
Abstract
Herbal and dietary supplements (HDS)-induced liver injury has been a great concern all over the world. Polygonum multiflorum Thunb., a well-known Chinese herbal medicine, is recently drawn increasing attention because of its hepatotoxicity. According to the clinical and experimental studies, P. multiflorum-induced liver injury (PM-DILI) is considered to be immune-mediated idiosyncratic liver injury, but the role of immune response and the underlying mechanisms are not completely elucidated. Previous studies focused on the direct toxicity of PM-DILI by using animal models with intrinsic drug-induced liver injury (DILI). However, most epidemiological and clinical evidence demonstrate that PM-DILI is immune-mediated idiosyncratic liver injury. The aim of this review is to assess current epidemiological, clinical and experimental evidence about the possible role of innate and adaptive immunity in the idiosyncratic hepatotoxicity of P. multiflorum. The potential effects of factors associated with immune tolerance, including immune checkpoint molecules and regulatory immune cells on the individual’s susceptibility to PM-DILI are also discussed. We conclude by giving our hypothesis of possible immune mechanisms of PM-DILI and providing suggestions for future studies on valuable biomarkers identification and proper immune models establishment.
Similar content being viewed by others

Introduction
Traditional herbal medicine has been extensively applied in many regions worldwide for thousands of years. With the popularity of herbal medicines as effective and affordable therapeutics for disease treatment, their adverse reactions have been drawn growing attention worldwide. Hepatotoxicity is one of the most common adverse reactions induced by herbal and dietary supplements (HDSs), and it can result in liver failure and even death. Data show that HDS‐induced liver injury accounts for 20% of cases of hepatotoxicity in the United States and 26.81% of that in mainland China [1, 2]. HDS-induced liver injury has increasingly become the most challenging drug safety problem worldwide.
Polygonum multiflorum Thunb. and its processed products are well-known herbal medicines that have been used for more than a thousand years in China and many other East Asian countries. Traditionally, raw P. multiflorum products are applied for detoxification, moistening the intestines and freeing the stools, while processed P. multiflorum products are used as tonic and antiaging agents. Previously, pharmacological studies and clinical practice have indicated its beneficial effects on treating hyperlipidemia, inflammation, learning and memory dysfunction, atherosclerosis and immune dysfunction [3, 4]. In recent years, however, liver injury induced by P. multiflorum and its preparations has been an increasing concern worldwide. Since the first case of P. multiflorum-induced liver injury (PM-DILI) was reported in Hong Kong in 1996, cases have successively emerged in many other countries and regions [5,6,7]. As a result, cautionary statements have been issued in China, Australia, Britain, Japan and Canada [8]. Great efforts have been made to investigate PM-DILI over the decades, but the underlying mechanisms remain poorly understood. In addition, the lack of reliable biomarkers makes it difficult to diagnose and predict the hepatotoxicity induced by P. multiflorum preparations.
In the past, studies on PM-DILI focused on the drug-related mechanism that the PM-DILI is the result of the direct toxicity from the ingredients or reactive metabolites of P. multiflorum [9, 10]. However, analyses of the mechanisms and clinical features associated with PM-DILI have revealed that there are still nonnegligible gaps in our understanding of its pathogenesis. Recent progress in clinical and basic studies has implied that PM-DILI is more likely to be immune-mediated idiosyncratic drug-induced liver injury (IDILI). Host-dependent mechanisms associated with immune factors, including individual immune stress status and immunogenetic variations, contribute to the risk of PM-DILI. With the great advancements in the understanding of the mechanisms of DILI in recent years, largely due to improvements in genetic studies and animal models, the crucial role of immune homeostasis in the pathogenesis of DILI has come into view [11] and may provide new insight into the mechanisms of PM-DILI. The aim of this review is to integrate the current knowledge on the mechanisms of PM-DILI and give our viewpoint on the possible immune mechanisms that determine the risk of PM-DILI.
Clinical characteristics of PM-DILI
As a well-known Chinese herbal medicine, P. multiflorum or its processed products are taken by many people to nourish the liver and kidney. Although there are many cases of liver injury induced by P. multiflorum, epidemiological studies have shown that the overall incidence is relatively low. It has been reported that PM-DILI is more common in females [12], which may be attributed to the fact that the rate of use of P. multiflorum products for health care is higher in the female population. The age distribution of PM-DILI patients is wide, with a median age ranging from 36 to 49 [13]. In general, the occurrence and severity of liver injury are not remarkably associated with the gender or age of the patients [14,15,16]. The pharmacological effects and uses of raw and processed P. multiflorum are different. According to traditional Chinese medicine theory, processed P. multiflorum is used more frequently in the clinic due to its tonic and antiaging effects, whereas raw P. multiflorum is used for treating constipation [17]. Additionally, processed forms of P. multiflorum display lower toxicity than raw P. multiflorum [16]. Clinical analyses have demonstrated that the incidence of liver injury caused by raw P. multiflorum and processed P. multiflorum products are 18.06% and 49.35%, respectively; this difference may be the result of the wider application and more frequent use of processed P. multiflorum products in the population. However, this still suggests that all forms of P. multiflorum products may lead to liver damage regardless of herbal processing [16, 18]. Most patients reported taking daily doses lower than the upper limit of the standard Chinese Pharmacopoeia dose of 3–6 g/day for raw P. multiflorum and 6–12 g/day for processed P. multiflorum [14, 16, 18], which indicated that overdose of raw or processed P. multiflorum may not be the main risk factor for PM-DILI. The delay between the start of P. multiflorum administration and the onset of liver injury ranges from 1 to 240 days. The median time of latency of PM-DILI is ~30–45 days, suggesting delayed-onset hepatotoxicity. It is interesting to note that some patients recovering from previous PM-DILI are easily rechallenged with P. multiflorum. The recurrence of PM-DILI always leads to a quick onset of more severe symptoms, and the median time of latency decreases from 39 days for the first onset to 18 days for the recurrence [16, 18,19,20]. In addition, a few patients have reported a family history of liver injury induced by P. multiflorum, and patients with immune-related diseases, such as psoriasis and system lupus erythematosus, have a relatively higher risk of liver damage after taking P. multiflorum products [21]. This evidence implies that host factors may modify an individual’s risk of PM-DILI.
Histological characterization of patients with PM-DILI show that hepatocellular liver injury is the most common type of liver injury, followed by cholestatic liver injury and mixed liver injury. The histological presentation of the liver in hepatocellular liver injury usually involves focal necrosis and infiltration and activation of immunocytes, such as lymphocytes, eosinophils, neutrophils and Kupffer cells (KCs), whereas cholestatic live injury includes lobular necroinflammation and cholestasis [5, 7, 14, 18, 22,23,24]. One common adverse reaction induced by P. multiflorum is skin rash [25, 26], which is a type of immune-mediated idiosyncratic adverse drug reaction. In a handful of patients, PM-DILI is also accompanied by some immunoallergic signs of drug hypersensitivity, such as rash, fever, and eosinophilia [12, 16]. Immune mechanisms explain the idiosyncratic nature of IDILI, and it has been widely accepted that most IDILI is immune-mediated. In conclusion, based on this evidence, PM-DILI is likely to be immune-mediated idiosyncratic drug-induced liver injury (IDILI) [7, 14, 15, 23].
Mechanisms of PM-DILI
Direct hepatotoxicity of P. multiflorum
Intrinsic hepatotoxicity, which usually refers to direct toxic stress leading to hepatic cell death, is commonly thought to be a form of predictable liver injury that can be reproduced in animal models and occurs in a dose-dependent manner [27]. PM-DILI was once considered intrinsic hepatotoxicity based on numerous animal and in vitro studies. Both water and ethanol extracts of P. multiflorum have shown hepatotoxicity in L02 human liver cells in some studies [28,29,30,31] but have shown no toxicity in hepatocytes in other studies [26]. Although ethanol extracts and raw P. multiflorum have been reported to exert higher hepatotoxicity than aqueous extracts and processed products, respectively, in many animal experiments [32], several studies have demonstrated that P. multiflorum does not induce obvious liver injury in rats [33,34,35] and that water extracts even elicit beneficial effects on the liver [36]. Liver injury only occurs in animals when P. multiflorum is administered at much higher doses than the clinical dose regardless of extraction and processing methods [32]. Chemical analyses have demonstrated that stilbenes and anthraquinones are the major characteristic constituents. 2,3,5,4′-Tetrahydroxystilbene-2-O-β-glucoside (TSG) and combined anthraquinones such as emodin-8-O-β-D-glucoside and physcion-8-O-β-D-glucoside are dominant in raw products, while TSG, emodin and physcion are the main components of processed products. It has been reported that the content of TSG is decreased by ~60%, whereas the content of emodin is increased by >30% [4]. However, another study demonstrated that the content of both TSG and emodin is remarkably decreased after processing [37]. Several studies have shown that the toxicity of ethanol extract is higher than that of water extract and that the content of anthraquinones is significantly higher in ethanol extract than in water extract [38, 39]. Many efforts have been made to explore the hepatotoxic components of P. multiflorum. It is widely accepted that anthraquinones, such as emodin and physcion, and their derivatives may be responsible for PM-DILI through direct cytotoxicity [40,41,42,43]. Emodin has been reported to inhibit mitochondrial complex function [44], induce apoptosis of hepatic cells through S phase arrest [28] and activate the NF-κB inflammatory pathway and the P53 apoptosis pathway [45]. Glucuronidation through UGT2B7 and elimination through MRP2 might result in the detoxification of emodin, and decreased UGT2B7 activity due to long-term use of emodin and the inhibition of UGTs and MRP2 by probenecid can cause emodin accumulation, thus leading to hepatotoxicity [46, 47]. Electrophilic physcion and emodin and their oxidative metabolites might display high reactivity with macromolecules and glutathione in hepatic cells, contributing to anthraquinone-induced direct hepatocyte toxicity [48,49,50].
Because the toxic effects of P. multiflorum in the liver are controversial, the mechanisms of the intrinsic hepatotoxicity of P. multiflorum remain elusive. In vitro studies have demonstrated the possible mechanisms, including the induction of apoptosis and the inhibition of liver cell proliferation [29, 38]. In vivo metabolomics analyses have indicated that the disruption of amino acid metabolism, lipid metabolism, bile acid metabolism and energy metabolism is involved in PM-DILI and that vitamin B6, tryptophan and citrate might be metabolic biomarkers [51,52,53]. A proteomics study revealed that PM-DILI is related to oxidative phosphorylation pathways and that NADH dehydrogenase family proteins and slc16a2 may be potential biomarkers [54]. However, these omics studies did not further explore the functions of these pathways and biomarkers in PM-DILI.
Although these studies suggest, to some extent, that drug-related factors of P. multiflorum may induce direct liver injury, it is still limited to explain the pathogenesis of PM-DILI based on the animal and hepatocyte models of intrinsic toxicity because the characteristics of these models are very different from the characteristics of human disease, such as low incidence, dose-independence and lymphocyte infiltration and activation. Clinical analyses have also suggested that PM-DILI appears to be dose-independent immune-mediated idiosyncratic drug-induced liver injury, which means intrinsic hepatotoxicity may only partly explain the liver injury induced by P. multiflorum. Given that most people taking P. multiflorum products at the recommended therapeutic dose do not develop liver injury, it is possible that the intake of direct toxic components is not enough to damage the liver but instead only initiates very mild hepatic cell stress, which can be resisted by the tissue regeneration, repair or antistress systems that maintain the normal structure and functions of hepatocytes exposed to low concentrations of toxic components and that idiosyncratic mechanisms may be the key to the pathogenesis of PM-DILI.
The role of immune stress in PM-DILI
With a few exceptions (e.g., acetaminophen-induced DILI), most cases of DILI in humans are considered idiosyncratic. However, the immune response may influence the dose-response curve towards sensitization to toxicity at therapeutic doses, blurring the boundary between these two types of DILI [55]; in some cases of acetaminophen (APAP)-induced hepatotoxicity, the immune response even contributes to individual susceptibility to liver injury [56, 57]. According to the clinical characteristics, PM-DILI is considered idiosyncratic drug-induced liver injury, and host immune factors play a key role in PM-DILI. Recent progress in animal models of immune stress and clinical pharmacogenomics studies has suggested that immunological stress and human leukocyte antigens mediate the idiosyncratic liver injury induced by P. multiflorum.
The association between hepatotoxicity and immunological stress, especially lipopolysaccharide (LPS)-mediated inflammatory stress, has drawn increasing attention in recent years. LPS activates a variety of cells in the liver, such as KCs, endothelial cells, stellate cells, and bile duct epithelial cells, by binding to Toll-like receptors (TLRs) and initiates inflammatory responses by producing and releasing numerous immune mediators [58]. Increasing evidence has shown the indispensable role of LPS and pre-existing inflammation in liver injury induced by xenobiotic agents [59]. Mice pretreated with a modest inflammatory dose of LPS became susceptible to intrinsic liver injury induced by nontoxic doses of APAP [60]. Several drugs that cause IDILI in humans, such as diclofenac, sulindac, ranitidine, amiodarone, halothane, and trovafloxacin, also induce idiosyncrasy-like liver injury in animals when coadministered with a nontoxic dose of LPS [61,62,63,64,65,66,67]. Therefore, it is widely acknowledged that inflammatory stress can sensitize the liver to injury from both intrinsic and idiosyncratic hepatotoxic agents.
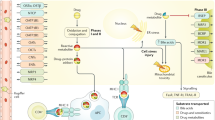
A PM-DILI model based on LPS-mediated immunological stress was recently established. In this model, even a dose of P. multiflorum 70-fold higher than the clinical equivalent dose does not induce liver injury in normal animals, whereas cotreatment with LPS at a nontoxic dose and P. multiflorum at the clinically equivalent dose causes obvious liver injury in a dose-dependent manner [68, 69]. Based on this model, recent studies have demonstrated the mechanisms of PM-DILI. A nontoxic dose of endotoxin induces mild inflammation without causing liver injury. Coexposure to LPS and P. multiflorum can induce the activation of the TLR4-NF-κB pathway, which leads to the activation and differentiation of inflammatory cells (Kupffer cells, monocytes, neutrophils, etc.) and the release of proinflammatory cytokines and chemokines (TNF-α, IL-1β, IL-6, MIP-2, MCP-1, etc.), disturb the immune balance and increase the susceptibility of the liver to toxins [21]. It has been shown that cis-2, 3, 5, 4′-tetrahydroxystilbene-2-O-β-D-glucoside (cis-TSG) displays idiosyncratic hepatotoxicity in LPS-treated rats by inhibiting the activation of the PPAR-γ pathway in liver tissues and that the immunopotentiation effect of trans-TSG might aggravate LPS/cis-TSG-induced liver injury [70,71,72]. In addition, a nontoxic dose of LPS can also potentiate the hepatotoxicity of emodin and its derivatives at safe doses [43, 73]. These results are inconsistent with the observations that only an excessive dose of anthraquinones can induce PM-DILI in intrinsic models. Metabolomics studies on the LPS-mediated mild immune stress model of PM-DILI have suggested that the TCA cycle, the urea cycle, sphingolipid metabolism, vitamin B6 metabolism and pyrimidine metabolism are disturbed [70, 74]. Significantly increased plasma levels of L-phenylalanine and L-glutamine might be related to acute phase protein synthesis under inflammatory stress. Elevated expression of inflammatory cytokines in the plasma, massive immunocyte infiltration and an obvious inflammatory response in liver tissues are observed [75]. According to these results, a hypothesis for the mechanism of the immune stress-mediated idiosyncratic hepatotoxicity of P. multiflorum was proposed recently (Fig. 1). When the body is in a state of immunological activation, components (e.g., trans-TSG) with immunoenhancing activity might increase an individual’s susceptibility to potential toxic components (e.g., cis-TSG and emodin), leading to extensive injury of hepatic cells and overexpression of inflammatory cytokines [76].

The possible mechanism of immune stress model of PM-DILI. Treatment with a standard dose of P. multiflorum does not cause liver injury, and nontoxic LPS only causes mild immune stress without immune cell infiltration. However, cotreatment can lead to significant immune stress by activating the TLR-NF-κB pathway and inhibiting the PPARγ pathway, which results in increased susceptibility of hepatic cells to components with toxic potential, such as cis-TSG and anthraquinones. In addition, trans-TSG can aggravate liver injury via its immune-enhancing function
Evidence, including stimulation of the TLR-NF-κB pathway, activation of inflammatory cells and increased expression of cytokines and chemokines, has indicated that the innate immune response may be involved in PM-DILI [77]. Although the fact that LPS-mediated mild inflammatory stress can lead to hepatotoxicity of a nontoxic dose of P. multiflorum has referable value, it is still questionable whether the pathogenesis of this model can accurately reflect that of hepatotoxicity in patients with PM-DILI. Is this model just an example of how inflammatory stress induced by a nonhepatotoxic dose of LPS causes a shift in the dose-response curve towards sensitization to the intrinsic hepatotoxicity of P. multiflorum within the therapeutic dose range? No epidemiological evidence has shown that bacteria-infected patients more easily develop PM-DILI. This “inflammagen model” developed using LPS cotreatment does not truly reflect idiosyncratic liver injury induced by P. multiflorum in the clinic, including its delayed onset, but it aims to simulate the clinical phenomenon of patients with immune activation, such as patients with immune diseases, being more susceptible to developing liver injury after taking P. multiflorum. Other host factors, such as adaptive immunity, are likely to better explain the difference in an individual’s susceptibility to immune-mediated idiosyncratic hepatotoxicity induced by P. multiflorum.
Human leukocyte antigen and PM-DILI
The adaptive immune response has been reported to play a key role in inducing inflammation and modulating an individual’s susceptibility to DILI [55]. Multiple clinical phenomena, including delayed-onset liver injury, rapid onset on rechallenge, lymphocyte activation and infiltration, suggest that adaptive immunity may be involved in IDILI induced by P. multiflorum. The idiosyncratic nature has long been regarded as evidence of a genetic predisposition to hepatotoxicity. Major histocompatibility complex (MHC) molecules, which play a central role in activating T cells, are responsible for initiating the inappropriate immune response that causes IDILI. Growing evidence has revealed that human leukocyte antigen (HLA) polymorphisms are associated with susceptibility to several idiosyncratic adverse reactions, especially IDILI [78]. The possible mechanisms underlying T cell-mediated drug hypersensitivity regulated by HLA have evolved into three hypotheses: the hapten/prohapten model, the pharmacological interaction (P-I) model and the altered peptide repertoire model [79,80,81]. Recently, a polymorphism in an HLA gene, HLA-B*35:01, was identified as a specific biomarker associated with PM-DILI [24]. Despite the small sample size, the case-control study showed that the HLA-B*35:01 allele had high specificity and sensitivity for diagnosing PM-DILI. In a prospective cohort study, HLA-B*35:01 carriers had an 8-fold higher risk of developing a 2-fold elevation of liver enzymes compared with noncarriers, indicating that it might have high value for both diagnosing and predicting PM-DILI. Similar to that of other HLA-associated forms of DILI and drug hypersensitivity, the mechanism of PM-DILI is related to the interaction between antigen-presenting cells (APCs) and T cells. Toxic components, such as emodin, physcion [48,49,50], and their reactive metabolites in the liver may be taken up by APCs, including dendritic cells (DCs); and act as haptens covalently binding to self-proteins to form adducts that are then proteolytically processed in the endoplasmic reticulum to trigger DC maturation. The adducts are presented at the cell surface by HLA-B*35:01 molecules to activate CD8+ T cells by interacting with T cell receptors (TCRs) [82, 83]. Other pathways through which toxic components/metabolites lead to the presentation of an altered peptide repertoire by binding HLA-B*35:01 molecules or through which toxic components/metabolites bind to TCRs to induce T cell activation via interactions with MHC I may be alternative mechanisms. Interactions between APCs and T cells lead to a cytotoxic immune response that damages liver cells and causes the release of toxic components, reactive metabolites, adducts, cytokines and damage-associated molecular patterns (DAMPs) [84], which aggravates the initial injury, forms a vicious cycle, and eventually results in liver injury or even liver failure.
Despite the strong association, the positive predictive value (PPV) of HLA-B*35:01 for PM-DILI is limited (37.5%). Similar PPVs have been observed in other drug-induced adverse reactions, such as the PPV of HLA-B*15:02 for carbamazepine-induced Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) and the PPV of HLA-B*57:01 for abacavir hypersensitivity syndrome and flucloxacillin-induced liver injury. For example, only a very low proportion of HLA-B*15:02 carriers (1%–5%) were shown to develop SJS/TEN [85], and 55% of HIV-infected HLA-B*57:01 individuals exposed to abacavir were shown to be tolerant to hypersensitivity reactions [86]. Additionally, 4.7% (3/64) of noncarriers of HLA-B*35:01 showed elevated levels of liver enzymes even after P. multiflorum treatment. These results suggest that there are factors other than MHC molecules that contribute to the pathogenesis of PM-IDILI, although HLA risk alleles may play a critical role in this pathogenesis.
The role of immune tolerance
Our recent prospective cohort study showed that 5 out of 8 HLA-B*35:01 carriers did not experience liver enzyme elevation after taking P. multiflorum, indicating that immune tolerance may be involved in this phenomenon. The liver is an immune-privileged organ with a strong capacity for immune tolerance [87]. Clinical adaptation and tolerance have been observed in many cases of idiosyncratic drug reactions, including IDILI [86, 88, 89]. Although many HLA alleles show strong associations with DILI, MHC molecules are not the only determinants of T cell-mediated adaptive immune response activation. Immune checkpoint molecules, regulatory immune cells and some other factors may also participate in the process of immune activation and tolerance, which contributes to the pathogenesis of DILI (Fig. 2). Despite the somewhat speculative nature of this evidence and the lack of more clinical proof, this mechanism has been demonstrated in some animal models and probably exists in humans.

Possible mechanisms of immune tolerance in DILI. Immune checkpoint molecules (PD-1, CTLA-4, etc.) negatively regulate the immune response mediated by antigen presentation (e.g., reactive drugs and metabolite hapten), resulting in immune tolerance. Immune tolerance can also be mediated by the inhibitory effects of immune cells, such as Kupffer cells, Tregs and MDSCs, on the maturation, differentiation, proliferation and cytokine production of APCs and T cells
The fact that most individuals with genetic HLA predisposition do not develop overt DILI indicates the subtle balance between immune activation and tolerance. The recognition of the peptide-MHC complexes of antigen-presenting cells (APCs) and hepatocytes by T cells refers to signal 1, and the interaction between immune checkpoint molecules refers to signal 2. Both signals are required for the immune response, and a weak immune response and low immune tolerance are likely induced without signal 2 [11, 90, 91]. Coinhibitory molecules, such as programmed cell death protein 1 (PD-1) and cytotoxic T lymphocyte associate protein 4 (CTLA-4), also negatively regulate the immune response via their immunosuppressive effects. Several studies have successfully produced animal models with impaired immune tolerance that are very similar to humans with IDILI by using PD-1−/− mice and anti-CTLA-4 antibodies. These models of impaired immune tolerance have been applied in a number of drug-induced hepatotoxicity reactions in mice and have not only unmasked the role of various immune cells in the immune process of IDILI but also determined the potential ability of drugs to cause IDILI [92,93,94,95]. Notably, immune checkpoint inhibitors (ICIs) can cause an imbalance in the immune system, leading to immune-related hepatitis, which mimics autoimmune conditions [96]. Several ICIs have recently been reported to induce DILI triggered by disrupted immune tolerance in cancer patients receiving immunotherapies [97,98,99], which supports the observations made in animal models of impaired tolerance. In addition to signal 2 molecules, some innate immune factors also contribute to immune homeostasis in DILI. HLA-B*57:01 transgenic mice administered abacavir do not show elevated levels of liver enzymes unless they are cotreated with CpG oligodeoxynucleotide, a TLR9 agonist, to overcome immune tolerance. This finding provides an explanation for why other infections may render most HIV patients carrying the HLA-B*57:01 allele more susceptible to abacavir-induced liver injury.
The functional status that depends on the balance between the activation and tolerance of immunocytes in the liver plays a vital role in regulating the immune response in the liver [69]. Some immune cells also regulate the process of immune tolerance. The effect of Tregs on tolerance in idiosyncratic drug adverse reactions has drawn growing attention in recent years. A recent study created a novel transgenic HLA-B*57:01 mouse model to overcome tolerance to abacavir hypersensitivity reactions induced by CD4+ T cell (probably CD4+ Treg) depletion, which results in the maturation of dendritic cells (DCs) and the induction of drug-hypersensitive effector CD8+ T cells [100]. Although HLA-B*57:01 was identified as a risk factor for flucloxacillin-induced liver injury, CD8+ T cells from both carrier and noncarrier donors can be primed with the drug [101]. Flucloxacillin-induced liver injury has been successfully induced in CD4+-deficient mice [102], which is an interesting model that shows the possible immunosuppressive effect of CD4+ T cells on flucloxacillin-induced immune-mediated liver injury in humans. The immunosuppressive cytokine IL-10, which is secreted by Tregs and Kupffer cells, as well as the interaction between Tregs and Kupffer cells, is very important for tolerance induction in toxin-induced liver injury in mice [103, 104]. The increase in Tregs in mice treated with amodiaquine prevents animals from progressing from mild to severe drug-induced liver injury [94]. The findings observed in mouse models were consistent with the observations made in humans with isoniazid-induced mild liver injury [105, 106]. The components of P. multiflorum show immunomodulatory effects, upregulating IL-10 levels in many in vivo models, including PM-DILI [43, 107,108,109], which provides hints on the possible mechanisms associated with P. multiflorum tolerance in humans. Myeloid-derived suppressor cells (MDSCs) act as immunosuppressive regulators involved in immune tolerance [110]. Halothane is an anesthetic drug that can induce self-limiting ALT elevations or severe liver injury. MDSCs show a significant immunosuppressive effect on T cells in halothane-treated mice. The depletion of MDSCs by anti-Gr1 antibodies prior to initial halothane treatment impairs immune tolerance, resulting in the development of halothane-induced allergic hepatitis via the CD4+ T cell-mediated immune response in mice [111]. Interfering with the functions of immune checkpoint molecules as well as regulatory immune cells seems to be a general model of IDILI; thus, its application in the study of the immune mechanisms of PM-DILI is worth considering.
Recently, the rs2476601 polymorphism in the protein tyrosine phosphatase nonreceptor type 22 (PTPN22) gene, which reduces the immune tolerance of T cells to promote autoimmunity, was identified to be associated with liver injury induced by multiple drugs. In European populations, rs2476601 doubles the risk of amoxicillin/clavulanic acid-induced liver injury among individuals with HLA-A*02:01 and HLA-DRB1*15:01 [112]. Although this allele may not be related to HLA-B*35:01-linked P. multiflorum tolerance due to its absence in Asian people, it may be valuable in Western countries. Polymorphisms in the PD-1 gene and CTLA- 4 gene, such as rs2227981, rs2227982 and rs231775, which do not suppress the immune response, can be considered potential targets, although no report has investigated susceptibility to PM-DILI. In addition, these animal models provide a framework for future studies on the effect of alterations in immune activation and tolerance on PM-DILI.
Conclusion and future perspective
Idiosyncratic drug-induced liver injury is a result of complex interactions between drugs/reactive metabolites and the host immune response. P. multiflorum-induced hepatotoxicity is considered immune-mediated idiosyncratic liver injury. Although in vitro and in vivo studies have indicated that the features of typical intrinsic liver injury only appear when an overdose of P. multiflorum is administered, most patients that develop DILI take standard doses of P. multiflorum. Therefore, it is reasonable to suppose that the intrinsic process only induces mild cellular stress and occurs at the early stage in the pathogenesis of PM-DILI. Both innate immunity and adaptive immunity are involved in the development and severity of PM-DILI, and an individual’s adaptive immune homeostasis is likely to play a more crucial part in the individual’s susceptibility to PM-DILI.
The mechanisms of PM-DILI are multifactorial. Interactions between drug and host factors determine the pathogenesis of PM-DILI. An overall hypothesis of the possible mechanisms of PM-DILI is proposed in this section (Fig. 3). P. multiflorum-derived anthraquinones, such as emodin and physcion, and their reactive metabolites can directly induce apoptosis of hepatic cells, cause cellular dysfunction and stress by chemically reacting with macromolecules and glutathione and damage mitochondria by inhibiting canalicular bile acid efflux transporters [113, 114], leading to hepatocyte stress. In most cases, when a normal clinical dose is consumed, the liver regeneration and repair systems maintain the normal structure and functions of liver cells that are exposed to low concentrations of toxic components, protecting the liver from injury induced by P. multiflorum. However, in some cases of overdose, hepatocyte stress progresses to massive hepatic cell death in a dose-dependent manner due to excessive exposure to toxic components, which reflects the intrinsic characteristics of PM-DILI observed in in vitro and in vivo models. On the other hand, the release of danger signals, such as damage-associated molecular patterns molecules (DAMPs) (e.g., HMGB1 [115] and HSP [116]) from injured hepatocytes, are recognized by TLRs located on immune cells in the liver, such as Kupffer cells, inducing the innate immune response through the activation and recruitment of inflammatory cells and the production of chemokines and cytokines. The inflammatory response promotes cell death and aggravates initial injury. This innate immune pathway of PM-DILI is supported by the endotoxin-mediated inflammatory stress model. Recently, HLA-restricted T cell responses indicated that adaptive immunity may dictate the risk of liver injury induced by P. multiflorum. P. multiflorum activates the immune response in susceptible individuals with the HLA-B*35:01 allele through the hapten pathway, the P-I pathway or altered peptide repertoire. The interaction between APCs/hepatocytes and drug-specific T cells via signal 1 and signal 2, the danger signals from injured cells, and the effects of regulatory immune cells and their cytokines are involved in the regulation of adaptive immune activation and tolerance, which provides a possible explanation for the interindividual differences in the immune response to PM-DILI among HLA-B*35:01 carriers. In addition, it is worth determining whether B cells also play a role in the immune pathogenesis of PM-DILI. B cells can also act as antigen-presenting cells to activate specific T cells. The cytotoxic immune process can trigger inflammation or the production and release of danger signals from injured hepatocytes and activate CD4+ helper T cells, leading to the stimulation of B cells, which can generate antibodies and form memory cells [84, 117]. A range of B cell-derived antidrug antibodies and autoantibodies has been detected in patients with liver injury induced by drugs, such as halothane and isoniazid [118, 119]. Thus, it would be helpful to determine the possible effects of B cells PM-DILI by detecting whether specific antibodies exist in patients with liver injury induced by P. multiflorum. Other HLA alleles, such as DRB5*01:01 and DRB5*01:02, which have been reported to be associated with DILI induced by amoxicillin/clavulanate [120], are also likely to be risk alleles for PM-DILI in larger sample sizes, which means that CD4+ T cells may also participate in the immune mechanism of PM-DILI if these MHC II alleles are proven to be associated with PM-DILI. In addition, polymorphisms in genes related to immunity and tolerance, including PD-1 and CTLA-4, are worth considering for increasing the predictive value of HLA-B*35:01. Pharmacogenomics is still a promising tool for studying interindividual differences in the drug response to P. multiflorum-induced hepatotoxicity.

Hypothesis of the pathogenesis of PM-DILI. Some components of P. multiflorum, such as emodin, and/or its reactive metabolites, cause cell stress and dysfunction by inducing oxidative stress, binding to cellular proteins and inhibiting bile transport. a In most cases, liver regeneration and repair systems prevent hepatocytes from being damaged by cell stress when the concentration of toxic components/metabolites in the liver is low. However, the continuous accumulation and overdose exposure to toxic components/reactive metabolites in hepatocytes result in massive hepatic cell death in a dose-dependent manner. This intrinsic feature of PM-DILI has been observed in cell and animal models. b The danger signals (e.g., DAMPs) released from the stressed hepatocytes are recognized by TLRs located on the immune cells in the liver, such as Kupffer cells, inducing an innate immune response through the activation and recruitment of inflammatory cells and the production of chemokines and cytokines. This inflammatory response is also possibly enhanced by trans-TSG in P. multiflorum. This inflammatory process results in hepatic cell death, adding further damage to the initial injury of liver cells. c Adaptive immunity plays a major role in the pathogenesis of idiosyncratic DILI of P. multiflorum, which is HLA-associated and only occurs in individuals with the susceptible HLA allele HLA-B*35:01. The combined action of signal 1 (interaction of TCRs and peptide-MHC complexes), signal 2 (immune checkpoint molecules) and regulatory immune cells contributes to the adaptive immune response in the pathogenesis of PM-DILI. In addition, danger signals, inflammatory cells and cytokines also contribute to immune homeostasis. The state of balance between immune activation and tolerance determines the clinical outcomes. Some individuals with susceptible HLA alleles who develop immune tolerance show mild or no injury, while others experience overt and severe liver injury because of immune activation
The mechanisms of innate and adaptive immunity in PM-DILI and the components that cause the immune response are still unclear. Moreover, other host factors that may participate in the immune response to PM-DILI need to be discovered and elucidated. For example, the gut microbiome may have an important influence. On one hand, the microbiome may metabolize the ingredients of P. multiflorum into toxic forms, resulting in increased exposure of the liver to toxins [121]. On the other hand, the microbiome shapes immunity and tolerance by engaging in crosstalk with the immune system [122]. For instance, repeated exposure to low concentrations of pathogen-associated molecular patterns (PAMPs) results in immune tolerance [123].
Current knowledge of drug–host interactions in PM-DILI is still limited. More valuable biomarkers need to be discovered and validated for accurate PM-DILI diagnosis and prediction. Appropriate immune-mediated models, particularly transgenic mouse models, must be established to investigate the mechanisms of the idiosyncratic hepatotoxicity of P. multiflorum and the toxic components of P. multiflorum [124]. All of these efforts will enable the development of personalized medicine for the prediction, diagnosis and treatment of P. multiflorum-induced liver injury.
References
Navarro VJ, Khan I, Bjornsson E, Seeff LB, Serrano J, Hoofnagle JH. Liver injury from herbal and dietary supplements. Hepatology. 2017;65:363–73.
Shen T, Liu YX, Shang J, Xie Q, Li J, Yan M, et al. Incidence and etiology of drug-induced liver injury in mainland China. Gastroenterology. 2019;156:2230–41.
Lin L, Ni B, Lin H, Zhang M, Li X, Yin X, et al. Traditional usages, botany, phytochemistry, pharmacology and toxicology of Polygonum multiflorum Thunb.: a review. J Ethnopharmacol. 2015;159:158–83.
Liu Y, Wang Q, Yang JB, Guo XH, Liu WX, Ma SC, et al. Polygonum multiflorum Thunb.: a review on chemical analysis, processing mechanism, quality evaluation, and hepatotoxicity. Front Pharmacol. 2018; 9: 364.
Park GJH, Mann SP, Ngu MC. Acute hepatitis induced by Shou-Wu-Pian, a herbal product derived from Polygonum multiflorum. J Gastroenterol Hepatol. 2001;16:115–7.
Battinelli L, Daniele C, Mazzanti G, Mastroianni CM, Lichtner M, Coletta S, et al. New case of acute hepatitis following the consumption of Shou Wu Pian, a Chinese herbal product derived from Polygonum multiflorum. Ann Intern Med. 2004;140:E589–E90.
Jung KA, Min HJ, Yoo SS, Kim HJ, Choi SN, Ha CY, et al. Drug-induced liver injury: twenty five cases of acute hepatitis following ingestion of Polygonum multiflorum Thunb. Gut Liver. 2011;5:493–9.
Yang JB, Liu Y, Wang Q, Ma SC, Wang AG, Cheng XL, et al. Characterization and identification of the chemical constituents of Polygonum multiflorum Thunb. by high-performance liquid chromatography coupled with ultraviolet detection and linear ion trap FT-ICR hybrid mass spectrometry. J Pharm Biomed Anal. 2019;172:149–66.
Hunt CM. Mitochondrial and immunoallergic injury increase risk of positive drug rechallenge after drug-induced liver injury: a systematic review. Hepatology. 2010;52:2216–22.
Boelsterli UA, Lee KK. Mechanisms of isoniazid-induced idiosyncratic liver injury: emerging role of mitochondrial stress. J Gastroenterol Hepatol. 2014;29:678–87.
Mak A, Uetrecht J. Immune mechanisms of idiosyncratic drug-induced liver injury. J Clin Transl Res. 2017;3:145–56.
Wei Y, Liu M, Liu J, Li H. Influence factors on the hepatotoxicity of polygoni multiflori radix. Evid Based Complement Alternat Med. 2019;2019:5482896.
Lai X, Wu J, Chen S, Lai P, Wang X, Wang Y, et al. Risk factors analysis and security application discussion of Polygonum multiflorum based on retrospective study. China J Chin Mater Med. 2018;43:3205–10.
Dong HH, Slain D, Cheng JC, Ma WH, Liang WF. Eighteen cases of liver injury following ingestion of Polygonum multiflorum. Complement Ther Med. 2014;22:70–4.
Wang JB, Ma ZJ, Niu M, Zhu Y, Liang QS, Zhao YL, et al. Evidence chain-based causality identification in herb-induced liver injury: exemplification of a well-known liver-restorative herb Polygonum multiflorum. Front Med-Prc. 2015;9:457–67.
Lei X, Chen J, Ren J, Li Y, Zhai J, Mu W, et al. Liver damage associated with Polygonum multiflorum Thunb.: a systematic review of case reports and case series. Evid Based Complement Altern Med. 2015;2015:459749.
Committee CP. Pharmacopoeia of the People’s Republic of China. Beijing: China Medical Science and Technology Press; 2015.
Zhu Y, Liu S, Wang J, Song H, Li Y, He T, et al. Clinical analysis of drug-induced liver injury caused by Polygonum multiflorum and its preparations. Chin J Integr Tradit West Med (Chin). 2015;35:1442–7.
Panis B, Wong DR, Hooymans PM, De Smet PA, Rosias PP. Recurrent toxic hepatitis in a Caucasian girl related to the use of Shou-Wu-Pian, a Chinese herbal preparation. J Pediatr Gastr Nutr. 2005;41:256–8.
Shao YL, Zhang SC, Wu JM, Guo FC, Huang ZY, Liu LG. Clinical features of drug-induced liver injury rechallenge with Polygonum multiflorum. Chin J Hepatol. 2018;26:686–9.
Wang J, Li C, Zhu Y, Song H, Bai Z, Xiao X. Integrated evidence chain-based identification of Chinese herbal medicine-induced hepatotoxicity and rational usage: exemplification by Polygonum multiflorum (He shou wu). Chin Sci Bull. 2016;61:971–80.
Cardenas A, Restrepo JC, Sierra F, Correa G. Acute hepatitis due to Shen-Min - A herbal product derived from Polygonum multiflorum. J Clin Gastroenterol. 2006;40:629–32.
Chun-Yu L, Qin H, Dan G, Rui-Yu L, Yun Z, Hui-Fang L, et al. Idiosyncratic drug-induced liver injury linked to Polygonum multiflorum: a case study by pharmacognosy. Chin J Integr Med. 2017;23:625–30.
Li CP, Rao T, Chen XP, Zou ZS, Wei AW, Tang JF, et al. HLA-B*35:01 allele is a potential biomarker for predicting polygonum multiflorum-induced liver injury in humans. Hepatology. 2019;70:346–57.
Ma J, Zheng L, He YS, Li HJ. Hepatotoxic assessment of Polygoni Multiflori Radix extract and toxicokinetic study of stilbene glucoside and anthraquinones in rats. J Ethnopharmacol. 2015;162:61–8.
Yu J, Xie J, Mao XJ, Wang MJ, Li N, Wang J, et al. Hepatoxicity of major constituents and extractions of Radix Polygoni Multiflori and Radix Polygoni Multiflori Praeparata. J Ethnopharmacol. 2011;137:1291–9.
Roth RA, Ganey PE. Intrinsic versus idiosyncratic drug-induced hepatotoxicity-two villains or one? J Pharmacol Exp Ther. 2010;332:692–7.
Zhang R, Zhang C, Sun Z, Deng Q. Damage effect of Polygonum multiflorum fractions on human normal liver cells L02 and liver cancer cells HepG2. China J Chin Mater Med. 2012;37:1830–5.
Zhang RC, Liu B, Sun ZX, Xu DY. Effects of extract of Polygonum multiflorum on cell cycle arrest and apoptosis of human liver cell line L02. Chin J Integr Med. 2010;8:554–61.
Lin LF, Lin HM, Zhang M, Ni BR, Yin XB, Qu CH, et al. A novel method to analyze hepatotoxic components in Polygonum multiflorum using ultra-performance liquid chromatography-quadrupole time-of-flight mass spectrometry. J Hazard Mater. 2015;299:249–59.
Han LF, Wang P, Wang YL, Zhao QY, Zheng F, Dou ZY, et al. Rapid discovery of the potential toxic compounds in Polygonum multiflorum by UHPLC/Q-Orbitrap-MS-based metabolomics and correlation analysis. Front Pharmacol. 2019; 10:329.
Li HL, Wang XB, Liu Y, Pan DF, Wang Y, Yang NA, et al. Hepatoprotection and hepatotoxicity of Heshouwu, a Chinese medicinal herb: context of the paradoxical effect. Food Chem Toxicol. 2017;108:407–18.
Li DK, Chen J, Ge ZZ, Sun ZX. Hepatotoxicity in rats induced by aqueous extract of polygoni multiflori radix, root of Polygonum multiflorum related to the activity inhibition of CYP1A2 or CYP2E1. Evid Based Complement Alternat Med. 2017;2017:9456785.
Wang T, Wang J, Jiang Z, Zhou Z, Li Y, Zhang L, et al. Study on hepatotoxicity of aqueous extracts of Polygonum multiforum in rats after 28-day oral administration-analysis on correlation of cholestasis. China J Chin Mater Med. 2012;37:1445–50.
He Y, Song M, Wang W, Lin P, Li Y, Gu W, et al. Chronic toxicity of both raw and processed Polygoni Multiflori Radix on rats. J Chin Pharmacol Sci. 2016;25:46–56.
Noda T, Yamada T, Ohkubo T, Omura T, Ono T, Adachi T, et al. Hot-water-extracts of Polygonum multiflorum do not induce any toxicity but elicit limited beneficial effects on the liver in mice. J Health Sci. 2009;55:720–5.
Ma ZJ, Li XF, Lv Y, Jiang BQ, Zhao YL, Zhang YM, et al. Comparative study on preparation of Polygoni Multiflori Radix based on hepatotoxic bioassay. China J Chin Mater Med. 2015;40:2325–9.
Lv GP, Meng LZ, Han DQ, Li HY, Zhao J, Li SP. Effect of sample preparation on components and liver toxicity of Polygonum multiflorum. J Pharmacol Biomed. 2015;109:105–11.
Yang JB, Li WF, Liu Y, Wang Q, Cheng XL, Wei F, et al. Acute toxicity screening of different extractions, components and constituents of Polygonum multiflorum Thunb. on zebrafish (Danio rerio) embryos in vivo. Biomed Pharmacother. 2018;99:205–13.
Li CL, Ma J, Zheng L, Li HJ, Li P. Determination of emodin in L-02 cells and cell culture media with liquid chromatography-mass spectrometry: application to a cellular toxicokinetic study. J Pharmacol Biomed Anal. 2012;71:71–8.
Dong X, Fu J, Yin X, Cao S, Li X, Lin L, et al. Emodin: a review of its pharmacology, toxicity and pharmacokinetics. Phytother Res. 2016;30:1207–18.
Wang CF, Wu XD, Chen M, Duan WG, Sun LX, Yan M, et al. Emodin induces apoptosis through caspase 3-dependent pathway in HK-2 cells. Toxicology. 2007;231:120–8.
Zhang L, Liu XY, Tu C, Li CY, Song D, Zhu JX, et al. Components synergy between stilbenes and emodin derivatives contributes to hepatotoxicity induced by Polygonum multiflorum. Xenobiotica 2019:1–11. https://doi.org/10.1080/00498254.2019.1658138.
Lin LF, Liu YL, Fu S, Qu CH, Li H, Ni J. Inhibition of mitochondrial complex function-the hepatotoxicity mechanism of emodin based on quantitative proteomic analyses. Cells-Basel. 2019;8:E263.
Quan Y, Gong L, He J, Zhou Y, Liu M, Cao Z, et al. Aloe emodin induces hepatotoxicity by activating NF-kappaB inflammatory pathway and P53 apoptosis pathway in zebrafish. Toxicol Lett. 2019;306:66–79.
Wu LL, Han WC, Chen YL, Zhang T, Liu JJ, Zhong SL, et al. Gender differences in the hepatotoxicity and toxicokinetics of emodin: the potential mechanisms mediated by UGT2B7 and MRP2. Mol Pharmacol. 2018;15:3931–45.
Wu LL, Chen YL, Liu H, Zhan ZK, Liang Z, Zhang T, et al. Emodin-induced hepatotoxicity was exacerbated by probenecid through inhibiting UGTs and MRP2. Toxicol Appl Pharmacol. 2018;359:91–101.
Qin X, Peng Y, Zheng J. In vitro and in vivo studies of the electrophilicity of physcion and its oxidative metabolites. Chem Res Toxicol. 2018;31:340–9.
Jiang LL, Zhao DS, Fan YX, Yu Q, Li P, Li HJ. Detection of emodin derived glutathione adduct in normal rats administered with large dosage of polygoni multiflori radix. Front Pharmacol. 2017;8:446.
Qin BY, Xu Y, Chen JM, Huang WL, Peng Y, Zheng J. Chemical reactivity of emodin and its oxidative metabolites to thiols. Chem Res Toxicol. 2016;29:2114–24.
Li YX, Gong XH, Liu MC, Peng C, Li P, Wang YT. Investigation of liver injury of polygonum multiflorum Thunb. in rats by metabolomics and traditional approaches. Front Pharmacol. 2017;8:791.
Xia XH, Yuan YY, Liu M. The assessment of the chronic hepatotoxicity induced by Polygoni Multiflori Radix in rats: a pilot study by using untargeted metabolomics method. J Ethnopharmacol. 2017;203:182–90.
Zhang CE, Niu M, Li Q, Zhao YL, Ma ZJ, Xiong Y, et al. Urine metabolomics study on the liver injury in rats induced by raw and processed Polygonum multiflorum integrated with pattern recognition and pathways analysis. J Ethnopharmacol. 2016;194:299–306.
Lin LF, Li H, Lin HM, Zhang M, Qu CH, Yan L, et al. Application of iTRAQ-based quantitative proteomics approach to identify deregulated proteins associated with liver toxicity induced by polygonum multiflorum in rats. Cell Physiol Biochem. 2017;43:2102–16.
Chen MJ, Suzuki A, Borlak J, Andrade RJ, Lucena MI. Drug-induced liver injury: interactions between drug properties and host factors. J Hepatol. 2015;63:503–14.
Fannin RD, Russo M, O’Connell TM, Gerrish K, Winnike JH, Macdonald J, et al. Acetaminophen dosing of humans results in blood transcriptome and metabolome changes consistent with impaired oxidative phosphorylation. Hepatology. 2010;51:227–36.
Harrill AH, Watkins PB, Su S, Ross PK, Harbourt DE, Stylianou IM, et al. Mouse population-guided resequencing reveals that variants in CD44 contribute to acetaminophen-induced liver injury in humans. Genome Res. 2009;19:1507–15.
Deng XM, Luyendyk JP, Ganey PE, Roth RA. Inflammatory stress and idiosyncratic hepatotoxicity: hints from animal models. Pharmacol Rev. 2009;61:262–82.
Barman PK, Mukherjee R, Prusty BK, Suklabaidya S, Senapati S, Ravindran B. Chitohexaose protects against acetaminophen-induced hepatotoxicity in mice. Cell Death Dis. 2016;7:e2224.
Maddox JF, Amuzie CJ, Li M, Newport SW, Sparkenbaugh E, Cuff CF, et al. Bacterial- and viral-induced inflammation increases sensitivity to acetaminophen hepatotoxicity. J Toxicol Environ Health A. 2010;73:58–73.
Zou W, Devi SS, Sparkenbaugh E, Younis HS, Roth RA, Ganey PE. Hepatotoxic interaction of sulindac with lipopolysaccharide: role of the hemostatic system. Toxicol Sci. 2009;108:184–93.
Shaw PJ, Hopfensperger MJ, Ganey PE, Roth RA. Lipopolysaccharide and trovafloxacin coexposure in mice causes idiosyncrasy-like liver injury dependent on tumor necrosis factor-alpha. Toxicol Sci. 2007;100:259–66.
Luyendyk JP, Maddox JF, Cosma GN, Ganey PE, Cockerell GL, Roth RA. Ranitidine treatment during a modest inflammatory response precipitates idiosyncrasy-like liver injury in rats. J Pharmacol Exp Ther. 2003;307:9–16.
Lu J, Jones AD, Harkema JR, Roth RA, Ganey PE. Amiodarone exposure during modest inflammation induces idiosyncrasy-like liver injury in rats: role of tumor necrosis factor-alpha. Toxicol Sci. 2012;125:126–33.
Dugan CM, MacDonald AE, Roth RA, Ganey PE. A mouse model of severe halothane hepatitis based on human risk factors. J Pharmacol Exp Ther. 2010;333:364–72.
Deng X, Stachlewitz RF, Liguori MJ, Blomme EA, Waring JF, Luyendyk JP, et al. Modest inflammation enhances diclofenac hepatotoxicity in rats: role of neutrophils and bacterial translocation. J Pharmacol Exp Ther. 2006;319:1191–9.
Deng X, Luyendyk JP, Zou W, Lu J, Malle E, Ganey PE, et al. Neutrophil interaction with the hemostatic system contributes to liver injury in rats cotreated with lipopolysaccharide and ranitidine. J Pharmacol Exp Ther. 2007;322:852–61.
Li CY, Li XF, Tu C, Li N, Ma Z, Pang J, et al. The idiosyncratic hepatotoxicity of Polygonum multiflorum based on endotoxin model. Acta Pharmacol Sin. 2015;50:28–33.
Fan X, Wang JB, Xie LH, Dong YS, Han G, Hu D, et al. A new animal model for Polygonum multiflorum Thunb-induced liver injury in rats and its potential mechanisms. Toxicol Res. 2015;4:1085–97.
Li CY, Niu M, Bai ZF, Zhang CG, Zhao YL, Li RY, et al. Screening for main components associated with the idiosyncratic hepatotoxicity of a tonic herb, Polygonum multiflorum. Front Med-Prc. 2017;11:253–65.
Meng YK, Li CY, Li RY, He LZ, Cui HR, Yin P, et al. Cis-stilbene glucoside in Polygonum multiflorum induces immunological idiosyncratic hepatotoxicity in LPS-treated rats by suppressing PPAR-gamma. Acta Pharmacol Sin. 2017;38:1340–52.
He LZ, Yin P, Meng YK, Tang JF, He TT, Niu M, et al. Immunological synergistic mechanisms of trans-/cis-stilbene glycosides in Heshouwu-related idiosyncratic liver injury. Sci Bull. 2017;62:748–51.
Tu C, Gao D, Li XF, Li CY, Li RS, Zhao YL, et al. Inflammatory stress potentiates emodin-induced liver injury in rats. Front Pharmacol. 2015;6:233.
Li CY, Tu C, Gao D, Wang RL, Zhang HZ, Niu M, et al. Metabolomic study on idiosyncratic liver injury induced by different extracts of Polygonum multiflorum in rats integrated with pattern recognition and enriched pathways analysis. Front Pharmacol. 2016;7:483.
Tu C, He Q, Li CY, Niu M, Han ZX, Ge FL, et al. Susceptibility-related factor and biomarkers of dietary supplement polygonum multiflorum-induced liver injury in rats. Front Pharmacol. 2019;10:335.
Bai ZF, Meng YK, He LZ, Tang JF, Xiao XH. Immune idiosyncratic liver injury induced by traditional non-toxic traditional Chinese medicine and a hypothesis of its mechanism. Chin Pharmacol J. 2017;52:1105–9.
Jaeschke H, Naisbitt DJ. Immune mechanisms in drug-induced liver injury. In: Drug-Induced Liver Toxicity. (Springer, 2018), p 511–31.
Kaliyaperumal K, Grove JI, Delahay RM, Griffiths WJH, Duckworth A, Aithal GP. Pharmacogenomics of drug-induced liver injury (DILI): Molecular biology to clinical applications. J Hepatol. 2018;69:948–57.
Yun J, Adam J, Yerly D, Pichler WJ. Human leukocyte antigens (HLA) associated drug hypersensitivity: consequences of drug binding to HLA. Allergy. 2012;67:1338–46.
Redwood AJ, Pavlos RK, White KD, Phillips EJ. HLAs: Key regulators of T-cell-mediated drug hypersensitivity. HLA. 2018;91:3–16.
Uetrecht J, Naisbitt DJ. Idiosyncratic adverse drug reactions: current concepts. Pharmacol Rev. 2013;65:779–808.
Megherbi R, Kiorpelidou E, Foster B, Rowe C, Naisbitt DJ, Goldring CE, et al. Role of protein haptenation in triggering maturation events in the dendritic cell surrogate cell line THP-1. Toxicol Appl Pharmacol. 2009;238:120–32.
Rodriguez-Pena R, Lopez S, Mayorga C, Antunez C, Fernandez TD, Torres MJ, et al. Potential involvement of dendritic cells in delayed-type hypersensitivity reactions to beta-lactams. J Allergy Clin Immun. 2006;118:949–56.
Grove JI, Aithal GP. Human leukocyte antigen genetic risk factors of drug-induced liver toxicology. Expert Opin Drug Metab Toxicol. 2015;11:395–409.
Amstutz U, Shear NH, Rieder MJ, Hwang S, Fung V, Nakamura H, et al. Recommendations for HLA-B*15:02 and HLA-A*31:01 genetic testing to reduce the risk of carbamazepine-induced hypersensitivity reactions. Epilepsia. 2014;55:496–506.
Mallal S, Phillips E, Carosi G, Molina JM, Workman C, Tomazic J, et al. HLA-B*5701 screening for hypersensitivity to abacavir. N Engl J Med. 2008;358:568–79.
Knolle PA, Gerken G. Local control of the immune response in the liver. Immunol Rev. 2000;174:21–34.
Metushi I, Uetrecht J, Phillips E. Mechanism of isoniazid-induced hepatotoxicity: then and now. Br J Clin Pharmacol. 2016;81:1030–6.
Dara L, Liu ZX, Kaplowitz N. Mechanisms of adaptation and progression in idiosyncratic drug induced liver injury, clinical implications. Liver Int. 2016;36:158–65.
Cho T, Uetrecht J. How reactive metabolites induce an immune response that sometimes leads to an idiosyncratic drug reaction. Chem Res Toxicol. 2017;30:295–314.
Uetrecht J. Mechanisms of idiosyncratic drug-induced liver injury. Adv Pharmacol (San Diego, Calif). 2019;85:133–63.
Mak A, Uetrecht J. The combination of anti-CTLA-4 and PD1−/− mice unmasks the potential of isoniazid and nevirapine to cause liver injury. Chem Res Toxicol. 2015;28:2287–91.
Mak A, Cho TE, Uetrecht J. The effects of immune modulators on amodiaquine-induced liver injury. Chem Res Toxicol. 2018;31:739–44.
Metushi IG, Hayes MA, Uetrecht J. Treatment of PD-1(-/-) mice with amodiaquine and anti-CTLA4 leads to liver injury similar to idiosyncratic liver injury in patients. Hepatology. 2015;61:1332–42.
Mak A, Kato R, Weston K, Hayes A, Uetrecht J. An impaired immune tolerance animal model distinguishes the potential of troglitazone/pioglitazone and tolcapone/entacapone to cause IDILI. Toxicol Sci. 2018;161:412–20.
Michot JM, Bigenwald C, Champiat S, Collins M, Carbonnel F, Postel-Vinay S, et al. Immune-related adverse events with immune checkpoint blockade: a comprehensive review. Eur J Cancer. 2016;54:139–48.
Zen Y, Yeh MM. Hepatotoxicity of immune checkpoint inhibitors: a histology study of seven cases in comparison with autoimmune hepatitis and idiosyncratic drug-induced liver injury. Mod Pathol. 2018;31:965–73.
Suzman DL, Pelosof L, Rosenberg A, Avigan MI. Hepatotoxicity of immune checkpoint inhibitors: An evolving picture of risk associated with a vital class of immunotherapy agents. Liver Int. 2018;38:976–87.
De Martin E, Michot JM, Papouin B, Champiat S, Mateus C, Lambotte O, et al. Characterization of liver injury induced by cancer immunotherapy using immune checkpoint inhibitors. J Hepatol. 2018;68:1181–90.
Cardone M, Garcia K, Tilahun ME, Boyd LF, Gebreyohannes S, Yano M, et al. A transgenic mouse model for HLA-B*57:01-linked abacavir drug tolerance and reactivity. J Clin Invest. 2018;128:2819–32.
Monshi MM, Faulkner L, Gibson A, Jenkins RE, Farrell J, Earnshaw CJ, et al. Human leukocyte antigen (HLA)-B*57:01-restricted activation of drug-specific T cells provides the immunological basis for flucloxacillin-induced liver injury. Hepatology. 2013;57:727–39.
Nattrass R, Faulkner L, Vocanson M, Antoine DJ, Kipar A, Kenna G, et al. Activation of flucloxacillin-specific CD8+ T-cells with the potential to promote hepatocyte cytotoxicity in a mouse model. Toxicol Sci. 2015;146:146–56.
Erhardt A, Biburger M, Papadopoulos T, Tiegs G. IL-10, regulatory T cells, and Kupffer cells mediate tolerance in concanavalin A-induced liver injury in mice. Hepatology. 2007;45:475–85.
Breous E, Somanathan S, Vandenberghe LH, Wilson JM. Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology. 2009;50:612–21.
Usui T, Meng XL, Saide K, Farrell J, Thomson P, Whitaker P, et al. Characterization of isoniazid-specific T-cell clones in patients with anti-tuberculosis drug-related liver and skin injury. Toxicol Sci. 2017;155:420–31.
Metushi IG, Zhu X, Chen X, Gardam MA, Uetrecht J. Mild isoniazid-induced liver injury in humans is associated with an increase in Th17 cells and T cells producing IL-10. Chem Res Toxicol. 2014;27:683–9.
Sharma R, Tiku AB. Emodin inhibits splenocyte proliferation and inflammation by modulating cytokine responses in a mouse model system. J Immunotoxicol. 2016;13:20–6.
Tong HF, Chen KJ, Chen H, Wu HY, Lin HD, Ni ZL, et al. Emodin prolongs recipient survival time after orthotopic liver transplantation in rats by polarizing the Th1/Th2 paradigm to Th2. Anat Rec. 2011;294:445–52.
Lin P, Lu JM, Wang YF, Gu W, Yu J, Zhao RH. Naturally occurring stilbenoid TSG reverses non-alcoholic fatty liver diseases via gut-liver axis. PLoS ONE. 2015;10:e0140346.
Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol. 2009;9:162–74.
Chakraborty M, Fullerton AM, Semple K, Chea LS, Proctor WR, Bourdi M, et al. Drug-induced allergic hepatitis develops in mice when myeloid-derived suppressor cells are depleted prior to halothane treatment. Hepatology. 2015;62:546–57.
Cirulli ET, Nicoletti P, Abramson K, Andrade RJ, Bjornsson ES, Chalasani N, et al. A missense variant in PTPN22 is a risk factor for drug-induced liver injury. Gastroenterology. 2019;156:1707.
Tujios S, Fontana RJ. Mechanisms of drug-induced liver injury: from bedside to bench. Nat Rev Gastroenterol Hepatol. 2011;8:202–11.
Kang L, Si L, Rao J, Li D, Wu Y, Wu S, et al. Polygoni Multiflori Radix derived anthraquinones alter bile acid disposition in sandwich-cultured rat hepatocytes. Toxicol Vitr. 2017;40:313–23.
Pang J, Bai Z, Niu M, Tu C, Ma Z, Zhao Y, et al. The toxic and protective effects of Polygonum multiflorum on normal and liver injured rats based on the symptom-based prescription theory. Acta Pharmacol Sin. 2015;50:973–9.
Yang X, Zhang Y, Liu Y, Chen C, Xu W, Xiao H. Emodin induces liver injury by inhibiting the key enzymes of FADH/NADPH transport in rat liver. Toxicol Res (Camb). 2018;7:888–96.
Williams CD, Jaeschke H. Role of innate and adaptive immunity during drug-induced liver injury. Toxicol Res. 2012;1:161–70.
Metushi IG, Sanders C, Acute Liver Study G, Lee WM, Uetrecht J. Detection of anti-isoniazid and anti-cytochrome P450 antibodies in patients with isoniazid-induced liver failure. Hepatology. 2014;59:1084–93.
Satoh H, Martin BM, Schulick AH, Christ DD, Kenna JG, Pohl LR. Human anti-endoplasmic reticulum antibodies in sera of patients with halothane-induced hepatitis are directed against a trifluoroacetylated carboxylesterase. Proc Natl Acad Sci U S A. 1989;86:322–6.
Wuillemin N, Terracciano L, Beltraminelli H, Schlapbach C, Fontana S, Krahenbuhl S, et al. T cells infiltrate the liver and kill hepatocytes in HLA-B*57:01-associated floxacillin-induced liver injury. Am J Pathol. 2014;184:1677–82.
Li H, He J, Jia W. The influence of gut microbiota on drug metabolism and toxicity. Expert Opin Drug Metab Toxicol. 2016;12:31–40.
Wu HJ, Wu E. The role of gut microbiota in immune homeostasis and autoimmunity. Gut Microbes. 2012;3:4–14.
Maddux AB, Douglas IS. Is the developmentally immature immune response in paediatric sepsis a recapitulation of immune tolerance? Immunology. 2015;145:1–10.
Zhang L, Liu X, Tu C, Li C, Song D, Zhu J, et al. Components synergy between stilbenes and emodin derivatives contributes to hepatotoxicity induced by by Polygonum multiflorum. Xenobiotica. 2019;1–11. https://doi.org/10.1080/00498254.2019.1658138.
Acknowledgements
This study was mainly funded by The National Natural Science Foundation of China (81803837) and The Nature Science Foundation of Hu-nan Province (2019JJ50839).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Rao, T., Liu, Yt., Zeng, Xc. et al. The hepatotoxicity of Polygonum multiflorum: The emerging role of the immune-mediated liver injury. Acta Pharmacol Sin 42, 27–35 (2021). https://doi.org/10.1038/s41401-020-0360-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-0360-3
Keywords
This article is cited by
-
Hippophae rhamnoides reverses decreased CYP2D6 expression in rats with BCG-induced liver injury
Scientific Reports (2023)
