 60 mL of toluene, followed by the addition of a mixture of an aqueous HCl (2.4 g, 1.5 M), F127 (EO106PO70EO106, polyethylene oxide–polypropylene oxide–polyethylene oxide, 0.15 g) and deionized water (0.6 g). Subsequently, the resultant mixture was homogenized at 5000 rpm for 15 min, leading to a Pickering emulsion. Then, 1.8 g of tetraethoxysilane (TEOS) was added to this Pickering emulsion system. After rotating at 40 °C for 24 h, this mixture was transferred into an autoclave and kept at 80 °C. After 24 h, the resultant precipitate was filtered, washed thoroughly with deionized water and ethanol, and then dried at 80 °C. Finally, the obtained solid powder was calcined in air at 550 °C for 5 h (ramping rate, 2 °C min−1).
CO2 adsorption/desorption in fixed-bed reactors
Typically, 0.5 g of adsorbent was packed into a quartz fixed-bed reactor. First, the sample was completely purged with N2 at 25 mL min−1 for 60 min at 100 °C. After the temperature was cooled to 75 °C, the reactor was switched to the test gas (15% CO2 in N2, v/v) at 25 mL min−1 for CO2 adsorption. After 60 min, the temperature was increased to 100 °C under N2 (25 mL min−1) and then kept for 60 min for desorption. The CO2 concentration at the outlet of the reactor was measured with an online gas analyzer.
Enantioselective trans-esterification of alcohols
A column with a sand filter and a valve at the bottom was used as a reactor (its interior diameter was 1.34 cm, and the pore diameter of the sand filter was 4.5–9 µm), and 0.8 g of CALB/hn@MSMs or 2.4 g of CALB/SBA-15 was packed in the column. A solution of 1-phenyl ethanol (0.4 M) and vinyl acetate (1.6 M) in n-octane was pumped into the column reactor and allowed to pass through the column at 45 °C. The outflow from the column bottom was sampled for gas chromatography (GC) analysis at intervals. The product was further confirmed with gas chromatography-mass spectrometer (GC-MS).
Results and discussion
Synthesis of multi-compartmentalized MSMs
Siliceous materials are targeted here because they are widely used in diverse fields, and their synthesis represents a typical sol–gel chemistry. Partially hydrophobic silica nanospheres with diameters of 40–60 nm were used as Pickering emulsifiers [transmission electron microscopy (TEM) images, N2 sorption isotherms, and energy-dispersive X-ray (EDX) spectra are displayed in Supplementary Figure S1]26. The fabrication began with the formulation of a water-in-oil Pickering emulsion by homogenizing a mixture of water, aqueous ammonia, and toluene (as the oil phase) in the presence of silica emulsifier and CTAC. The bulk water phase was thereby compartmentalized into numerous water droplets that were dispersed in toluene. When TMOS, a silica precursor, was introduced into this Pickering emulsion, interfacial sol–gel growth around the water droplets and surfactant assembly-directed sol–gel process within the droplet-confined space occurred simultaneously, forming the desired microspheres (Fig. 1). After removing the surfactants, the mesopores in the outer shell and in the inner compartments were evacuated. This protocol is fairly simple and scalable since the whole process mainly involves a one-step synthesis.
Characterization of multi-compartmentalized MSMs exemplified by hn@MSMs
The obtained materials were first checked with scanning electron microscopy (SEM). For example, as shown in Fig. 2a, b, the hn@MSM sample consists of discrete and regular microspheres with a mean diameter of 25 μm. Interestingly, many hollow nanospheres ca. 1.3 μm in size (Fig. 2c, d) are present within the cavities of the microspheres, as seen from a deliberately broken sample. In the magnified SEM image (Fig. 2e), one can find that the microsphere has an outer shell ca. 120–130 nm thick. Interestingly, the SEM image of the surface of hn@MSMs and the TEM image of a fragment from the outer shell in Supplementary Figure S2 show that the initially added emulsifier nanoparticles are randomly distributed over the external surface of the shell, indicating that the shell forms due to condensation of the siliceous precursor at the inner surfaces of the water droplets. On the shell, there exist abundant, relatively uniform pores 8.5 nm in size (Fig. 2f).

a SEM image. b High-magnification SEM image showing a single particle. c SEM image showing the interior structure of a microsphere. d SEM image showing the interior hollow nanospheres. e SEM image showing the outer shell. f TEM image showing the pores on the outer shell. g TEM image showing the interior nanospheres. h TEM image showing mesopores on the shell of the internal hollow nanospheres. i Small-angle XRD pattern. j Nitrogen adsorption–desorption isotherm. k BJH pore size distribution
The TEM images in Fig. 2g, h further reveal the hollow internal structure of the nanospheres. On the shell of the internal nanospheres, radially aligned mesopores are observed. The mesoporous structure of the obtained microspheres was confirmed by small-angle XRD and N2 sorption measurements (Fig. 2i, j), which show a diffraction peak at 0.88° and a type-IV isotherm. The specific surface area and pore volume were measured to be 760 m2 g−1 and 1.52 cm3 g−1, respectively. The average pore size is as high as 9 nm (Fig. 2k), much higher than that of common MCM-4130, due to the micelle swelling caused by toluene during self-assembly. Notably, the bulk density of hn@MSMs was determined to be as low as 0.1 g cm−1, which agrees with the presence of large void spaces within the microspheres.
Tuning the interior structure and particle size of multi-compartmentalized MSMs
As expected, the interior architecture of MSMs and the particle sizes could be tailored by simply changing the synthesis conditions, such as the amounts of CTAC, aqueous ammonia and TMOS, and the droplet size.
When the amount of CTAC was varied from 3.75 to 7.5 wt% while the amounts of aqueous ammonia and TMOS were fixed at 5 wt% (with respect to the water phase) and 0.75 g, respectively, n@MSMs were obtained. The thickness of the outer shell increased from 120 to 160 nm (Fig. 3a) because more CTAC molecules adsorbed at the interface benefit the interfacial sol–gel process. Meanwhile, the diameter of the interior nanospheres increased from 460 to 560 nm (TEM images in Supplementary Figure S3). Interestingly, when the amount of CTAC increased to 10 wt%, hn@MSMs were obtained, the thickness of the outer shell increased to 190 nm, and the inner compartments evolved to hollow mesoporous nanospheres with a diameter of 740 nm due to the formation of multiple emulsions (see “Discussion of mechanism” section). When the amount of CTAC was further increased to 15 wt%, the thickness of the outer shell began to decrease (down to ca. 110 nm), and the interior hollow mesoporous nanospheres changed to irregular plate-like particles (Fig. 3a). Despite the change in the CTAC amount, all these samples not only present a multi-compartmentalized spherical structure but also exhibit a mesoporous structure because a type-IV isotherm of N2 sorption and an XRD diffraction peak of approximately 0.7–1.0° are present (Supplementary Figure S4).

a Changing the amount of CTAC. b Changing the amounts of TMOS and aqueous ammonia
The aqueous ammonia amount also significantly impacts the interior architecture of MSMs (the amounts of CTAC and TMOS were fixed at 10 wt% and 0.75 g, respectively). When using 0.5 wt% aqueous ammonia, “solid” MSMs (without multi-compartmentalized structures) were obtained (Fig. 3b and Supplementary Figure S5). When using 2.5 wt% aqueous ammonia, n@MSMs were obtained, and radially aligned mesopores were present on their inner nanospheres (TEM image in Fig. 3b). Increasing the amount of aqueous ammonia from 5 to 10 wt% led to hn@MSMs (TEM image in Fig. 3b).
Another key factor for the structural evolution of MSMs is the amount of TMOS (the amounts of CTAC and aqueous ammonia were fixed at 10 and 5 wt%, respectively). As the amount of TMOS increased from 0.375 to 3.0 g, the interior of the hollow microspheres became gradually filled with sub-compartments, and their outer shell thickness increased considerably from 127 to 370 nm. Interestingly, despite the TMOS amount, these inner compartments are always hollow nanospheres with sizes ranging from 800 to 1000 nm (Fig. 3b and Supplementary Figure S6).
The silica microspheres inherit not only the morphology of the water droplets but also the droplet size. This makes it possible to facilely control their size. According to the established knowledge of Pickering emulsions, the droplet size can be tuned by varying the concentration of the nanoparticle emulsifier. As shown in Supplementary Figure S7, microspheres with a mean size of 97 and 140 μm were successfully fabricated using 2 and 1 wt% silica emulsifier. Such ability to easily adjust the size of the microspheres is relatively difficult to realize for commonly used surfactant-based emulsions19,20.
Discussion of mechanism
Based on the above results, we propose a mechanism for the formation of multi-compartmentalized MSMs (Fig. 1). SiO2 particles, as emulsifiers, cover the surface of the water droplets, thus guaranteeing high stability against their coalescence. Such droplet surfaces serve as a spherical template for the growth of silicate shells through an interfacial sol–gel process because the presence of an oil/water interface is favorable for heterogeneous nucleation31,32,33. This is supported by control experiments in which the absence of SiO2 nanoparticles or an oil phase led to irregular particles (Supplementary Figure S8). The template effects of droplets are further verified by the above observation that the resultant microspheres are essentially equal to the droplets in size. This interfacial growth occurs mainly at the inner surface of the water droplets, as evidenced by the SEM images (Fig. 2e). During this course, surfactants also play important roles in the formation of spherical morphology and mesopores. Without the addition of CTAC, the resultant materials not only have cracks on the external surface but also lack mesopores on the shell (Supplementary Figure S9).
TMOS in toluene gradually diffuses to the oil/water interface and is hydrolyzed to silicates when contacting water molecules. The silicates cross the interface with the aid of CTAC. In addition to a portion of the silicates being used for shell growth, others further enter the water droplets. Then, they co-assemble with CTAC through electrostatic interactions S+I−34, forming nanomicelle–silicate composites. At low base concentrations, hydrolysis of TMOS is incomplete, and the resultant nanomicelle–silicate composites bear fewer charges. At the same time, due to the slow hydrolysis, only a small amount of the composite is available for nucleation. These factors work together, leading to a monolithic structure within the water droplets. When the base concentration increases, more nanomicelle–silicate composites are available for nucleation and growth. As a result, numerous nanospheres are formed within the water droplets. These only occur in the cases of low concentrations of CTAC (less than 10 wt%) or low concentrations of base (less than 5 wt%). When the CTAC concentration is above 10 wt%, excess CTAC can stabilize smaller toluene droplets within the water droplets, forming oil-in-water-in-oil multiple emulsions. This was confirmed by a fluorescent experiment (Supplementary Figure S10). Due to the presence of toluene droplets within the water droplets, the nucleation and growth of nanomicelle–silicate composites occur around the toluene droplets through the aforementioned heterogeneous nucleation31,32,33, yielding hollow nanospheres. It should be noted that due to the high Laplace pressure20, the inner toluene droplets are unstable, coalesce over time, and eventually escape from the interior of the water droplets. At low concentrations of the base, the nucleation and growth are very slow. Before shell formation, toluene escapes from the interior of the water droplets. This scenario also leads to nanospheres instead of hollow nanospheres within the water droplets.
The diversity of the internal structure is attributed to our unique synthesis protocol, in which the particle emulsifier and the molecular surfactant differ in nature, avoiding their mutual interference. As a consequence, the assembly of particles on the surface of the water droplets and the assembly of surfactant molecules within the droplets can thereby be tuned independently. Such judicious combination enables the simultaneous control of the inner compartments and outer shells, which is unachievable for the existing synthesis systems.
Extension of the synthesis method
The above mechanism implies that our Pickering emulsion-based protocol is flexible and extendable. To confirm this, a nonionic surfactant Pluronic F127 was used to direct the interior structures, which co-assemble with silicates through (S0H+)(X−I+) interactions under acidic conditions34. The synthesis process is similar to that of the SiO2 emulsifier and CTAC except for the use of acid as a catalyst. Despite the TEOS amount, the obtained microspheres present a regular spherical morphology and are the same size (Fig. 4a–c), again indicating that the morphology and size of the microspheres are governed by the parent droplets. Interestingly, depending on the TEOS amount, their interior structures differ remarkably, even when other synthesis conditions were unchanged. When using 0.9–3.6 g TEOS, various hollow MSMs were obtained. When using 0.9 g TEOS, the obtained MSMs were nearly completely hollow in the interior (hollow MSMs). When increasing the TEOS amount to 1.35 and further to 1.8 and 2.25 g, sub-compartments appeared within the hollow microspheres (Fig. 4d and its insets, n@MSMs), and the number of inner compartments increased with the TEOS amount. Abundant mesopores are present on the inner compartments of n@MSMs (Fig. 4g), and their mesoporous structure is further confirmed by N2 sorption measurements (Fig. 4h). Interestingly, as the TEOS amount increased to 3.6 g, a single large sphere was observed inside the hollow microspheres (Fig. 4e, y@MSMs), and wormlike pores were present in the inner spheres (Supplementary Figure S11a). Further increasing the TEOS amount to 5.4 g leads to “solid” MSMs with disordered mesopores (Fig. 4f and Supplementary Figure S11b). The textural parameters, such as the specific surface area, pore size, and pore volume, also change considerably with the TEOS amount (Supplementary Table S1, and Supplementary Figure S11c and d). Such a remarkable variation confirms that the interior structure can be tuned by altering the sol–gel conditions. These results further highlight the excellent ability of our synthesis protocol to tune the interior structures of microspheres.

a–c SEM images. d–f and insets of d SEM images showing the interior compartments. g TEM images showing the mesopores of the inner compartments within n@MSMs synthesized with 1.8 g TEOS. h Nitrogen adsorption–desorption isotherm of n@MSMs synthesized with 1.8 g TEOS
Stability and permeability
The mechanical stability of microspheres is crucial to their practical applications. To examine the stability against mechanical compression, hn@MSMs were packed in a fixed-bed reactor and then subjected to treatment with a stream of liquid (n-octane) at 60 mL h−1 under 1 MPa. It was found that their morphology and structure were well preserved after 10 h of treatment, and no significant structural collapse occurred, as evidenced from the SEM images (Supplementary Figure S12a–c). Their mechanical stability was also examined with flowing gas (N2) at 25 mL min−1 under 2 MPa. After 4 h of treatment, their morphology and structure remained the same as the fresh materials (Supplementary Figure S12d–f). These results confirm that these microspheres possess a mechanical strength high enough to sustain the flow of both liquid and gas under relatively high pressures, which is unattainable for the existing artificial cells. This is an implication that they are amenable to practical catalysis and gas adsorption.
The permeability that is a feature of natural cells is of paramount importance for artificial analogous materials. The Nile red-labeled enzyme (lipase CALB, molecules sizes: 3.0 × 4.0 × 5.0 nm3) was used as a fluorescent probe to examine the permeability of microspheres. To increase the affinity to the probe molecules, hn@MSMs as a representative sample were modified with hydrophobic n-octyltrimethoxysilane prior to the examination. The time-dependent fluorescent images for the permeation process were recorded. As displayed in Fig. 5a, once hn@MSMs were brought into contact with a solution of the fluorescent probe, the fluorescent molecules passed through the outer shell within 2.4 s, as evidenced by the presence of an annular fluorescent pattern. After 10 min, the concave-shaped fluorescent pattern gradually changed to a bulge-shaped pattern, implying that fluorescent molecules reached the center of the microspheres and that the adsorption was complete. During this process, the fluorescence intensity in the center was observed to gradually increase with time and level off (Fig. 5b). These findings provide sound evidence that hn@MSMs are highly permeable toward external molecules.

a 3D confocal fluorescence microscopy images showing the transport of Rhodamine B-labeled lipase CALB into hn@MSMs as a function of time. b Fluorescence intensity in the center of hn@MSMs as a function of time
Applications-I: CO2 adsorption
In view of the high mechanical stability and good permeability of the microspheres, we set out to examine their application in CO2 capture. For example, after impregnating hn@MSMs with tetraethylenepentamine (TEPA, 78.9 wt% with respect to hn@MSMs), the solid sorbent TEPA/hn@MSMs were obtained. EDS elemental mapping revealed that TEPA molecules are homogeneously distributed throughout the microspheres (Fig. 6a). We first employed thermogravimetric analysis (TGA) to measure the CO2 adsorption kinetics of TEPA/hn@MSMs and their benchmark solid sorbent TEPA/SBA-15 (78.9 wt% TEPA was loaded onto SBA-15 microspheres with an average size of 7.0 μm, as shown in Supplementary Figure S13; TEPA molecules are also homogeneously distributed within the interior of the SBA-15 particles, which is confirmed by the EDS elemental mapping in Supplementary Figure S14). The adsorption tests were conducted using simulated flue gas (15% CO2 in N2, v/v) at 75 °C. As shown in Fig. 6b, for TEPA/hn@MSMs, a sharp weight gain was observed within the first 15 min due to CO2 adsorption, and at this point, the adsorption capacity corresponds to 4.2 mmol g−1 and then slowly increases to 5.1 mmol g−1 within the subsequent 120 min. Further prolonging the adsorption time to 180 min does not lead to a further remarkable increase in the adsorption capacity (leveling off at 5.3 mmol g−1). This adsorption capacity can be considered as the upper limit. In contrast, the CO2 adsorption capacity of TEPA/SBA-15 is as low as 0.52 mmol g−1 within the first 15 min and reaches only 2.0 mmol g−1 after 180 min. Such a striking comparison highlights the merits of hn@MSMs. The enhanced adsorption rate and capacity are attributed to the unique structures of hn@MSMs. The spatially separated nanocompartments within hn@MSMs are helpful to disperse the TEPA molecules and thus allow the TEPA molecules to be more accessible to CO2, leading to the high adsorption capacity. The large void spaces among the nanocompartments within the hn@MSMs facilitate CO2 diffusion, resulting in a high adsorption rate.

a Elemental mapping (from SEM) of a single particle of TEPA/hn@MSMs. b CO2 adsorption kinetics over TEPA/hn@MSMs and TEPA/SBA-15 determined by TG. c Consecutive adsorption in a fixed-bed reactor. CO2 adsorption conditions: sorbent activation or desorption at 100 °C under 1 atm N2 at 25 mL min−1 for 120 min (with TG) or 60 min (in a fixed-bed reactor); CO2 capture at 75 °C under 1 atm 15% CO2 (in N2, v/v) at 25 mL min−1 for 180 min (with TG) or 60 min (in a fixed-bed reactor)
To further assess the potential practical applications of TEPA/hn@MSMs, repeated cycles of CO2 capture and desorption were carried out in a fixed-bed reactor. As micron-sized particles, TEPA/hn@MSMs can be packed directly in the fixed-bed column reactor without the addition of any binder (Fig. 6c). The capture process was implemented at 75 °C, and the CO2 content in the outflow was monitored by a gas analyzer. The CO2 adsorption capacity of TEPA/hn@MSMs was as high as 5.6 mmol g−1, which was slightly greater than the value determined by TG. After adsorption saturation, the simulated flue gas was switched to N2, and the reactor was heated to 100 °C to release CO2. In the second cycle, the adsorption capacity was still as high as 5.59 mmol g−1. Even after 20 adsorption–desorption cycles, the CO2 adsorption capacity could still reach 5.0 mmol g−1, which is only slightly lower than the value of the fresh sorbent. The decrease in the CO2 adsorption capacity is mainly attributed to the decrease in the TEPA content in TEPA/hn@MSMs during recycling because 10.6 wt% TEPA was lost after 20 cycles, according to the TG measurement results (Supplementary Figure S15). Such an adsorption capacity is even higher than that obtained by analogous nanometer-sized silica-based sorbents35 and is comparable to that of sorbents derived from silica nanocapsules or mesoporous silica foam under similar conditions36,37. Notably, because the particles can be as large as dozens of microns in size, our TEPA/hn@MSM sorbent does not need any binder to conglutinate fine particles and does not require the addition of sieved sands or glass wools for packing the fixed beds, which is often necessary for nanoparticle adsorbents. This is also beneficial for practical applications.
Applications-II: enzymatic reaction
We next tested the application of hn@MSMs in enzymatic reactions, where highly porous materials are desired to be used as a support to immobilize enzymes. The above lipase CALB (without labeling) was selected. Following the above procedure, CALB was immobilized onto hn@MSMs, leading to catalyst CALB/hn@MSMs (Fig. 7a). The loading of CALB was determined to be as high as 54.4 mg g−1. The EDS elemental mapping of CALB/hn@MSMs reveals that CALB molecules are homogeneously distributed throughout the microspheres (Fig. 7b). N2 sorption experiments confirmed that CALB molecules enter the pores of hn@MSMs because their pore size decreased from 8.8 (octyl-modified hn@MSMs) to 8.0 nm (Supplementary Figure S16). To further confirm the CALB location inside the pores of hn@MSMs, we labeled CALB with Au clusters for TEM observation (Supplementary Figure S17a)38. The Au-CALB composites were found to be uniformly present inside the pores of hn@MSMs, indicating that CALB molecules are uniformly distributed inside the pores of hn@MSMs. Enantioselective trans-esterification of alcohols, where one of the alcohol enantiomers is selectively converted to a chiral ester while leaving the other enantiomer unreacted, was chosen to evaluate the performances of CALB/hn@MSMs. This enzymatic reaction is of practical importance for obtaining both chiral alcohols and chiral esters but often suffers from low conversions caused by product inhibition effects39,40. The continuous-flow reaction is desired because the continuous flow probably mitigates the unwanted product inhibition effect27,41.

a Schematic illustration of enzymatic reactions within a CALB/hn@MSM particle. b Elemental mapping of a single particle of CALB/hn@MSMs. c The ee values of 1-phenylethyl alcohol with time over CALB/hn@MSMs and over CALB/SBA-15 in continuous-flow reactions. Reaction conditions: 0.2 g of CALB/hn@MSMs (the CALB loading is 54.4 mg g−1) or 0.6 g of CALB/SBA-15 (the CALB loading is 18.1 mg g−1), 45 °C, 6 mL h−1, and the column is 0.8 cm in diameter. d Specific activities of CALB/hn@MSMs and CALB/SBA-15 calculated on the basis of the ee values at steady state. e The ee values with time for the enantioselective trans-esterification of 1-phenylethyl alcohol. f The ee values with time for the enantioselective trans-esterification of 4-phenyl-2-butanol. g The ee values with time for the enantioselective trans-esterification of 1-indanol. Reaction conditions for e–g: 0.8 g of CALB/hn@MSMs (the CALB loading is 54.4 mg g−1), 45 °C, 8–10 mL h−1, 0.4 M alcohol in n-octane, 1.6 M vinyl acetate in n-octane, and 1.34 cm column diameter
After packing the microspheres of the CALB/hn@MSM catalyst in a column reactor, a solution of racemic 1-phenylethyl alcohol and vinyl acetate (acylation reagent) in n-octane was continuously fed into this column reactor at 6.0 mL h−1, and the product-containing stream was collected from the outlet of the reactor and analyzed by GC at different times. The determined enantiomeric excesses (ee values) of alcohol are plotted as a function of time (Fig. 7c). The ee value of the generated alcohol rapidly rose up to 95%. According to the ee values of alcohol at a steady state, the specific activity of CALB/hn@MSMs is calculated to be 1.67 U mg−1 (Fig. 7d). For comparison, CALB was also loaded onto the aforementioned SBA-15 microspheres in the same way as CALB/hn@MSMs, yielding a benchmark catalyst CALB/SBA-15 (as shown in Supplementary Figure S17c, the experiment of labeling CALB with Au clusters reveals that CALB molecules are homogeneously located inside the pores of SBA-15). Under identical conditions, CALB/SBA-15 gave ee values of only 30% for alcohol at a steady state (Fig. 7c). Its specific activity is estimated to be 0.78 U mg−1 (Fig. 7d), which is less than half that of CALB/hn@MSMs. The significantly boosted catalytic efficiency should also be attributed to the high dispersion of enzymes in the spatially separated nanocompartments, which benefits molecule transport within the interior of the microspheres. Moreover, CALB/hn@MSMs exhibited excellent operational stability. Over a period of 360 h, the ee values of alcohol and ester were always maintained above 98% at a flow velocity of 8–10 mL h−1 (Fig. 7e), and the specific activity had no significant loss. The morphology and structure of most CALB/hn@MSMs were largely preserved (Supplementary Figure S18). Enantioselective trans-esterification of other substrates, including 4-phenyl-2-butanol and 1-indanol, with high activity and long-term stability, further highlights their potential for practical applications (Fig. 7f, g).
Conclusions
We successfully developed a novel protocol to unprecedentedly fabricate micron-sized multi-compartmentalized mesoporous silica spheres based on a Pickering emulsion strategy. Due to its sufficiently high stability, Pickering droplets can not only create oil/water interfaces for growing mesoporous shells but also provide confined spaces to optionally construct multi-compartmentalized interior structures through surfactant assembly. Such a protocol was proven to be flexible and extendable by varying the surfactants and other synthesis conditions. It was demonstrated that a family of novel microspheres, such as hollow MSMs, hn@MSMs, n@MSMs, y@MSMs, and “solid” MSMs, were successfully fabricated, which constitute important supplements to the existing artificial cells. Interestingly, these microspheres exhibit excellent permeability to extraneous molecules due to their porous shell and large interior void space. Such structural feature accounts for their superiority in CO2 capture and enzymatic reactions. Due to the high dispersion of TEPA and enzyme in the spatially separated nanocompartments, the resultant CO2 sorbents and catalysts exhibited significantly enhanced CO2 capture efficiency and catalysis efficiency. Meanwhile, owing to the encapsulation of these nanocompartments inside the hollow micron-sized spheres, these CO2 sorbents and catalysts can be packed directly in fixed-bed reactors, which is unattainable for nanoparticle materials. Moreover, they exhibit long-term operational stability owing to their relatively high mechanical strength. Our method and the underlying novel synthesis mechanism provide a new platform for engineering large-sized inorganic spheres with cell-like structures for innovative practical applications.
References
Prieto, G. et al. Hollow nano- and microstructures as catalysts. Chem. Rev. 116, 14056–14119 (2016).
Sun, M. H. et al. Applications of hierarchically structured porous materials from energy storage and conversion, catalysis, photocatalysis, adsorption, separation, and sensing to biomedicine. Chem. Soc. Rev. 45, 3479–3563 (2016).
Schoonen, L. & van Hest, J. C. M. Compartmentalization approaches in soft matter science: from nanoreactor development to organelle mimics. Adv. Mater. 28, 1109–1128 (2016).
Wang, X. J., Feng, J., Bai, Y. C., Zhang, Q. & Yin, Y. D. Synthesis, properties, and applications of hollow micro-/nanostructures. Chem. Rev. 116, 10983–11060 (2016).
Trantidou, T. et al. Engineering compartmentalized biomimetic micro- and nanocontainers. ACS Nano 11, 6549–6565 (2017).
Qian, Q. P., Huang, X. P., Zhang, X. Y., Xie, Z. G. & Wang, Y. P. One-step preparation of macroporous polymer particles with multiple interconnected chambers: a candidate for trapping biomacromolecules. Angew. Chem. Int. Ed. 52, 10625–10629 (2013).
Marguet, M., Bonduelle, C. & Lecommandoux, S. Multicompartmentalized polymeric systems: towards biomimetic cellular structure and function. Chem. Soc. Rev. 42, 512–529 (2013).
Weiss, M. et al. Sequential bottom-up assembly of mechanically stabilized synthetic cells by microfluidics. Nat. Mater. 17, 89–96 (2018).
Villar, G., Heron, A. J. & Bayley, H. Formation of droplet networks that function in aqueous environments. Nat. Nanotechnol. 6, 803–808 (2011).
Shen, L. et al. Formation of nickel cobalt sulfide ball-in-ball hollow spheres with enhanced electrochemical pseudocapacitive properties. Nat. Commun. 6, 7694 (2015).
Yang, T. Y. et al. A synthetic strategy for carbon nanospheres impregnated with highly monodispersed metal nanoparticles. NPG Asia Mater. 8, e240 (2016).
Carne-Sanchez, A., Imaz, I., Cano-Sarabia, M. & Maspoch, D. A spray-drying strategy for synthesis of nanoscale metal-organic frameworks and their assembly into hollow superstructures. Nat. Chem. 5, 203–211 (2013).
Waldron, K., Wu, Z. X., Zhao, D. Y., Chen, X. D. & Selomulya, C. On the improvement of pore accessibility through post-synthesis hydrothermal treatments of spray dried SBA-15 microspheres. Chem. Eng. Sci. 127, 276–284 (2015).
Wang, B. et al. Macroporous materials: microfluidic fabrication, functionalization and applications. Chem. Soc. Rev. 46, 855–914 (2017).
Scholz, S. & Lercher, J. A. Hierarchically structured millimeter-sized (organo) silica spheres with a macroporous shell and a meso/microporous core. Chem. Mater. 23, 2091–2099 (2011).
Bollhorst, T., Rezwan, K. & Maas, M. Colloidal capsules: nano- and microcapsules with colloidal particle shells. Chem. Soc. Rev. 46, 2091–2126 (2017).
Li, M., Harbron, R. L., Weaver, J. V., Binks, B. P. & Mann, S. Electrostatically gated membrane permeability in inorganic protocells. Nat. Chem. 5, 529–536 (2013).
Nollet, M., Depardieu, M., Destribats, M., Backov, R. & Schmitt, V. Thermo-responsive multi-cargo core shell particles. Part. Part. Syst. Charact. 30, 62–66 (2013).
Oh, C. et al. Distribution of macropores in silica particles prepared by using multiple emulsions. J. Colloid Interface Sci. 254, 79–86 (2002).
Wu, S. H., Hung, Y. & Mou, C. Y. Compartmentalized hollow silica nanospheres templated from nanoemulsions. Chem. Mater. 25, 352–364 (2013).
Du, G. et al. One-step synthesis of hydrophobic multicompartment organosilica microspheres with highly interconnected macro-mesopores for the stabilization of liquid marbles with excellent catalysis. Langmuir 33, 5223–5235 (2017).
Liu, J. et al. A facile vesicle template route to multi-shelled mesoporous silica hollow nanospheres. J. Mater. Chem. 20, 4595 (2010).
Kim, T. S. et al. Carbon-decorated iron oxide hollow granules formed using a silk fibrous template: lithium-oxygen battery and wastewater treatment applications. NPG Asia Mater. 9, e450 (2017).
Guan, B. Y., Yu, L. & Lou, X. W. Chemically assisted formation of monolayer colloidosomes on functional particles. Adv. Mater. 28, 9596–9601 (2016).
Fornasieri, G. et al. Mesoporous and homothetic silica capsules in reverse-emulsion microreactors. Adv. Mater. 16, 1094–1097 (2004).
Zhang, M. et al. Compartmentalized droplets for continuous flow liquid–liquid interface catalysis. J. Am. Chem. Soc. 138, 10173–10183 (2016).
Zhang, M. et al. Ionic liquid droplet microreactor for catalysis reactions not at equilibrium. J. Am. Chem. Soc. 139, 17387–17396 (2017).
Yang, Y., Ning, Y., Wang, C. & Tong, Z. Capsule clusters fabricated by polymerization based on capsule-in-water-in-oil Pickering emulsions. Polym. Chem. 4, 5407 (2013).
Xu, X. W. et al. One-pot colloidal chemistry route to homogeneous and doped colloidosomes. J. Am. Chem. Soc. 135, 12928–12931 (2013).
Kresge, C. T., Leonowicz, M. E., Roth, W. J., Vartuli, J. C. & Beck, J. S. Ordered mesoporous molecular-sieves synthesized by a liquid-crystal template mechanism. Nature 359, 710–712 (1992).
Baillot, M., Bentaleb, A., Laurichesse, E., Schmitt, V. & Backov, R. Triggering the mechanical release of mineralized Pickering emulsion-based capsules. Langmuir 32, 3880–3889 (2016).
Destribats, M. et al. Tailored silica macrocellular foams: combining limited coalescence-based Pickering emulsion and sol–gel process. Adv. Funct. Mater. 22, 2642–2654 (2012).
Shen, D. et al. Biphase stratification approach to three-dimensional dendritic biodegradable mesoporous silica nanospheres. Nano Lett. 14, 923–932 (2014).
Zhao, D. Y., Huo, Q. S., Feng, J. L., Chmelka, B. F. & Stucky, G. D. Nonionic triblock and star diblock copolymer and oligomeric surfactant syntheses of highly ordered, hydrothermally stable, mesoporous silica structures. J. Am. Chem. Soc. 120, 6024–6036 (1998).
Singh, B. & Polshettiwar, V. Design of CO2 sorbents using functionalized fibrous nanosilica (KCC-1): insights into the effect of the silica morphology (KCC-1 vs. MCM-41). J. Mater. Chem. A 4, 7005–7019 (2016).
Qi, G. G. et al. High efficiency nanocomposite sorbents for CO2 capture based on amine-functionalized mesoporous capsules. Energy Environ. Sci. 4, 444–452 (2011).
Qi, G., Fu, L. & Giannelis, E. P. Sponges with covalently tethered amines for high-efficiency carbon capture. Nat. Commun. 5, 5796 (2014).
Engström, K. et al. Co-immobilization of an enzyme and a metal into the compartments of mesoporous silica for cooperative tandem catalysis: an artificial metalloenzyme. Angew. Chem. Int. Ed. 52, 14006–14010 (2013).
Koeller, K. M. & Wong, C. H. Enzymes for chemical synthesis. Nature 409, 232–240 (2001).
Sandig, B. & Buchmeiser, M. R. Highly productive and enantioselective enzyme catalysis under continuous supported liquid–liquid conditions using a hybrid monolithic bioreactor. ChemSusChem 9, 2917–2921 (2016).
Lach, S., Yoon, S. M. & Grzybowski, B. A. Tactic, reactive, and functional droplets outside of equilibrium. Chem. Soc. Rev. 45, 4766–4796 (2016).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (21733009, 21573136, and U1510105), the Program for New Century Excellent Talents in University (NECT-12-1030), and the Doctoral Education Innovation Project in Shanxi Province (2017–5).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wei, L., Yan, S., Wang, H. et al. Fabrication of multi-compartmentalized mesoporous silica microspheres through a Pickering droplet strategy for enhanced CO2 capture and catalysis. NPG Asia Mater 10, 899–911 (2018). https://doi.org/10.1038/s41427-018-0083-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41427-018-0083-9
This article is cited by
-
Pickering emulsion droplets and solid microspheres acting synergistically for continuous-flow cascade reactions
Nature Catalysis (2024)
-
Multi-compartmental MOF microreactors derived from Pickering double emulsions for chemo-enzymatic cascade catalysis
Nature Communications (2023)
-
Effectual and rapid synthesis of bis(pyrazolyl)methanes and pyrazolopyranopyrimidines catalyzed by nano-[TSPSED][Cl]2 and investigating their biological activity
Research on Chemical Intermediates (2023)
-
Liquid marble-derived solid-liquid hybrid superparticles for CO2 capture
Nature Communications (2019)
-
Recent Advances in CO2 Adsorption from Air: a Review
Current Pollution Reports (2019)
