
Abstract
Gliflozins are known as SGLT2 inhibitors, which are used to treat diabetic patients by inhibiting glucose reabsorption in kidney proximal tubules. Recent studies show that gliflozins may exert other effects independent of SGLT2 pathways. In this study we investigated their effects on skeletal muscle cell viability and paracrine function, which were crucial for promoting revascularization in diabetic hindlimb ischemia (HLI). We showed that treatment with empagliflozin (0.1−40 μM) dose-dependently increased high glucose (25 mM)-impaired viability of skeletal muscle C2C12 cells. Canagliflozin, dapagliflozin, ertugliflozin, ipragliflozin and tofogliflozin exerted similar protective effects on skeletal muscle cells cultured under the hyperglycemic condition. Transcriptomic analysis revealed an enrichment of pathways related to ferroptosis in empagliflozin-treated C2C12 cells. We further demonstrated that empagliflozin and other gliflozins (10 μM) restored GPX4 expression in high glucose-treated C2C12 cells, thereby suppressing ferroptosis and promoting cell viability. Empagliflozin (10 μM) also markedly enhanced the proliferation and migration of blood vessel-forming cells by promoting paracrine function of skeletal muscle C2C12 cells. In diabetic HLI mice, injection of empagliflozin into the gastrocnemius muscle of the left hindlimb (10 mg/kg, every 3 days for 21 days) significantly enhanced revascularization and blood perfusion recovery. Collectively, these results reveal a novel effect of empagliflozin, a clinical hypoglycemic gliflozin drug, in inhibiting ferroptosis and enhancing skeletal muscle cell survival and paracrine function under hyperglycemic condition via restoring the expression of GPX4. This study highlights the potential of intramuscular injection of empagliflozin for treating diabetic HLI.
Similar content being viewed by others


Introduction
Gliflozins are known as sodium–glucose co-transporter-2 (SGLT2) inhibitors. Their oral administration can lower blood glucose levels of patients with type 2 diabetes by blocking glucose reabsorption in kidney proximal tubules [1,2,3,4,5,6,7,8,9]. However, recent reports showed that orally administered gliflozins, such as empagliflozin, canagliflozin, and dapagliflozin, could also exert other functions, including improving cardiac functions and treating acute heart failure in diabetic and non-diabetic patients, in an SGLT2-independent manner [10,11,12,13,14]. This indicates that their functions are underestimated, and they might be potential new drugs for treating other diseases in an SGLT2-independent manner.
Peripheral artery disease (PAD) is strongly associated with increased morbidity and mortality in both non-diabetic and diabetic patients [15, 16]. In 2015, 237 million people were estimated to have PAD, and this trend is still increasing [17]. Lower limb ischemia, the most common manifestation of PAD, is the pathological change in blood vessels that results in insufficient blood supply to the lower extremities [18]. In severe cases, lower limb ischemia can lead to amputation and mortality [19]. As one of the main risk factors for PAD, hyperglycemia leads to impaired angiogenesis and healing processes in chronic wounds, thereby causing worse outcomes in diabetic lower limb ischemia patients [20, 21]. Due to the larger surface of ischemic damage and higher relapse rate, patients with severe lower limb ischemia, including those with diabetes, are not appropriate for standard revascularization strategies, such as by-pass and/or vascular stents. Thus, they are currently condemned as “no-option for therapy”. For approximately two decades, therapeutic angiogenesis, which aims to induce the formation of new and functional blood vessels at ischemic sites, has been regarded as a promising strategy for treating lower limb ischemia, including the severe type seen in diabetic patients [22]. However, as angiogenesis is a complex, multi-step process involving various angiogenic factors and multiple cell types, there is currently no effective small-molecule drug for therapeutic angiogenesis.
Skeletal muscle, as the largest paracrine and endocrine organ, is a prospective target for the treatment of many diseases, including sarcopenia and muscle wasting [23]. Targeting skeletal muscle has also become an attractive potential therapeutic strategy for treating diabetic lower limb ischemia as it can secrete various angiogenic factors. These paracrine signals, in turn, affect the vascular endothelial and smooth muscle cells—the two main types of blood vessel-forming cells that initiate the formation of inner vessel tubes and promote vascular maturation, respectively [16, 24,25,26,27,28]. However, skeletal muscle is severely affected in diabetic patients. Hyperglycemic conditions suppress its viability and secretory functions, thereby impairing its angiogenic functions—another main cause of poor prognosis in patients with diabetic lower limb ischemia [29]. Increasing skeletal muscle cell viability and secretory function are the main hurdles for therapeutic angiogenesis targeting skeletal muscle cells in patients with diabetes [24, 30]. However, the detailed mechanism of hyperglycemic-induced skeletal muscle cell death has not been completely elucidated [31]. Hence, there is an urgent need to elucidate the molecular mechanism underlying hyperglycemia-induced skeletal muscle cell death and to develop small-molecule drugs to increase their viability and paracrine function [26, 28, 30].
In this study, we examined the effect of empagliflozin on skeletal muscle cell viability and cell death under hyperglycemia and performed a systematic analysis using RNA-sequencing to unravel the molecular mechanism. We found that hyperglycemia suppresses skeletal muscle cell viability by inducing ferroptosis, a type of non-apoptotic programmed cell death caused mainly by the disruption of redox homeostasis due to the dysregulation of iron ion metabolism, disruption in the de novo synthesis of glutathione (GSH), and/or aberrant lipid peroxidation reduction process by lipid repair enzymes [32]. We further revealed that hyperglycemia suppresses the expression level of glutathione peroxidase 4 (GPX4), thereby induces skeletal muscle cells ferroptosis; while empagliflozin restores skeletal muscle cell viability and paracrine potential by inhibiting ferroptosis through promoting its accumulation in an SGLT2-independent manner. Furthermore, we explored the possibility of using intramuscularly injected empagliflozin as a new small-molecule drug for potential therapeutic angiogenesis in diabetic hindlimb ischemia (HLI).
Materials and methods
Cell lines and cell culture
C2C12, HUVECs, MOVAS and HEK293T cell lines were all purchased from the American Type Culture Collection (ATCC). Cells were cultured with Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Life Technologies, Grand Island, NY, USA) added with 10% fetal bovine serum (FBS; Biological Industries, Beit Haemek, Israel) in a humidified incubator (37 °C, 5% CO2). Routine detection for mycoplasma was performed by Mycoplasma Detection Kit-QuickTest (Biotool, Houston, TX, USA). Prior to use, C2C12 cells were differentiated into myotubes using DMEM with 2% horse serum (Biological Industries) for five days [33]. For experiments under hyperglycemic condition, cells were cultured in DMEM with 25 mM glucose (final concentration), while for experiments under normoglycemic condition, cells were cultured in DMEM with 7.5 mM glucose (final concentration). Cells were then exposed to hypoxic condition in a hypoxia chamber (Anaeropouch Box, 0.1% O2, Mitsubishi Gas Chemical, Tokyo, Japan) for indicated time. Transfection was performed using Lipofectamine 2000 (Invitrogen Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instruction.
Plasmids and constructs
For constructing shRNA expression vectors against murine GPX4 (NM_001037741.4), shRNA target sites prediction and shRNA expression vectors construction were performed as described previously (target sites: 5′-GCAGGAGCCAGGAAGTAAT-3′ for shGPX4-1; and 5′-GCCAGGAAGTAATCAAGAA-3′ for shGPX4-2) [34, 35]. For control vector (shCon), a vector containing a stretch of seven thymines downstream to the U6 promoter was used.
Animal experiment
Male C57BL/6 mice aged 6 weeks (body weight 15–20 g) were purchased from Chongqing Medical University (Chongqing, China). Animal studies were approved by the Laboratory Animal Welfare and Ethics Committee of Chongqing Medical University, and carried out in the Chongqing Medical University. All animal studies conformed to the approved Guidelines for the Care and Use of Laboratory Animals of Chongqing Medical University. All efforts were made to minimize suffering.
For diabetes induction, C57BL/6 mice were fed with high fat diet (HFD) for 3 weeks (20% kcal protein, 20% kcal carbohydrate, and 60% kcal fat). Intraperitoneal administration of 60 mg/kg body weight streptozotocin (STZ, Sigma-Aldrich, St Louis, MO, USA) diluted in sodium citrate buffer was then performed for the following six days. Mice were fasted overnight prior to each STZ injection and blood glucose level measurement. Blood glucose level was evaluated using Accu-Check Integra (Roche Diagnostics, Shanghai, China). Mice with blood glucose level above 16.6 mM were assumed as diabetic mice, and were used for establishing diabetic HLI model as described previously [28]. Briefly, mice were anesthetized using ketamine/xylazin (80 mg/kg body weight and 50 mg/kg body weight, respectively). Femoral artery of the left hindlimb was excised, while the corresponding right hindlimb was left without surgery and used as control. Mice were then grouped randomly. Empagliflozin dissolved in 10% DMSO (empagliflozin final concentration: 10 mg/kg body weight) or 10% DMSO, as well as shCon or shGPX4, were injected into the gastrocnemius muscle of the left hindlimb of the diabetic HLI mice. Treatments were done every three days for 21 days, starting from 24 h post-surgery. Damage caused by ischemia was evaluated with visual examination and scored at indicated time points as described previously (0 = no difference with control, 1 = mild change in color, 2 = moderate change in color, 3 = severe change in color, necrosis, loss of subcutaneous tissue, and 4 = lower extremity amputation) [28, 36]. Blood perfusion in the hindlimb was visualized and analyzed by a Laser Doppler Imager (Moor Instruments Ltd, Axminster, Devon, UK) at indicated times. Prior to visualization, mice were anesthetized and the fur of the hindlimb area was depilated. Blood perfusion ratio was acquired by calculating the ratio between ischemic hindlimb (left) to corresponding control (right hindlimb) as described previously [26, 28, 37].
Transmission electron microscopy
Skeletal muscle cells treated as described above and tissue samples from the gastrocnemius muscle of the left hindlimb of HLI mice were pre-fixed with a 3% glutaraldehyde. Cells and tissues were then post-fixed in 1% osmium tetroxide, dehydrated in series acetone, infiltrated, and embedded in Epox 812, before being cut into ultra-thin sections. Sections were stained with uranyl acetate and lead citrate prior to being examined with JEM-1400-FLASH Transmission Electron Microscope (JEOL, Tokyo, Japan).
RNA extraction, quantitative reverse transcription PCR (qRT-PCR), and Western blotting
Total RNA was extracted using TRIzol (Invitrogen) according to manufacturer’s instruction. Total RNA (1 μg) was reverse-transcribed into cDNA according to manufacturer’s instruction, and qRT-PCR was performed using SYBR Premix Ex Taq (Takara Bio, Dalian, China). The sequences of the primers used were listed in Supplementary Table S1. Cells were lysed with radioimmunoprecipitation assay (RIPA) lysis buffer with protease inhibitor and phosphatase inhibitor cocktail (complete cocktail, Roche Applied Science, Indianapolis, IN, USA) according to manufacturer’s instruction. Antibodies used in Western blotting are listed in Supplementary Table S2. Protein quantification was analyzed with Quantity One software (Thermo Scientific, Waltham, MA, USA). β-Actin was used as the loading control for qRT-PCR and Western blotting. Data were represented as relative to the control, which was assumed as 1.
Statistical analysis
Statistical analyses were performed using SPSS Statistics (v17.0) and GraphPad Prism (v8.4). Quantitative data were expressed as mean ±SDs from three independent experiments unless further indicated. Statistical analysis of the blood perfusion ratio between time points in vivo was carried out by using repeated-measures ANOVA, while difference between treatment groups was evaluated using one-way ANOVA. Limb morphological assessment was performed by a nonparametric Mann–Whitney test. Two-tailed unpaired Student’s t test was performed when there were only two groups. Other statistical analysis was performed using one-way ANOVA. *P < 0.05 was considered as statistically significant.
Results
Gliflozins promote skeletal muscle cell survival under hyperglycemic conditions
To evaluate whether gliflozins can enhance skeletal muscle cell viability under hyperglycemic conditions, we first examined their effects on C2C12 cell viability. We induced mature C2C12 cells as described previously [33]. Their maturation was confirmed by increased levels of myogenic differentiation 1 (MyoD1) and myogenin (MyoG), the two myogenic regulatory factors, in differentiated skeletal muscles (Supplementary Fig. S1a). We then treated differentiated C2C12 cells with different doses of empagliflozin under hyperglycemic conditions. While hyperglycemia reduced the viability of C2C12 cells, empagliflozin significantly increased their viability in a dose-dependent manner (Fig. 1a). Notably, there was no significant difference between the effects of 10 μM and higher doses of empagliflozin; hence, we used 10 μM empagliflozin in further experiments. We next investigated the effects of other aryl-C-glucosides drugs, canagliflozin, dapagliflozin, ertugliflozin, ipragliflozin, and tofogliflozin [1], on C2C12 cell viability to examine if this class of drugs has similar effects on skeletal muscle cells cultured under hyperglycemic conditions. Like empagliflozin, these drugs enhanced C2C12 cell viability under hyperglycemic conditions (Fig. 1b). Furthermore, gliflozins robustly suppressed hyperglycemia-induced cell death (Fig. 1c), while increasing C2C12 cells proliferation under hyperglycemia (Supplementary Fig. S1b, c). These results show that gliflozins could protect skeletal muscle cells by reducing hyperglycemia-induced cell death, thus increasing their viability.

a Cell viability of C2C12 cells treated with indicated concentration of empagliflozin. b Cell viability of C2C12 cells treated with 10 μM gliflozins. c Cell death rate of C2C12 cells treated with 10 μM gliflozins, as examined using PI staining and flow cytometry. d RNA-sequencing analysis of C2C12 treated with 10 μM empagliflozin. Heat-map represents the result of clustering analysis of differentially expressed genes with fold-change ≥1.5 and P-value < 0.05. e GO analysis of differentially expressed genes with fold-change ≥1.5 and P-value < 0.05. Enriched GO biological processes were identified and listed according to their enrichment scores (-log10 P-value). f KEGG enrichment analysis of differentially expressed genes with fold-change ≥1.5 and P-value < 0.05. C2C12 cells cultured under hyperglycemia were used as control. All experiments were performed under hyperglycemia unless further indicated. Data were presented as mean ± SD (n = 3). NG normoglycemia, HG hyperglycemia, NS not significant, *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
To systematically examine the mechanism by which gliflozins suppress skeletal muscle cell death and induce their viability under hyperglycemic conditions, we performed RNA sequencing and transcriptomic analysis of empagliflozin-treated C2C12 cells. We observed that empagliflozin treatment increased the expression of 3687 genes and decreased that of 5049 genes in C2C12 cells (Fig. 1d). Gene Ontology (GO) analysis showed enrichment in angiogenesis and response to hypoxia, indicating the possibility of using empagliflozin as a therapeutic angiogenesis agent for treating HLI (Fig. 1e). Furthermore, lipid metabolic process and response to oxidative stress were also enriched by empagliflozin treatment. Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis showed enrichment of pathways related to ferroptosis and GSH metabolism (Fig. 1f). Given that ferroptosis is a type of cell death induced by lipid peroxidation and aberrant GSH metabolism, these results indicate that empagliflozin might affect skeletal muscle cell viability by suppressing ferroptosis.
Empagliflozin inhibits ferroptosis in skeletal muscle cells under hyperglycemic conditions
Ferroptosis is a recently identified form of non-apoptotic programmed cell death caused by the disruption of redox homeostasis due to the dysregulation of iron, GSH synthesis, and lost activity of lipid repair enzymes (Fig. 2a). To examine whether hyperglycemia could induce ferroptosis in skeletal muscle cells, we first examined lipid peroxidation in C2C12 cells cultured under hyperglycemic conditions. Indeed, we observed an increase in lipid oxygen reactive species (ROS; Supplementary Fig. S2a), lipid peroxidation (Supplementary Fig. S2b), and 4-hydroxynonenal (4-HNE, a lipid peroxidation-specific aldehyde; Supplementary Fig. S2c) levels in C2C12 cells cultured under hyperglycemic conditions. Hyperglycemia also led to mitochondrial shrinkage (Supplementary Fig. S2d), a typical characteristic of ferroptosis caused by lipid peroxidation-induced disruption of the mitochondrial membrane [38]. Furthermore, treatment with ferrostatin-1, a small-molecule cellular ROS scavenger that can suppress ferroptosis [38], conspicuously restored C2C12 cell viability suppressed by hyperglycemia (Supplementary Fig. S2e). These results clearly demonstrate that hyperglycemia induces ferroptosis in skeletal muscle cells.

a Schematic diagram of molecular mechanism of ferroptosis. b Lipid ROS level in empagliflozin-treated C2C12 cells, as examined using C11-BODIPY staining and flow cytometry. c, d Lipid peroxidation ratio in empagliflozin-treated C2C12 cells, as analyzed using C11-BODIPY staining. Representative images (c; scale bars: 100 μm) and quantification results (d; n = 6) were shown. e, f 4-HNE level in C2C12 cells treated with empagliflozin, as examined using Western blotting. Representative images (e) and quantification results (f) were shown. β-Actin was used as loading control. g Transmission electron microscopy images of the mitochondria in C2C12 cells treated with empagliflozin. Red arrows: mitochondria; scale bars: 200 nm. All experiments were performed under hyperglycemia. Data were presented as mean ± SD (n = 3, unless further indicated). HG hyperglycemia, Empa: 10 μM empagliflozin; **P < 0.01; ****P < 0.0001.
Next, we examined the effect of empagliflozin on hyperglycemia-induced ferroptosis. To this end, we treated mature C2C12 cells with 10 μM empagliflozin. Empagliflozin suppressed the increase in lipid ROS and lipid peroxidation levels under hyperglycemic conditions, thereby reducing the lipid peroxidation ratio (Fig. 2b–d). Furthermore, empagliflozin also suppressed the levels of 4-HNE (Fig. 2e, f) and malondialdehyde (MDA; Supplementary Fig. S2f), and significantly restored the morphology of mitochondria in C2C12 cells cultured under hyperglycemic conditions (Fig. 2g). These results suggest that empagliflozin protects the mitochondria from lipid peroxidase-induced damage and ameliorates ferroptosis.
Gliflozins inhibit hyperglycemia-induced ferroptosis in a GPX4-dependent manner
Maintenance of iron ion homeostasis, de novo synthesis of GSH and reduction of lipid ROS by GSH are the main pathways that maintain ferroptosis homeostasis. To reveal the molecular mechanism of empagliflozin regulation of hyperglycemia-induced ferroptosis, we analyzed the fold change of the differentially expressed genes related to ferroptosis in empagliflozin-treated skeletal muscle cells cultured under hyperglycemia using RNA-seq data, namely ferritin light chain 1 (FTL1), GPX4, glutamate-cysteine 3 ligase, modifier subunit (GCLM), glutathione synthetase (GSS), solute carrier family 3, member 2 (SLC3A2), glutamate-cysteine ligase, catalytic subunit (GCLC), ferritin heavy chain 1 (FTH1), solute carrier family 7, member 11 (SLC7A11), and solute carrier family 40 (iron-regulated transporter), member 1 (SLC40A1). As shown in Fig. 3a, b, the level of GPX4, an enzyme that facilitates GSH to reduce lipid hydroperoxides to lipid alcohols [39], was most significantly affected. Similar results were obtained from the validation of its protein expression in empagliflozin-treated skeletal muscle cells (Fig. 3c, d). It is noteworthy that empagliflozin did not significantly affect the expression of FTH1, which is crucial for iron homeostasis; and SLC7A11, the core subunit of system xc−, a cystine-glutamate antiporter that mediates the exchange of extracellular cystine with intracellular glutamate and crucial for de novo GSH synthesis [39, 40]. Meanwhile, empagliflozin promoted the levels of FTL1 and SLC40A1, which are involved in iron homeostasis; as well as GCLM, GSS, SLC3A2, and GCLC, which are regulators of de novo GSH synthesis; however, the increase in the expression of these factors was significantly lower than that of GPX4. Furthermore, we observed a decrease of GPX4 expression in skeletal muscle cells exposed to hyperglycemia compared to those cultured under normoglycemia (Supplementary Fig. S3a, b), which is in accordance with ArrayExpress: E-GEOD-31553 (https://www.ebi.ac.uk/arrayexpress/experiments/E-GEOD-31553). These results suggest that hyperglycemia might induce skeletal muscle cells ferroptosis by suppressing the level of GPX4, thereby disrupting skeletal muscle cells reduction potential. Together, these results suggest that empagliflozin suppressed hyperglycemia-induced ferroptosis in skeletal muscle cells mainly by regulating the reduction process of lipid peroxidation. We further confirmed these results by treating C2C12 cells with erastin, which inhibits GSH synthesis [38], and (1S, 3R)-Ras-selective lethal small molecule 3 (RSL3), which inhibits lipid ROS reduction [32]. Treatment with both erastin and RSL3 reduced the viability of C2C12 cells in a dose-dependent manner (Supplementary Fig. S3c, d). However, empagliflozin restored the cell viability suppressed by RSL3, but not erastin (Fig. 3e, f). Concomitantly, empagliflozin did not affect the protein level of SLC7A11 (Supplementary Fig. S3e, f), and failed to cancel the suppressive effect of erastin on SLC7A11 expression (Supplementary Fig. S3g, h).

a Heatmap showing ferroptosis-related genes which are differently expressed genes in C2C12 cells cultured under hyperglycemia and treated with 10 μM empagliflozin obtained by RNA-seq. Values are scaled as indicated (1.0 to −1.5). b mRNA expression levels of ferroptosis-related genes in C2C12 cells cultured under hyperglycemia and treated with 10 μM empagliflozin. c, d GPX4 protein expression in C2C12 cells cultured under hyperglycemia and treated with 10 μM empagliflozin, as examined using Western blotting. Representative images (c) and quantification results (d) were shown. Cell viability of C2C12 cells treated with 10 μM empagliflozin and 100 nM RSL3 (e) or 500 nM erastin (f). g, h GPX4 protein expression in C2C12 cells treated with 10 μM empagliflozin and 100 nM RSL3, as examined using Western blotting. Representative images (g) and quantification results (h) were shown. i, j Lipid peroxidation ratio in C2C12 cells treated with 10 μM empagliflozin and 100 nM RSL3, as analyzed using C11-BODIPY staining. Representative images (i; scale bars: 100 μm) and quantification results (j; n = 6) were shown. k Cell death rate of C2C12 cells treated with 10 μM empagliflozin and 100 nM RSL3 as examined using PI staining and flow cytometry. β-Actin was used for qRT-PCR normalization and as Western blotting loading control. All experiments were performed under hyperglycemia. Data were presented as mean ± SD (n = 3, unless further indicated). HG hyperglycemia, NS not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Next, we analyzed the effect of RSL3 treatment on empagliflozin-treated C2C12 cells. Treatment with RSL3 clearly suppressed empagliflozin-induced GPX4 protein expression (Fig. 3g, h) and concomitantly canceled the suppressive effect of empagliflozin on lipid peroxidation in C2C12 cells (Fig. 3i, j). Subsequently, we observed that RSL3 attenuated the inhibitory effect of empagliflozin on C2C12 cell death (Fig. 3k).
To further confirm the role of GPX4 in empagliflozin inhibition of hyperglycemia-induced ferroptosis, we knocked down GPX4 expression in C2C12 cells using two shRNA expression vectors targeting different sites of GPX4 (Supplementary Fig. S4a–c). We used shGPX4-2 for further experiments as its knockdown effect was stronger. The absence of GPX4 canceled the effect of empagliflozin on GPX4 mRNA (Fig. 4a) and protein (Fig. 4b, c) expression. Concomitantly, GPX4 knockdown abolished the effects of empagliflozin on suppressing hyperglycemia-induced lipid peroxidation (Fig. 4d, e). Subsequently, we observed that empagliflozin failed to promote viability and suppress cell death of GPX4-knocked down C2C12 cells cultured under hyperglycemic conditions (Fig. 4f, g). It is also noteworthy that empagliflozin could slightly promote GPX4 expression in C2C12 cells under normoglycemia (Supplementary Fig. S5a, b); suggesting that empagliflozin could also enhance GPX4 expression under normoglycemia, although the effect was significantly lower than in hyperglycemia.

GPX4 mRNA (a) and protein (b, c) expression in GPX4-knocked down C2C12 cells treated with 10 μM empagliflozin, as examined using qRT-PCR and Western blotting, respectively. Representative images (b) and quantification results (c) were shown. β‐Actin was used for qRT-PCR normalization and as Western blotting loading control. d, e Lipid peroxidation ratio in GPX4-knocked down C2C12 cells treated with 10 μM empagliflozin, as analyzed using C11-BODIPY staining. Representative images (d; scale bars: 100 μm) and quantification results (e; n = 6) were shown. f Cell viability in GPX4-knocked down C2C12 cells treated with 10 μM empagliflozin. g Cell death rate in GPX4-knocked down C2C12 cells treated with 10 μM empagliflozin, as examined using PI staining and flow cytometry. All experiments were performed under hyperglycemia. Data were presented as mean ± SD (n = 3, unless further indicated). **P < 0.01; ***P < 0.001; ****P < 0.0001.
To assess whether other gliflozins also promote skeletal muscle cell viability through the GPX4/ferroptosis pathway, we examined whether they could increase GPX4 expression. Treatment with canagliflozin, dapagliflozin, ertugliflozin, ipragliflozin, and tofogliflozin also induced the expression of GPX4 mRNA and protein under hyperglycemic conditions (Fig. 5a–c). Furthermore, knocking-down GPX4 clearly suppressed the effect of gliflozins in suppressing C2C12 cell death (Fig. 5d), and subsequently attenuated their effect in increasing C2C12 cell viability (Fig. 5e). This effect is most plausibly achieved by suppressing hyperglycemia-induced ferroptosis (Fig. 5f), as gliflozins reduced hyperglycemia-induced 4-HNE and MDA levels in skeletal muscle cells (Fig. 5g, h).

mRNA (a) and protein (b, c) expression of GPX4 in C2C12 cells treated with 10 μM of indicated gliflozins, as examined using qRT-PCR and Western blotting, respectively. Representative images (b) and quantification results (c) were shown. β‐Actin was used for qRT-PCR normalization and as Western blotting loading control. d Cell death rate in GPX4-knocked down C2C12 cells treated with 10 μM of indicated gliflozins, as examined using PI staining and flow cytometry. e Cell viability in GPX4-knocked down C2C12 cells treated with 10 μM of indicated gliflozins. f Lipid ROS level in C2C12 cells treated with 10 μM of indicated gliflozins, as examined using C11-BODIPY staining and flow cytometry. 4-HNE (g) and MDA (h) levels in C2C12 cells treated with 10 μM of indicated gliflozins, as analyzed using ELISA and Lipid Peroxidation MDA Assay Kit, respectively. All experiments were performed under hyperglycemia. Data were presented as mean ± SD (n = 3). HG hyperglycemia; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 versus shCon; while #P < 0.05; ##P < 0.01; ###P < 0.001; ####P < 0.0001 versus shGPX4.
Together, these results demonstrate that empagliflozin and other gliflozins could suppress hyperglycemia-induced ferroptosis in skeletal muscle cells by promoting GPX4 expression.
Empagliflozin promotes skeletal muscle cells’ angiogenic potential in a GPX4-dependent manner
As demonstrated in previous studies and confirmed in Supplementary Fig. S6, the expression of angiogenic factors in skeletal muscle cells decreased under hyperglycemic conditions [16, 28, 41]. GO analysis revealed the enrichment of “angiogenesis” in empagliflozin-treated C2C12 cells under hyperglycemic conditions. Skeletal muscle cells contribute to angiogenesis through their secretome to vascular endothelial cells (HUVECs) and smooth muscle cells (MOVAS), the two major types of blood vessel-forming cells [24, 42, 43]. Our results showed that empagliflozin enhanced the mRNA expression of angiogenin-1 (ANG1), fibroblast growth factor 2 (FGF2), hepatocyte growth factor (HGF), platelet-derived growth factor-BB (PDGF-BB), and vascular endothelial growth factor-A (VEGF-A), which are angiogenic factors crucial for the formation and maturation of blood vessels, in C2C12 cells (Fig. 6a). However, these effects were abolished by GPX4 silencing. Similar results were obtained for protein expression (Fig. 6b). Concomitantly, empagliflozin promoted the secretion of ANG1, PDGF-BB, and VEGF-A from C2C12 cells under hyperglycemic conditions, but not from GPX4-knocked down cells (Fig. 6c).

mRNA (a) and protein (b) expression of angiogenic factors in GPX4-knocked down C2C12 cells treated with 10 μM empagliflozin, as examined using qRT-PCR and Western blotting, respectively. β‐Actin was used for qRT-PCR normalization and as Western blotting loading control. c Secreted amount of ANG1, PDGF-BB and VEGF-A in the culture medium of C2C12 cells treated with 10 μM empagliflozin, as analyzed using ELISA. Proliferation potential of HUVECs (d, e) and MOVAS cells (f, g) cultured with indicated conditioned media collected from GPX4-knocked down C2C12 cells treated with 10 μM empagliflozin, as examined using EdU incorporation assay. Representative images (d and f; scale bars: 100 μm) and quantification results (e and g; n = 6) were shown. Migration potential of HUVECs (h, i) and MOVAS cells ( j, k) cultured with indicated conditioned media, as analyzed using transwell migration assay. Representative images (h and j; scale bars: 200 μm) and quantification results (i and k; n = 6) were shown. All experiments were performed under hyperglycemia. Data were presented as mean ± SD (n = 3, unless further indicated). CM-shCon and CM-shGPX4: conditioned media obtained from shCon-transfected or GPX4-knocked down C2C12 cells; CM-shCon+Empa and CM-shGPX4+Empa: conditioned media obtained from shCon- or shGPX4-transfected C2C12 cells treated with 10 μM empagliflozin. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Next, we obtained conditioned media from C2C12 cells transfected with shCon or shGPX4 under hyperglycemic conditions, and treated them with 10% dimethylsulfoxide (DMSO; CM-shCon and CM-shGPX4, respectively), or with empagliflozin (CM-shCon+Empa and CM-shGPX4+Empa). Then, we examined their effects on blood vessel-forming cells. Compared to those cultured with CM-shCon, the ratio of EdU-positive cells was significantly increased in HUVECs cultured with CM-shCon+Empa. This indicates that empagliflozin enhanced HUVEC proliferation potential through the skeletal muscle cell secretome (Fig. 6d, e). However, this effect could not be achieved using CM-shGPX4+Empa. Similar tendencies were observed in MOVAS cells (Fig. 6f, g). These results clearly showed that empagliflozin could enhance vascular endothelial and smooth muscle cell proliferation potential through the skeletal muscle cell secretome. In addition, they also showed that GPX4 in skeletal muscle cells is crucial for this effect.
Migration potential is another characteristic of the angiogenic function of blood-vessel-forming cells, which is crucial for their migration towards the ischemic site. Migration potential of HUVECs cultured with CM-shCon+Empa, but not with CM-shGPX4+Empa, was increased robustly, as there were more cells migrated to the lower compartment of the transwell chamber than those cultured with the control (CM-shCon, Fig. 6h, i). Similarly, MOVAS cell migration potential was also significantly enhanced by CM-shCon+Empa (Fig. 6j, k). Thus, empagliflozin, most plausibly through its effect in promoting skeletal muscle cells GPX4, robustly promotes the migration potential of blood vessel-forming cells.
Notably, treating HUVECs and MOVAS cells directly with empagliflozin did not enhance their proliferation (Supplementary Fig. S7a–d) nor their migration potential (Supplementary Fig. S7e–h). Together, our results indicate that empagliflozin enhances the proliferation and migration potentials of blood vessels-forming cells through enhanced skeletal muscle cell angiogenesis potential by inhibiting ferroptosis and promoting their viability in a GPX4-dependent manner, but not through its direct effect on them.
Empagliflozin promotes revascularization in diabetic HLI model mice in a GPX4-dependent manner
Finally, we examined the effect of empagliflozin on promoting revascularization in diabetic HLI mice. Diabetic HLI model mice were established by excising the left hindlimb femoral artery of diabetic mice. Empagliflozin was intramuscularly injected at the gastrocnemius muscle near the ischemic location, that is, near the excision location. Blood perfusion in both the left and corresponding right hindlimb, which was used as a control, was measured using a Laser Doppler Imager. The control group comprising of mice treated with shCon and 10% DMSO showed only 30% blood perfusion recovery at day 21 after surgery. In contrast, the recovery rate in the ischemic hindlimb of diabetic HLI mice treated intramuscularly with empagliflozin reached more than 70% at day 21 after surgery (Fig. 7a, b). Meanwhile, GPX4-knockdown clearly abolished the effect of empagliflozin on promoting the blood perfusion recovery in diabetic HLI mice. The morphological assessment further confirmed the positive effect of empagliflozin on revascularization and the crucial role of GPX4 in empagliflozin-induced revascularization (Fig. 7c). Mice in the empagliflozin-treated group showed better hindlimb morphologies, and most of them scored 1. In contrast, mice in the control group scored 2 or 3. Furthermore, GPX4 silencing abolished the effect of empagliflozin on the morphology of the ischemic hindlimb. These results indicate the efficacy of empagliflozin in enhancing the blood perfusion recovery in diabetic HLI mice and the essential role of GPX4 in the therapeutic effect of empagliflozin in diabetic HLI mice. It is also noteworthy that in mice treated with only shGPX4, the blood perfusion recovery was even slower than that of the control, and most of the mice scored 3 from day 14 after surgery for morphological assessment, further confirming that ferroptosis disrupts the angiogenic potential of diabetic HLI mice.

a, b Blood perfusion in the ischemic hindlimbs of diabetic HLI mice intramuscularly injected with empagliflozin (10 mg/kg body weight) and shCon or shGPX4 vectors at indicated time points. Representative images (a) and quantification data of blood perfusion ratio were shown (b; n = 7). c Morphological assessment of ischemic hindlimbs in diabetic HLI mice intramuscularly injected with empagliflozin (10 mg/kg body weight) and shCon or shGPX4 vectors at indicated time points (n = 7). d, e Immunofluorescence against PECAM-1 (green) and α-SMA (red) in ischemic hindlimbs tissue of diabetic HLI mice intramuscularly injected with empagliflozin (10 mg/kg body weight) and shCon or shGPX4 vectors at day 21 after surgery. Representative images (d; scale bars: 50 μm) and quantification results (e) were shown. f Representative images of immunohistochemical staining against GPX4 and 4-HNE in ischemic hindlimbs tissues of diabetic HLI mice intramuscularly injected with empagliflozin (10 mg/kg body weight) and shCon or shGPX4 vectors at day 21 after surgery (scale bars: 50 μm). g Transmission electron microscopy images of the mitochondria in the ischemic hindlimbs tissues of diabetic HLI mice intramuscularly administered with empagliflozin (10 mg/kg body weight) and shCon or shGPX4 vectors at day 21 after surgery. Red arrows: mitochondria; scale bars: 200 nm. Data were presented as mean ± SD (n = 6). NS not significant; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Then, we examined the potential mechanism of intramuscularly injected empagliflozin in promoting the blood perfusion recovery in diabetic HLI mice. Immunofluorescence staining of platelet derived cell adhesion molecule-1 (PECAM-1), a marker of vascular endothelial cells, and alpha-smooth muscle actin (α-SMA), a marker of smooth muscle cells, showed that empagliflozin treatment noticeably increased the number of both PECAM-1- and α-SMA-positive cells (shown in green and red, respectively; Fig. 7d). Intramuscular injection of empagliflozin also increased the number of PECAM-1-positive tube-like structures covered by α-SMA-positive cells (merged in yellow), which indicate mature blood vessels in the ischemic hindlimb of diabetic HLI mice (Fig. 7d, e). Furthermore, in accordance with the blood perfusion recovery and morphological analysis results, GPX4 silencing clearly reduced the number of PECAM-1- and α-SMA-positive cells as well as the number of mature blood vessels induced by empagliflozin.
We next examined GPX4 expression in the gastrocnemius muscle of diabetic HLI mice. As shown in Fig. 7f, empagliflozin treatment promoted GPX4 accumulation. Concomitantly, immunohistochemical staining for 4-HNE also showed that empagliflozin significantly reduced lipid peroxidation in the gastrocnemius muscle of diabetic HLI mice. However, GPX4 silencing abolished these effects (Fig. 7f). Furthermore, we observed mitochondrial morphology in tissues obtained from the gastrocnemius muscle of diabetic HLI mice. The control group had smaller, shrunken mitochondria, while the mitochondria in the empagliflozin-treated group were larger with increased mitochondrial crista (Fig. 7g). However, GPX4 silencing canceled the effect of empagliflozin in protecting mitochondrial morphology.
Furthermore, H&E staining showed that there was no obvious damage to the heart, liver, spleen, lung, or kidney of mice injected with empagliflozin intramuscularly (Supplementary Fig. S8). Moreover, there was no significant difference in the body weight between the groups (Supplementary Table S3). Although further detailed investigations are needed, these results preliminarily indicated that there was no significant toxicity induced by intramuscular administration of empagliflozin.
Orally administered gliflozins have been known to suppress blood glucose levels by inhibiting SGLT2. However, we did not observe any significant difference in the blood glucose levels among the different diabetic HLI mice treated with empagliflozin or 10% DMSO (Supplementary Table S4). These results indicated that gliflozins administered directly to skeletal muscle cells might exert functions different from those administered orally. ArrayExpress (https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-2836) showed that SGLT2 is highly expressed in renal cells but not in skeletal muscle cells [44]. Our results further confirmed this, as we could clearly detect the expression of SGLT2 in HEK293T cells but not in C2C12 cells (Supplementary Fig. S9). Hence, gliflozins exert their suppressive effect on skeletal muscle cell ferroptosis through a mechanism distinct from their anti-hyperglycemic function.
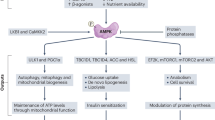
Taken together, our results show that empagliflozin inhibits ferroptosis by promoting GPX4 accumulation to enhance skeletal muscle cell survival. Furthermore, it also protects their paracrine function, thereby triggering vascular endothelial and smooth muscle cells’ proliferation and migration capabilities, and subsequently promotes revascularization through enhanced formation of mature blood vessels in diabetic HLI model mice (Fig. 8). Hence, promoting skeletal muscle cell viability by inhibiting ferroptosis is a promising strategy for the future application of empagliflozin in treating diabetic HLI.

Empagliflozin inhibits hyperglycemia-induced ferroptosis in skeletal muscle cells by enhancing GPX4 expression, thus promoting revascularization in diabetic HLI mice.
Discussion
Therapeutic angiogenesis, which aims to stimulate revascularization to enhance tissue viability and recover blood perfusion, has recently become a prospective strategy for lower limb ischemia patients [24, 25]. Skeletal muscle was reported to be a prospective target to promote therapeutic angiogenesis as its paracrine function is essential to form new blood vessels [16, 24, 26, 28]. However, impaired blood flow in the lower extremities in lower limb ischemia reduces skeletal muscle cell viability, thus demonstrating impaired capillary density and muscle oxygen delivery to an extreme extent [45]. This condition is worse in lower limb ischemia patients with diabetes, as hyperglycemia increases ROS and produces advanced glycation end products (AGEs), which reduces the mitochondrial efficiency of skeletal muscle myotubes and the expression of myogenic regulatory factors, leading to skeletal muscle cell death and reduced muscular strength in diabetic patients [46, 47]. Furthermore, hyperglycemia-induced systemic impairment also affects skeletal muscle cell cellular functions, including paracrine function, as well as the proliferation and migration potential of blood vessel-forming cells. These lead to poor angiogenic potential in diabetic lower limb ischemia, and subsequently, to severe outcomes and poor prognosis with a wider damaged area and high reoccurrence rate. Together, these factors constitute the main obstacle for treating diabetic lower limb ischemia patients. In addition to being inappropriate for standardized revascularization therapy, it is difficult to induce effective therapeutic angiogenesis in these patients [16, 20, 24]. Thus, there is an urgent need to develop novel small-molecule drugs to treat diabetic lower limb ischemia.
Oral administration of gliflozins has recently been found to exert positive effects on renal outcomes by inhibiting SGLT2 [7,8,9] as well as improving cardiac functions and treating acute heart failure in an SGLT2-independent manner [10,11,12,13,14]. In this study, we clearly showed that empagliflozin could increase the viability of skeletal muscle cells suppressed by hyperglycemia by promoting the expression of GPX4, whose expression is decreased under hyperglycemia. This, in turn, leads to the suppression of skeletal muscle cell ferroptosis, and subsequently, to an increase in the secretion of angiogenic factors, such as VEGF-A, PDGF-BB, and ANG1, from skeletal muscle cells, which then promotes the proliferation and migration potentials of blood vessel-forming cells. VEGF-A contributes to the formation of tube-like structures of endothelial cells and is recognized as an initiator of angiogenesis [48], while PDGF-BB and ANG1 are crucial for vascular maturation [16, 49]. Subsequently, intramuscular treatment with empagliflozin might restore the angiogenic potential of diabetic HLI model mice, enhance the formation of mature and functional blood vessels, and improve blood perfusion recovery. Hence, our study provides a potential novel therapeutic strategy for diabetic HLI by using empagliflozin.
Gliflozins exert their function of lowering blood glucose by inhibiting SGLT2 to block glucose reabsorption in the kidney [1,2,3]. However, SGLT2 expression is specific to renal and colorectal cells [44] and is not expressed in skeletal muscle cells. Furthermore, localized intramuscular administration of empagliflozin did not affect blood glucose levels. Thus, the mechanism by which empagliflozin suppresses skeletal muscle cell ferroptosis and subsequently increases therapeutic angiogenesis in diabetic HLI mice might occur through a completely distinct pathway than the one that suppresses blood glucose levels. Moreso, it is independent of SGLT2. Notably, oral administration of canagliflozin has been reported to attenuate myocardial infarction in mice models and impair blood reperfusion in the ischemic hindlimb by inhibiting mitochondrial functions, thus increasing the risk of limb amputation [50, 51]. However, our results showed that direct treatment of skeletal muscle cells with gliflozins, including canagliflozin, suppresses ferroptosis and increases skeletal muscle cell viability by enhancing the expression of GPX4 and decreasing the level of lipid ROS caused by hyperglycemia. These distinct effects might be owing to the different administration methods of gliflozins. Gliflozins taken orally might be metabolized and distributed systematically, whereas intramuscular injection provides a more localized and direct effect on target cells.
Ferroptosis is a recently identified form of non-apoptotic cell death caused by dysregulation of iron and lipid metabolism due to the loss of activity of lipid repair enzymes. This causes subsequent accumulation of lipid hydroperoxides [32, 52], which may lead to diseases such as neurodegenerative diseases, brain injuries, stroke, and ischemic heart injuries [53, 54]. Furthermore, a recent study showed an increase in ferroptosis in skeletal muscle cells of aged mice, suppressing skeletal muscle cell proliferation and regeneration [55]. Hence, our findings, which reveal the potential of gliflozins as novel anti-ferroptosis small-molecule drugs, may provide new therapeutic strategies for treating these diseases.
Despite their potential as new therapeutic strategies for treating diabetic HLI, there are still some limitations in this study that need to be solved. While the fact that empagliflozin is a well-known approved orally delivered drug whose safety has already been proven might benefit the translational process of these findings to clinical application of intramuscular injection of empagliflozin; it needs to be re-tested for intramuscular administration in patients. Furthermore, despite our preliminary experiments demonstrated that intramuscular empagliflozin treatment does not cause significant toxicity, as there is no significant difference in the body weight and morphologies in different organs of diabetic HLI mice intramuscularly injected with empagliflozin, further investigations and clinical studies are needed. Moreover, it should be noted that while all these gliflozins are aryl-C-glucosides with the same core structures and belong to the same class of drugs, differences in their chemical structures might regulate hyperglycemia-induced ferroptosis in skeletal muscle cells to different extents, thus further pre-clinical and clinical studies are needed before the clinical applications of gliflozins as anti-ferroptosis small-molecule drugs.
In conclusion, our study uncovered a novel role of empagliflozin as an anti-ferroptosis small-molecule drug that promotes the viability of skeletal muscle cells under hyperglycemic conditions by enhancing lipid ROS clearance through GPX4. Furthermore, we observed effective revascularization in diabetic HLI mice following intramuscular administration of empagliflozin. While further preclinical and clinical studies are needed, this study highlights a promising therapeutic strategy for intramuscular administration of empagliflozin to inhibit ferroptosis to promote therapeutic angiogenesis in diabetic HLI.
References
Washburn WN. Development of the renal glucose reabsorption inhibitors: a new mechanism for the pharmacotherapy of diabetes mellitus type 2. J Med Chem. 2009;52:1785–94.
Neumiller JJ, White JR Jr, Campbell RK. Sodium-glucose co-transport inhibitors: progress and therapeutic potential in type 2 diabetes mellitus. Drugs. 2010;70:377–85.
Isaji M. Sglt2 inhibitors: molecular design and potential differences in effect. Kidney Int Suppl. 2011;120:S14-9.
Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. 2017;377:644–57.
Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. 2019;380:347–57.
Kullmann S, Hummel J, Wagner R, Dannecker C, Vosseler A, Fritsche L, et al. Empagliflozin improves insulin sensitivity of the hypothalamus in humans with prediabetes: a randomized, double-blind, placebo-controlled, phase 2 trial. Diabetes Care. 2022;45:398–406.
Cannon CP, Pratley R, Dagogo-Jack S, Mancuso J, Huyck S, Masiukiewicz U, et al. Cardiovascular outcomes with ertugliflozin in type 2 diabetes. N Engl J Med. 2020;383:1425–35.
Kaku K, Watada H, Iwamoto Y, Utsunomiya K, Terauchi Y, Tobe K, et al. Efficacy and safety of monotherapy with the novel sodium/glucose cotransporter-2 inhibitor tofogliflozin in japanese patients with type 2 diabetes mellitus: a combined phase 2 and 3 randomized, placebo-controlled, double-blind, parallel-group comparative study. Cardiovasc Diabetol. 2014;13:65.
Wilding JP, Ferrannini E, Fonseca VA, Wilpshaar W, Dhanjal P, Houzer A. Efficacy and safety of ipragliflozin in patients with type 2 diabetes inadequately controlled on metformin: a dose-finding study. Diabetes Obes Metab. 2013;15:403–9.
Lahnwong S, Palee S, Apaijai N, Sriwichaiin S, Kerdphoo S, Jaiwongkam T, et al. Acute dapagliflozin administration exerts cardioprotective effects in rats with cardiac ischemia/reperfusion injury. Cardiovasc Diabetol. 2020;19:91.
Jiang K, Xu Y, Wang D, Chen F, Tu Z, Qian J, et al. Cardioprotective mechanism of SGLT2 inhibitor against myocardial infarction is through reduction of autosis. Protein Cell. 2022;13:336–59.
Griffin M, Rao VS, Ivey-Miranda J, Fleming J, Mahoney D, Maulion C, et al. Empagliflozin in heart failure: diuretic and cardiorenal effects. Circulation. 2020;142:1028–39.
Quagliariello V, De Laurentiis M, Rea D, Barbieri A, Monti MG, Carbone A, et al. The SGLT-2 inhibitor empagliflozin improves myocardial strain, reduces cardiac fibrosis and pro-inflammatory cytokines in non-diabetic mice treated with doxorubicin. Cardiovasc Diabetol. 2021;20:150.
Voors AA, Angermann CE, Teerlink JR, Collins SP, Kosiborod M, Biegus J, et al. The SGLT2 inhibitor empagliflozin in patients hospitalized for acute heart failure: a multinational randomized trial. Nat Med. 2022;28:568–74.
Criqui MH, Aboyans V. Epidemiology of peripheral artery disease. Circ Res. 2015;116:1509–26.
Liu C, Han J, Marcelina O, Nugrahaningrum DA, Huang S, Zou M, et al. Discovery of salidroside-derivated glycoside analogues as novel angiogenesis agents to treat diabetic hind limb ischemia. J Med Chem. 2022;65:135–62.
Song P, Rudan D, Zhu Y, Fowkes FJI, Rahimi K, Fowkes FGR, et al. Global, regional, and national prevalence and risk factors for peripheral artery disease in 2015: an updated systematic review and analysis. Lancet Glob Health. 2019;7:e1020–e30.
Criqui MH, Matsushita K, Aboyans V, Hess CN, Hicks CW, Kwan TW, et al. Lower extremity peripheral artery disease: contemporary epidemiology, management gaps, and future directions: a scientific statement from the american heart association. Circulation. 2021;144:e171–e91.
Jude EB, Oyibo SO, Chalmers N, Boulton AJ. Peripheral arterial disease in diabetic and nondiabetic patients: a comparison of severity and outcome. Diabetes Care. 2001;24:1433–7.
Hazarika S, Dokun AO, Li Y, Popel AS, Kontos CD, Annex BH. Impaired angiogenesis after hindlimb ischemia in type 2 diabetes mellitus: differential regulation of vascular endothelial growth factor receptor 1 and soluble vascular endothelial growth factor receptor 1. Circ Res. 2007;101:948–56.
Falanga V. Wound healing and its impairment in the diabetic foot. Lancet. 2005;366:1736–43.
Annex BH. Therapeutic angiogenesis for critical limb ischaemia. Nat Rev Cardiol. 2013;10:387–96.
Benoit B, Meugnier E, Castelli M, Chanon S, Vieille-Marchiset A, Durand C, et al. Fibroblast growth factor 19 regulates skeletal muscle mass and ameliorates muscle wasting in mice. Nat Med. 2017;23:990–96.
Tateno K, Minamino T, Toko H, Akazawa H, Shimizu N, Takeda S, et al. Critical roles of muscle-secreted angiogenic factors in therapeutic neovascularization. Circ Res. 2006;98:1194–202.
Wu S, Nishiyama N, Kano MR, Morishita Y, Miyazono K, Itaka K, et al. Enhancement of angiogenesis through stabilization of hypoxia-inducible factor-1 by silencing prolyl hydroxylase domain-2 gene. Mol Ther. 2008;16:1227–34.
Wu S, Zhang J, Huang C, Jia H, Wang Y, Xu Z, et al. Prolyl hydroxylase domain-2 silencing induced by hydrodynamic limb vein injection enhances vascular regeneration in critical limb ischemia mice through activation of multiple genes. Curr Gene Ther. 2015;15:313–25.
Ariyanti AD, Sisjayawan J, Zhang J, Zhang JQ, Wang GX, Miyagishi M, et al. Elevating VEGF-A and PDGF-BB secretion by salidroside enhances neoangiogenesis in diabetic hind-limb ischemia. Oncotarget. 2017;8:97187–205.
Zhang J, Kasim V, Xie YD, Huang C, Sisjayawan J, Dwi Ariyanti A, et al. Inhibition of PHD3 by salidroside promotes neovascularization through cell-cell communications mediated by muscle-secreted angiogenic factors. Sci Rep. 2017;7:43935.
Caporali A, Meloni M, Nailor A, Mitic T, Shantikumar S, Riu F, et al. P75(NTR)-dependent activation of nf-kappab regulates microrna-503 transcription and pericyte-endothelial crosstalk in diabetes after limb ischaemia. Nat Commun. 2015;6:8024.
Zhang J, Nugrahaningrum DA, Marcelina O, Ariyanti AD, Wang G, Liu C, et al. Tyrosol facilitates neovascularization by enhancing skeletal muscle cells viability and paracrine function in diabetic hindlimb ischemia mice. Front Pharmacol. 2019;10:909.
Batista TM, Jayavelu AK, Wewer Albrechtsen NJ, Iovino S, Lebastchi J, Pan H, et al. A cell-autonomous signature of dysregulated protein phosphorylation underlies muscle insulin resistance in type 2 diabetes. Cell Metab. 2020;32:844–59.e5.
Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–76.
Lawson MA, Purslow PP. Differentiation of myoblasts in serum-free media: effects of modified media are cell line-specific. Cells Tissues Organs. 2000;167:130–7.
Miyagishi M, Taira K. Strategies for generation of an sirna expression library directed against the human genome. Oligonucleotides. 2003;13:325–33.
Nugrahaningrum DA, Marcelina O, Liu C, Wu S, Kasim V. Dapagliflozin promotes neovascularization by improving paracrine function of skeletal muscle cells in diabetic hindlimb ischemia mice through PHD2/HIF-1alpha axis. Front Pharmacol. 2020;11:1104.
Stabile E, Burnett MS, Watkins C, Kinnaird T, Bachis A, la Sala A, et al. Impaired arteriogenic response to acute hindlimb ischemia in CD4-knockout mice. Circulation. 2003;108:205–10.
Tanii M, Yonemitsu Y, Fujii T, Shikada Y, Kohno R, Onimaru M, et al. Diabetic microangiopathy in ischemic limb is a disease of disturbance of the platelet-derived growth factor-BB/protein kinase C axis but not of impaired expression of angiogenic factors. Circ Res. 2006;98:55–62.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, et al. Ferroptosis: process and function. Cell Death Differ. 2016;23:369–79.
Li Y, Li J, Li Z, Wei M, Zhao H, Miyagishi M, et al. Homeostasis imbalance of YY2 and YY1 promotes tumor growth by manipulating ferroptosis. Adv Sci. 2022;9:e2104836.
Luo LL, Han JX, Wu SR, Kasim V. Intramuscular injection of sotagliflozin promotes neovascularization in diabetic mice through enhancing skeletal muscle cells paracrine function. Acta Pharmacol Sin. 2022;43:2636–50.
Carmeliet P. Mechanisms of angiogenesis and arteriogenesis. Nat Med. 2000;6:389–95.
Giudice J, Taylor JM. Muscle as a paracrine and endocrine organ. Curr Opin Pharmacol. 2017;34:49–55.
Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419.
McDermott MM, Ferrucci L, Gonzalez-Freire M, Kosmac K, Leeuwenburgh C, Peterson CA, et al. Skeletal muscle pathology in peripheral artery disease: a brief review. Arterioscler Thromb Vasc Biol. 2020;40:2577–85.
Chiu CY, Yang RS, Sheu ML, Chan DC, Yang TH, Tsai KS, et al. Advanced glycation end-products induce skeletal muscle atrophy and dysfunction in diabetic mice via a RAGE-mediated, AMPK-down-regulated, AKT pathway. J Pathol. 2016;238:470–82.
Rai AK, Jaiswal N, Maurya CK, Sharma A, Ahmad I, Ahmad S, et al. Fructose-induced AGEs-RAGE signaling in skeletal muscle contributes to impairment of glucose homeostasis. J Nutr Biochem. 2019;71:35–44.
Takeshita S, Zheng LP, Brogi E, Kearney M, Pu LQ, Bunting S, et al. Therapeutic angiogenesis. A single intraarterial bolus of vascular endothelial growth factor augments revascularization in a rabbit ischemic hind limb model. J Clin Invest. 1994;93:662–70.
Xie H, Cui Z, Wang L, Xia Z, Hu Y, Xian L, et al. PDGF-BB secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis. Nat Med. 2014;20:1270–8.
Matthews DR, Li Q, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, et al. Effects of canagliflozin on amputation risk in type 2 diabetes: the canvas program. Diabetologia. 2019;62:926–38.
Lin Y, Nan J, Shen J, Lv X, Chen X, Lu X, et al. Canagliflozin impairs blood reperfusion of ischaemic lower limb partially by inhibiting the retention and paracrine function of bone marrow derived mesenchymal stem cells. EBioMedicine. 2020;52:102637.
Wang H, An P, Xie E, Wu Q, Fang X, Gao H, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. 2017;66:449–65.
Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22:266–82.
Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59:298–308.
Ding H, Chen S, Pan X, Dai X, Pan G, Li Z, et al. Transferrin receptor 1 ablation in satellite cells impedes skeletal muscle regeneration through activation of ferroptosis. J Cachexia Sarcopenia Muscle. 2021;12:746–68.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (31871367 and 81872273), the Natural Science Foundation of Chongqing (CSTB2022NSCQ-MSX0611 and CSTB2022NSCQ-MSX0612).
Author information
Authors and Affiliations
Contributions
SRW and VK arranged the research, designed the experiments, examined and construed the data, provided financial support and guided all the experiments. JXH did literature research, performed the cellular and animal experiments, examined and construed the data, prepared the figures and wrote the manuscript. LLL performed part of the cellular and animal experiments, examined and construed the data, and assisted in writing the manuscript. YCW performed part of the cellular and animal experiments. MM designed, constructed, and examined the shRNA expression vectors.
Corresponding authors
Ethics declarations
Competing interests
All authors declare no competing interests. The results of this study have been patented in China (No. ZL202010701077.7).
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Han, Jx., Luo, Ll., Wang, Yc. et al. SGLT2 inhibitor empagliflozin promotes revascularization in diabetic mouse hindlimb ischemia by inhibiting ferroptosis. Acta Pharmacol Sin 44, 1161–1174 (2023). https://doi.org/10.1038/s41401-022-01031-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-022-01031-0
