
Abstract
With treatment benefits in both the central nervous system and the peripheral system, the medical use of cannabidiol (CBD) has gained increasing popularity. Given that the therapeutic mechanisms of CBD are still vague, the systematic identification of its potential targets, signaling pathways, and their associations with corresponding diseases is of great interest for researchers. In the present work, chemogenomics-knowledgebase systems pharmacology analysis was applied for systematic network studies to generate CBD-target, target-pathway, and target-disease networks by combining both the results from the in silico analysis and the reported experimental validations. Based on the network analysis, three human neuro-related rhodopsin-like GPCRs, i.e., 5-hydroxytryptamine receptor 1 A (5HT1A), delta-type opioid receptor (OPRD) and G protein-coupled receptor 55 (GPR55), were selected for close evaluation. Integrated computational methodologies, including homology modeling, molecular docking, and molecular dynamics simulation, were used to evaluate the protein-CBD binding modes. A CBD-preferred pocket consisting of a hydrophobic cavity and backbone hinges was proposed and tested for CBD-class A GPCR binding. Finally, the neurophysiological effects of CBD were illustrated at the molecular level, and dopamine receptor 3 (DRD3) was further predicted to be an active target for CBD.
Similar content being viewed by others


Introduction
There is a history of >4000 years of medical and recreational uses of cannabis by humans [1]. In addition to the adoption of cannabis by young adults for recreational purposes, the medical use of cannabis has drawn increasing attention from the public. California passed Proposition 215 in 1996, as the first state in the US to legalize the medical use of cannabis. According to “State Medical Marijuana Laws” from the National Conference of State Legislatures published on 9/14/2017, currently 29 states in the US plus the District of Columbia, Guam, and Puerto Rico allow comprehensive public medical marijuana programs.
Delta-9-tetrahydrocannabinol (THC) is the major contributor in cannabis preparations that causes psychoactive effects [2]. THC targets cannabinoid receptors including cannabinoid receptor 1 (CB1) and cannabinoid receptor 2 (CB2) in the brain [3,4]. The psychoactive effects associated with THC include schizophrenia-like positive and negative symptoms, increased anxiety, and euphoria [2]. Unlike THC, cannabidiol (CBD) shows limited affinities towards cannabinoid receptors [5,6,7]. Instead, CBD is the major nonpsychoactive ingredient in cannabis, and studies have shown beneficial effects of CBD in neurological diseases [8]. Recently, Aso et al. [9] showed a therapeutic profile of CBD for treating neurodegenerative diseases in AβPP/PS1 mouse models. Cheng et al. [10] reported that CBD reversed the cognitive deficits of Alzheimer’s Disease transgenic mice. Devinsky [11] studied the relevance of epilepsy and the neuroprotective effects of CBD. Costa [12] demonstrated that CBD alleviated inflammation pain in a rat model.
One of the major research obstacles regarding CBD is the unclear mechanisms behind its promising treatment potentials. Given that CBD has a low-binding affinity towards traditional cannabinoid receptors (CB1 and CB2), the determination of the major active target(s) of CBD is a demanding and challenging task. Great efforts have been devoted towards this area of study by researchers [7,8,13,14,15,16], and further progress is still in urgent demand. In the present work (Fig. 1), chemogenomics-knowledgebase systems pharmacology analysis was performed for CBD. Both the data from the in silico results and the reported experimental validations were combined to generate a network of CBD targets. Pathway analysis was performed to bridge the gap between the targets and specific diseases. Integrated computational methodologies, including homology modeling, molecular docking, and molecular dynamics simulation were then used to evaluate the protein-CBD binding modes. According to the network analysis, three human neuro-related rhodopsin-like (class A) G protein-coupled receptors (GPCRs), 5-hydroxytryptamine receptor 1A (UniProt ID: 5HT1A_HUMAN or 5HT1A as an abbreviation), delta-opioid receptor (UniProt ID: OPRD_HUMAN or OPRD as an abbreviation), and G protein-coupled receptor 55 (UniProt ID: GPR55_HUMAN or GPR55 as an abbreviation), were closely evaluated. A hydrophobic cavity and backbone hinge model was proposed and tested for CBD-class A GPCR binding. Dopamine receptor 3 was further predicted to be a potential target for CBD.

A schematic representation of the workflow in this study
Materials and methods
Chemogenomics databases and tools
Chemogenomics databases including the Cannabinoid Ligand Database (http://cbligand.org/cbid), Chemogenomics Database for Alzheimer’s Disease [17] (http://cbligand.org/AD/), and Chemogenomics Database for Drug Abuse Research [18] (http://cbligand.org/CDAR/) were used for target prediction. Specifically, the incorporated chemogenomics tools used for CBD target mapping were the integrated TargetHunter [19] and HTDocking programs. TargetHunter is an in silico target identification tool for predicting therapeutic targets and off-targets of small molecules. The inquiry compound was cannabidiol (2-[(1R,6R)-6-isopropenyl-3-methylcyclohex-2-en-1-yl]-5-pentylbenzene-1,3-diol), with R confirmation for both chiral centers exhibited by CBD extracted from the marijuana plant. The fingerprint type was set as Extended-connectivity fingerprints 6 (ECFP6) [20]. The threshold for 2D similarity was set to be 60%. HTDocking is an in silico algorithm to perform high-throughput molecular docking studies between a compound and the protein targets in the chemogenomics databases with available crystal structures. CBD was the inquiry compound, and its affinity towards a total of 607 protein targets was screened and evaluated.
Cytoscape 3.5.1 [21] was used for constructing the networks by combining the in silico results and the reported experimental validations. The layout format was set to the Degree Sorted Circle layout.
Homology modeling
Modeller 9.19 [22,23] was used for building protein models for GPR55 and 5HT1A given that no crystal structures were available for them. A multiple template-based modeling module was adopted with loop refinement. Templates were selected based on three criteria, E-values [24], sequence similarity, and resolution. The conserved residues were marked according to the Ballesteros–Weinstein numbering system. The constructed models were evaluated with the Discrete Optimized Protein Energy (DOPE) [25] measurement and Ramachandran plot [26]. DOPE reflects the energy profile of each individual residue, making a comparison between the templates and the target easier to perform. The Ramachandran plot measures the dihedral angles ψ against φ of amino acid residues on the backbone structure of the protein. The Ramachandran plot was created through the web server RAMPAGE (http://mordred.bioc.cam.ac.uk/~rapper/rampage.php) [27].
Protein sequence information for target proteins GPR55 and 5HT1A was acquired from UniProt (http://www.uniprot.org) [28]. For GPR55, residues 1–10 and 299–319 were truncated to reduce the length of irrelevant loops on both the extracellular and intracellular parts. For 5HT1A, residues 1–30, 220–340, and 407–422 were truncated to reduce the length of irrelevant loops. The crystal structures of human OPRD (PDB entry: 4N6H; resolution, 1.8 Å; method, X-ray diffraction) [29], human dopamine D3 receptor (PDB entry: 3PBL; resolution, 2.89 Å; method, X-ray diffraction) [30], human adenosine A2A receptor (PDB entry: 5IU4; resolution, 1.72 Å; method, X-ray diffraction) [31], and human CXCR chemokine receptor type 4 (PDB entry: 3ODU; resolution, 2.5 Å; method, X-ray diffraction) [32], as well as 11 template protein structures for building GPR55 and 5HT1A models, were downloaded from the Protein Data Bank (http://www.rcsb.org). SYBYL-X 1.3 was used for the preparation of the crystal structure, including energy minimization and residue repair. SYBYL-X 1.3 and PyMol (http://www.pymol.org) were used for molecular visualization, structural superimposition, and data analysis following our usual operation routine [33].
Molecular docking between CBD and GPR55, 5HT1A, OPRD1, DRD3, ADORA2A, and CXCR4 proteins
Molecular docking studies were performed using Surflex-Dock GeomX, the suite implemented in SYBYL-X 1.3. A conserved orthosteric binding pocket among rhodopsin-like GPCRs was defined by selecting corresponding surrounding residues. The Kollman all atom approach was used to calculate atomic charges for the protein [34] and the Gasteiger–Hückel approach for the ligand [35] following our usual operation routine [33]. The hydrogen atoms of the protein were allowed to move. The number of additional starting conformations per molecule was set to 20. The distance to expand search grid was set to 6 Å. The maximum number of poses per ligand was set to 200, and the minimum RMSD between final poses was set to 0.05 Å. The optimized pose was selected and reported based on six criteria, hydrophobic (positive atomic contacts and atomic interpenetration), polar (hydrogen bonds and salt bridges), repulsive (unfavorable polar contacts), entropic (loss of translational and rotational entropy), solvation (difference between the potential and actual numbers of hydrogen bond), and crash (inappropriate penetration into the protein). The naltrindole was first extracted from the OPRD-naltrindole crystal structure (PDB entry: 4N6H) and then docked back to the binding pocket to function as a control to validate the docking algorithm. The naltrindole fits well inside the binding pocket (Supplementary Figure 1). The hydrogen bonds with ASP128 and TYR129 were able to recur, as well as the hydrophobic interactions towards VAL281 and TRP274. There was a good overlap between the docking result and the crystallized complex with the RMSD of ~ 0.2 Å, indicating a reliable docking process.
Enrichment test for GPR55 and 5HT1A models
Approximately 500 compounds were randomly selected from Clean Drug-like Zinc database (http://zinc.docking.org) [36]. In total 10 active compounds for the corresponding targets were downloaded from ChEMBL (https://www.ebi.ac.uk/chembl/) [37]. Random compounds and active compounds were mixed together to be screened using the homology protein models of GPR55 and 5HT1A. Surflex-Dock Screen, the suite implemented in SYBYL-X 1.3, was used for the in silico screening. The system settings were the same as those in the previous section except for the following: the movements of hydrogen atoms of the protein were not allowed; no additional starting conformation was allowed for the ligand; the distance to expand the search grid was set to 6 Å; the maximum number of poses per ligand was set to 3; the minimum RMSD between final poses was set to 0.05 Å.
Molecular dynamics simulation
The Membrane builder in CHARMM-GUI [38,39] was used for building up the CBD-protein-membrane complex. The ratio for DOPC: DOPE: CHL was set to 2:2:1 following the established simulation membrane systems [40,41,42]. The membrane was placed on the X–Y plane, and the Z axis was normal to the membrane. The water thickness was at least 17.5 Å on the top and bottom of the system. Na+ and Cl− ions were added to make a 0.15 M ion concentration. The whole system for the CBD-GPR55 complex included the GPR55 homology model, CBD, 80 DOPC molecules, 79 DOPE molecules, 41 CHL molecules, 13512 water molecules, 36 Na+ ions, and 42 Cl− ions. The size of the box for this system was ~85 × 85 × 103 Å3. The disulfide bond between CYS84 and CYS158 was specified. The whole system for the CBD-5HT1A complex included the 5HT1A homology model, CBD, 82 DOPC molecules, 80 DOPE molecules, 40 CHL molecules, 12317 water molecules, 33 Na+ ions, and 35 Cl− ions. The size of the box for this system was ~85 × 85 × 107 Å3. The disulfide bond between CYS79 and CYS157 was specified. The whole system for the CBD–OPRD complex included the crystal OPRD model, CBD, 82 DOPC molecules, 81 DOPE molecules, 41 CHL molecules, 14633 water molecules, 39 Na+ ions, and 55 Cl− ions. The size of the box for this system was ~86 × 86 × 107 Å3. The disulfide bond between CYS121 and CYS198 was specified.
The AMBER ff14SB force field [43] was applied to proteins, and the AMBER Lipid17 force field [44] was applied to lipids. Water molecules were treated with the TIP3P water model [45]. The partial atomic charges of ligands were derived by the restrained electrostatic potential (RESP) method [46] to fit the HF/6-31G* electrostatic potentials generated using the GAUSSIAN 16 software package [47]. The other force field parameters came from GAFF in AMBER16 [48]. The residue topologies for ligands were prepared using the ANTECHAMBER module [49].
The MD simulations were carried out using the PMEMD.mpi and PMEMD.cuda modules in the AMBER16 [50,51,52] package. First, the systems were relaxed to remove possible steric crashes by a set of minimization steps. The water and ions were relaxed first, followed by the lipids and then the protein and ligand complex. The harmonic restraint force constants decreased from 20 to 10, 5, and 1 kcal/mol/Å2, progressively. Finally, the whole systems were further minimized without any restraints. After the minimization stages, each system was gradually heated from 0 K to 300 K at the heating stages, and then the temperature was kept at 300 K at the following equilibrium and production stages. A time step of 1 fs was used for the heating and the first part of equilibrium and 2 fs for the rest of equilibrium and the following production stages. The periodic boundary condition was employed to produce the constant temperature and pressure (NPT) ensembles. The pressure was set at 1 atm and was controlled by the anisotropic (x-, y-, z-) pressure scaling protocol applied in AMBER [50] with a pressure relaxation time of 1 ps. The temperature was regulated using Langevin dynamics with the collision frequency of 5 ps−1 [53,54]. The Particle Mesh Ewald (PME) method [55,56] was adopted to handle the long-range electrostatics, and a 10 Å cutoff was set to treat real-space interactions. All the covalent bonds involving hydrogen atoms were constrained with the SHAKE algorithm [56]. The simulations time for each system was 200 ns, and the coordinates of simulated systems were saved every 0.1 ns.
Results
CBD target mapping
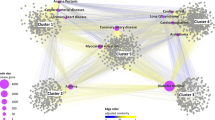
Three chemogenomics databases and two chemogenomics tools, TargetHunter and HTDocking, were used here for retrieving the information for protein targets as detailed in the Materials and Methods. Twenty-seven targets were predicted to be associated with CBD, as shown in Fig. 2. The size of each balloon represents the predicted affinity of CBD towards the corresponding target. The larger the size, the stronger the predicted affinity. Literature reviews were used to distinguish known, experimentally identified, and purely predicted targets [5,7,8,15,16,57,58,59,60,61,62,63,64]. Table 1 gives the annotation for the abbreviated protein representatives, the activity data from the literature for known compounds, and the information source for these targets. Bilin-binding protein (BBP), beta-amylase (AMYB), UDP-N-acetylglucosamine 1-carboxyvinyltransferase (MURA), dihydropteroate synthase (DHPS), and neuraminidase (NRAM) were identified as protein targets for non-human species. CBD has effects in numerous species, but the effects on the human body remain the major focus of this paper. Therefore, the following pathway and disease analysis exclude the non-human targets.

Chemogenomics-based targets mapping for CBD. The size of the balloons is correlated with the predicted affinity between the target and CBD. Balloons with pink color represent the purely predicted targets. Balloons with green color represent the experimentally validated targets. Balloons with yellow color represent the known targets
Signaling pathways and processes associated with the targets were analyzed to bridge the gap between the CBD targets and the involvement in specific diseases (Fig. 3). As a result, a total of 33 nodes of pathways or processes were found to be connected with the targets through 62 edges. The network exhibits polypharmacology of CBD, which is consistent with the wide spectrum of physiological responses that can be triggered by CBD. Specific signaling is specified for immune response and inflammation signals if possible. Nine pathways or processes with three or more edges connected are placed in the center for emphasis. Four of them (cannabinoid signaling, nicotine signaling, GPCR signaling, and neurophysiological process) are associated with neuro-related normal functions or disorders. CNS regulation can be a promising application of CBD. On the other hand, neurological side effects can be a major concern for CBD treatment.

CBD target-pathway network. Blue nodes represent the targets for CBD identified through target mapping. Yellow and orange nodes represent signaling pathways or processes. In particular, orange nodes are shared by three or more targets and placed in the center for emphasis. Targets are connected with pathways or processes with edges in the corresponding color
A target-disease network was constructed with the bridge of signaling pathways and processes (Fig. 4). In total, 25 nodes of diseases were connected with targets through 62 edges. Specific cancer types are detailed for carcinoma if possible. Diseases in both the peripheral and in CNS were observed. Diseases range from cardiovascular and inflammatory diseases to cancer, neuro-related diseases, etc. Neuro-related diseases are placed in the center for emphasis. These diseases are mainly associated with delta-type opioid receptor (OPRD), dopamine receptor 3 (DRD3), G protein-coupled receptor 55 (GPR55), hemoglobin subunit alpha beta (HBA), transient receptor potential cation channel subfamily V member 1 (TRPV1), cannabinoid receptor 1 (CNR1), cannabinoid receptor 2 (CNR2), and 5-hydroxytryptamine receptor 1A (5HT1A). Given that CBD exhibits limited affinities towards CB1 and CB2 receptors, the neurological effects of CBD may be a result of a “single molecule, multiple targets” effect.

CBD target-disease network. Blue nodes represent the targets for CBD identified through target mapping. Salmon and pink nodes represent different diseases. Pink nodes are neurological-related diseases and are placed in the center for emphasis. Targets are connected with diseases with edges in the corresponding color
To further probe into the mechanism by which CBD triggers neurophysiological effects, we selected three targets, GPR55, 5HT1A, and OPRD, for detailed binding mode evaluation. First, we sought an explanation for the treatment benefits of CBD on neuro-related diseases at the molecular level. Second, we expected to identify the conserved binding mode between CBD and rhodopsin-like GPCR targets. The reasons for selecting these three targets were that (i) they are all experimentally validated targets for CBD; (ii) they are all neural disease-related targets at both pathway and disease levels; (iii) they are all rhodopsin-like (Class A) GPCRs. Other receptors were not selected due to various reasons. For example, BBP, HBA, etc. may have better affinities with CBD according to the prediction compared with the affinities of the selected three proteins; however, either they are non-human proteins, or there is currently no experimental data to support the CBD-protein interaction. Among the proteins that were experimentally validated, FABPL, CP1A1, CP2C9, etc. are depicted with relatively larger balloons compared with OPRD and 5HT1A. However, they were not selected since they are metabolism enzymes, which may affect the pharmacokinetic profiles of the compounds but will not directly trigger neuro-related physiological responses. For proteins that can have neuro-related effects, transient receptor potential cation channel subfamily V member 1 and 2 (TRPV1 and TRPV2) have been experimentally validated to interact with CBD and reported to be associated with pain relief. However, they are transporters, the structures of which vary dramatically compared to the structures of rhodopsin-like GPCRs.
Homology modeling for GPR55 and 5HT1A
The crystal structure for OPRD is currently available from the Protein Data Bank (PDB entry: 4N6H). For GPR55 and 5HT1A, there are no available crystal structures. Homology modeling with Modeller was used to build the protein models. Sequences for GPR55 and 5HT1A were acquired from the UniProt database.
Non-redundant PDB sequences at 95% sequence identity were considered for the template search. Sequences with fewer than 30 or >4000 residues were discarded. For the GPR55 model, six templates were selected, including human protease-activated receptor 1 (PDB entry: 3VW7, sequence identity: 37%), human CXCR4 chemokine receptor (PDB entry: 3ODU, sequence identity: 31%), human kappa-opioid receptor (PDB entry: 4DKL, sequence identity: 30%), house mouse mu-opioid receptor (PDB entry: 4DJH, sequence identity: 48%), human kappa-opioid receptor (PDB entry: 4EJ4, sequence identity: 45%), and human CC chemokine receptor 9 (PDB entry: 5LWE, sequence identity: 44%). For the 5HT1A model, five templates were selected, including human beta-2 adrenergic receptor (PDB entry: 3KJ6, sequence identity: 48%), wild turkey beta-1 adrenergic receptor (PDB entry: 4AMJ, sequence identity: 45%), wild turkey beta-1 adrenoceptor (PDB entry: 4BVN, sequence identity: 44%), human 5HT1B receptor (PDB entry: 4IAR, sequence identity: 43%), and human A2a receptor (PDB entry: 4UG2, sequence identity: 41%).
Align2d, a dynamic programming algorithm that considers the structural information from the templates, was used to align the templates and the target sequences. Automodel class was followed to create one or more target model(s) according to the structures of templates and the alignment file. Supplementary Figure 2a, b shows the sequence alignments between the templates and the targets. The conserved residues were marked with an asterisk. Gaps can be observed in the loop regions and the extracellular and intracellular terminus. DOPE evaluation revealed no positive energies for individual residues (Supplementary Figure 3a, b). In addition, good correlations between the templates and the targets were found, and gaps could be observed for the positions with truncated sequences. The dihedral angles ψ against φ of amino acid residues on the backbone structure of the protein were evaluated and reported in the Ramachandran plot (Supplementary Figure 3c, d). For GPR55, 269 residues (93.7%) are in the favored region, 15 residues (5.2%) are in the allowed region, and 3 (1%) residues are in the outlier region. Three outliers, TRP43, SER78, and TYR156, are located on ICL1, ECL1, and ECL2 respectively, which are not involved in the orthosteric binding pocket formation. For 5HT1A, 244 residues (96.1%) are in the favored region, 8 residues (3.1%) are in allowed region, and 2 (0.8%) residues are in outlier region. Two outliers, PHE189 and ARG190, are located on ICL3, which is also not involved in the binding pocket formation.
Model validation with enrichment test
Enrichment tests were conducted to determine whether these models could distinguish active compounds from random compounds. Virtual screening using Surflex-Dock Screen was performed to determine the binding affinity between small molecules and protein models.
For the GPR55 model, 10 active compounds were acquired from the ChEMBL database, and 500 random compounds were randomly selected from the Zinc database, as specified in Materials and methods section (Supplementary Table 1). In total, 510 compounds were ranked according to their docking scores. The compound with highest docking score was ranked No.1, and the compound with the lowest docking score was ranked No. 510. In total, 10 active compounds ranked among the top 41 (top 8%), which meant that the active GPR55 compounds are favored by this GPR55 protein model (Supplementary Figure 4a). The distribution of docking scores revealed that active compounds tended to achieve higher docking scores than random compounds (Supplementary Figure 4b). The result of the Mann-Whitney test showed that the asymptotic significance (two-tailed) was <0.001. Compared with random compounds, the active compounds had a distribution of docking scores that was significantly right shifted.
For the 5HT1A model, the same 500 random compounds from the Zinc database were retained, and another 10 active compounds for 5HT1A were acquired from the ChEMBL database (Supplementary Table 2). Similarly, all of the compounds were ranked according to their docking scores. Ten active compounds ranked among the top 67 (top 13%), which means that the 5HT1A receptor can distinguish the active compounds from the random ones (Supplementary Figure 4c). The distribution of docking scores demonstrated that the active compounds tended to have higher docking scores than the random compounds (Supplementary Figure 4d). In addition, the Mann–Whitney test showed that the asymptotic significance (two-tailed) was <0.001.
Static CBD-protein binding mode analysis
The homology models of GPR55 and 5HT1A and the crystal structure of OPRD (PDB entry: 4N6H) were used for evaluating CBD–protein interactions. The static evaluation was first performed with molecular docking studies. Given the conservation in sequences and structural features among the rhodopsin-like (class A) GPCRs, the orthosteric binding pocket is conserved at the transmembrane (TM) helical bundles facing towards the extracellular matrix.
In GPR55, residues from TM3, TM4, TM5, TM6, TM7, and ECL2 were found to be involved in orthosteric binding pocket formation (Fig. 5a). Specifically, the surface of residues GLU88, PHE92, SER143, ILE146, PHE159, THR166, PHE172, PHE240, ARG243, and LEU260 defined the boundary of the pocket (Fig. 5b). CBD fit well inside the binding pocket with a cyclohexene group facing the extracellular matrix and a lipophilic chain facing the intracellular matrix. One hydrogen bond was observed between the residue THR166 on ECL2 and the hydroxyl group connected to the benzene ring of CBD. Hydrophobic interactions were observed between the aliphatic chain of CBD and the lipophilic chain and ring systems from PHE92 on TM3, PHE172 on TM5, PHE240 on TM6, and ILE146 on TM4 (Supplementary Figure 5a).

Simulated binding mode of CBD on GPR55. The protein is shown as a cartoon. CBD (cyan) and critical residues (yellow) involved in interactions and pocket formation are shown in sticks. H-bonds are marked as red dashes, and hydrophobic interactions are marked as blue dashes. a CBD-GPR55 binding follows a hydrophobic cavity and backbone hinge model. Cyan spheres represent the location of the hydrophobic cavity. Red spheres represent the location of the hinge formation. b Static docking pose and ligand-residue interactions between the GPR55 protein model and CBD. c Pose and ligand-residue interactions between the GPR55 protein model and CBD for the coordinate at 120 ns (equilibrium stage) during the molecular dynamics simulation
In 5HT1A, residues from TM2, TM3, TM5, TM6, TM7, and ECL2 were found to be involved in orthosteric binding pocket formation (Fig. 6a). To be specific, the surface of residues TYR66, PHE82, ASP86, CYS90, ILE159, ALA173, TRP208, PHE211, PHE212, and ASN236 defined the boundary of the pocket (Fig. 6b). CBD exhibited a similar binding pose as in GPR55 described above. The cyclohexene group faced up towards the extracellular region, and the aliphatic chain stretched inside the pocket. Two hydrogen bonds were observed between the hydroxyl groups on CBD and ASP86 on TM3 and ASN236 on TM7. Hydrophobic interactions were observed between the aliphatic chain of CBD and the lipophilic ring systems from TRP208, PHE211, and PHE212 on TM6 (Supplementary Fig. 5b).

Simulated binding mode of CBD on 5HT1A. The protein is shown as a cartoon. CBD (cyan) and critical residues (yellow) involved in interactions and pocket formation are shown in sticks. H-bonds are marked as red dashes, and hydrophobic interactions are marked as blue dashes. a CBD-5HT1A binding follows a model of hydrophobic cavity and backbone hinge. Cyan spheres represent the location of the hydrophobic cavity. Red spheres represent the location of the hinge formation. b Static docking pose and ligand-residue interactions between the 5HT1A protein model and CBD. c Pose and ligand-residue interactions between the 5HT1A protein model and CBD for the coordinate at 100 ns (equilibrium stage) during the molecular dynamics simulation
In OPRD, residues from TM3, TM6, and TM7 were found to be involved in orthosteric binding pocket formation (Fig. 7a). The surface of residues LEU125, ASP128, TYR129, MET132, ILE277, ILE304, and TYR308 defined the boundary of the pocket (Fig. 7b). CBD fit well inside the pocket. Three hydrogen bonds were observed between hydroxyl groups on CBD and ASP128 and TYR129 on TM3 and TYR308 on TM7. Hydrophobic interactions were observed between the aliphatic chain of CBD and the lipophilic chain and ring systems from ILE277 on TM6, ILE304, and TYR308 on TM7 (Supplementary Figure 5c).

Simulated binding mode of CBD on OPRD. The protein is shown as a cartoon. CBD (cyan) and critical residues (yellow) involved in interactions and pocket formation are shown in sticks. H-bonds are marked as red dashes, and hydrophobic interactions are marked as blue dashes. a CBD-OPRD binding follows a model of hydrophobic cavity and backbone hinge. Cyan spheres represent the location of the hydrophobic cavity. Red spheres represent the location of the hinge formation. b Static docking pose and ligand-residue interactions between the OPRD protein model and CBD. c Pose and ligand-residue interactions between the OPRD protein model and CBD for the coordinate at 30 ns (first equilibrium stage) during the molecular dynamics simulation. d Extracellular view of pose and ligand-residue interactions between the OPRD protein model and CBD for the coordinate at 160 ns (second equilibrium stage) during the molecular dynamics simulation
As shown in Figs. 5a, 6a, and 7a, a model of hydrophobic cavity and backbone hinge was proposed by summarizing all three molecular docking studies. Backbone donors and acceptors on ECL2, TM3, and TM7 function as hinges to fix and locate CBD, while hydrophobic cavities formed by residues on TM5, TM6, and TM7 hold the aliphatic chain of CBD.
Dynamic CBD-protein binding mode analysis
The model of hydrophobic cavity and backbone hinge was proposed through the molecular docking studies in a static manner. We then sought to test whether this mode can be maintained in dynamics studies. Therefore, 200-ns molecular dynamics simulation was performed for each of the CBD-protein complexes with lipids, water molecules, and ions, as described in Materials and methods.
For the CBD-GPR55 system (Fig. 8a), the receptor-ligand interactions between CBD and the surrounding residues were proven to be stable. The GPR55 model was equilibrated at ~40 ns, and CBD was equilibrated at ~100 ns (Fig. 8b). The overall root mean square deviation (RMSD) for the protein was ~2.6 Å, which was majorly contributed by the loop regions. The coordinate of the complex at 120 ns was selected to study the potential interactions and the detailed binding mode, given that the equilibrium of the system was maintained after 100 ns. CBD generally preserved the static binding pose with the cyclohexene group facing the extracellular matrix and the aliphatic chain facing the intracellular matrix (Fig. 5c). Due to the flipping and movement of ECL2, the hydrogen bond between CBD and THR166 was no longer maintained. Another residue, MET162, on ECL2 formed a hydrogen bond with CBD to function as the hinge. Hydrophobic interactions with TM3, TM5, and TM6 were preserved. PHE92, PHE172, PHE159, and PHE240 were involved in the formation of the hydrophobic cavity for holding the aliphatic chain (Supplementary Figure 6a). Overall, the proposed binding mode was preserved for the CBD-GPR55 complex in the dynamic view.

The 200-ns molecular dynamics simulation for the CBD-protein complexes. a Simulation system for the CBD-GPR55 protein model complex, with water in red spots, chlorine ions in green balls, sodium ions in yellow balls, membrane in cyan sticks, protein in purple cartoon, and CBD in cyan sticks. b RMSD change for both CBD and the GPR55 protein model. c Simulation system for the CBD-5HT1A protein model complex. d RMSD change for both CBD and the 5HT1A protein model. e Simulation system for the CBD-OPRD protein model complex. f RMSD change for both CBD and the OPRD protein model
For the CBD-5HT1A system (Fig. 8c), stable receptor-ligand interactions between CBD and 5HT1A were observed. The protein model was equilibrated at ~30 ns, and CBD was equilibrated at ~65 ns (Fig. 8d). The fluctuation of RMSD for CBD was observed at ~140 ns, which was mainly caused by the rotation of the cyclohexene group. The overall RMSD for the protein was ~2.1 Å, and the loop regions were the major contributor. The coordinate of the complex at 100 ns was selected to study the potential interactions and the detailed binding mode since the overall equilibrium of the system was consistent after 65 ns. The hydrophobic cavity and backbone hinge model was preserved for the protein–ligand complex (Fig. 6c). The hydrogen bond between CBD and ASN236 was maintained on TM7. An additional hydrogen bond for CBD with GLY232 on TM7 was also observed. Meanwhile, the other hydroxyl group could have hydrophilic interactions with SER160 on ECL2 through a water bridge. TRP208, PHE211, and PHE212 on TM6 were involved in hydrophobic cavity formation, and the hydrophobic interactions for CBD with these residues were preserved (Supplementary Figure 6b).
For the CBD-OPRD system (Fig. 8e), a relatively stable complexwas formed. Two equilibrium stages were observed for OPRD (Fig. 8f). The protein achieved the first equilibrium stage after ~5 ns and the second equilibrium stage after ~70 ns. CBD flipping could be found at the beginning of the second equilibrium stage of the protein, mainly caused by the outside rotation of TM1, TM6, and TM7 at the extracellular portion. The orthosteric binding site was increased after the rotation of helix bundles, which minimized the restrictions of the movement of CBD and allowed for more drastic movements of CBD. After the optimized pose was found, CBD possessed a stable RMSD profile after 160 ns. The coordinates of the complex at 30 ns, as the representative for the first equilibrium stage, was first selected to determine the detailed CBD binding pose. The cyclohexene group of CBD faced the extracellular matrix, and its aliphatic group faced the intracellular matrix. Hydrogen bonds were formed between CBD molecules and residues ASP128 (TM3) and LEU200 (ECL2) through a water bridge, and hydrophobic interactions were formed between CBD and residues TYP129 and MET132 on TM3 (Fig. 7c and Supplementary Figure 6c). The outward rotation of TM6 and TM7 made it difficult for CBD to interact with residues on these helical bundles. Instead, CBD interacted closely with TM3. The overall model of hydrophobic cavity and backbone hinge was preserved for this equilibrium stage. The coordinates of the complex at 160 ns, as the representative for the second equilibrium stage, were then selected for detailed examination. Through the extracellular view of the complex (Fig. 7d), CBD can clearly be seen to lie in the middle of the pocket parallel to the cell membrane. Considering the outward rotation of TM3, TM6, and TM7, the binding pocket was large enough to hold CBD in a parallel pose. Nevertheless, the hydrogen bond of CBD with ASP128 on TM3 was maintained to function as the hinge. TRP249, ILE277, and PHE280 on TM6 and LEU300 and ILE304 on TM7 were involved in the formation of the hydrophobic cavity and its interaction with the aliphatic chain of CBD (Supplementary Figure 6d). Despite the change in the confirmation of CBD from vertical to parallel, the overall model of hydrophobic cavity and backbone hinge was still preserved for this equilibrium stage.
The 200-ns molecular dynamics simulations for these three systems verified the proposed model of hydrophobic cavity and backbone hinge. Consistent binding modes between CBD and three active rhodopsin-like GPCR targets were observed for both static molecular docking studies and dynamic molecular simulations.
Prediction of dopamine receptor 3 as an active target for CBD
Dopamine receptor 3 (DRD3) was predicted to be a potential target for CBD through chemogenomics-based target mapping (Fig. 2). The pathway and process analysis associated DRD3 with multiple neuro-related disorders. Currently, there is no reported experimental activity for CBD with DRD3. Given that DRD3, which associates with multiple normal neuro-related functions, is also a rhodopsin-like GPCR, we tested whether CBD-DRD3 interactions could fit the proposed hydrophobic cavity and backbone hinge model.
A molecular docking study between CBD and DRD3 (PDB entry: 3PBL) was performed. Residues from TM2, TM3, TM5, TM6, TM7, and ECL2 were found to be involved in orthosteric binding pocket formation (Fig. 9a). Especially, the surface of residues VAL86, PHE106, ASP110, CYS181, ILE183, VAL189, SER193, PHE345, PHE346, and TYR365 defined the boundary of the pocket. Two hydrogen bonds, functioning as hinges, were observed between the residue ASP110 on TM3, TYR365 on TM7, and the two hydroxyl groups on CBD. Hydrophobic interactions were observed between the benzene rings from PHE345 and PHE346 on TM6 and the hydrophobic center (benzene ring and aliphatic chain) of CBD. The proposed binding mode was observed.

CBD-D3 protein model binding pose and negative controls. Proteins are shown in cartoon format in green. CBD is marked in cyan. Critical residues involved in interactions and pocket formation are marked in yellow. a Membrane and extracellular views of docking pose and ligand-residue interactions between the D3 protein model and CBD. b Membrane and extracellular views of docking pose and ligand-residue interactions between the A2A protein model and CBD. c Membrane and extracellular views of docking pose and ligand–residue interactions between the CXCR4 protein model and CBD
Furthermore, we examined whether this proposed model of hydrophobic cavity and backbone hinge holds true for the interaction of CBD with all rhodopsin-like GPCRs. Two rhodopsin-like GPCRs with crystal structures, adenosine A2A receptor (ADORA2A) and C–X–C chemokine receptor type 4 (CXCR4), were randomly selected for a test of their interaction profiles with CBD using molecular docking studies. For ADORA2A (PDB entry: 5IU4), residues from TM1, TM2, TM6, TM7, and ECL2 were involved in orthosteric binding pocket formation (Fig. 9b). The surface of residues TYR9, ALA63, SER67, PHE168, ASP170, LEU249, ASN253, MET270, and ILE274 defined the boundary of the pocket. CBD fit inside with the aliphatic chain facing up towards the extracellular space, while the cyclohexene group faced down. PHE168 on ECL2 formed one hydrogen bond with CBD. However, the proposed binding mode was not observed for the protein–CBD complex. For CXCR4 (PDB entry: 3ODU), residues from TM1, TM2, TM3, TM7, and ECL2 were involved in orthosteric binding pocket formation (Fig. 9c). The surface of residues TYR45, TRP94, ASP97, CYS109, HIS113, TYR116, CYS186, ARG188, SER285, and GLU288 defined the cavity of the binding pocket. Again, no hydrophobic cavity was formed by these surrounding residues to hold the aliphatic chain. One hydrogen bond was formed between CYS109 on ECL2 and CBD. Therefore, the proposed binding mode was not observed.
Conclusion
Considering the promising treatment potentials of CBD on multiple neuro-related diseases, efforts are in demand to reveal the acting target(s) for CBD and the unclear mechanisms behind the therapeutic potentials. In this study, chemogenomics-knowledgebase systems pharmacology was combined with the literature to report a target network analysis on CBD with both experimentally validated and computationally predicted molecular targets. Pathway analysis was performed to bridge the gap between the targets and their involvement in diseases. Based on the results from the CBD-target, target-pathway, and target-disease networks, we focused on three reported neuro-related rhodopsin-like GPCR targets, GPR55, 5HT1A, and OPRD. A hydrophobic cavity and backbone hinge model was proposed and tested with both static and dynamic evaluations to describe the interactions between CBD and active rhodopsin-like GPCR targets. The proposed model can guide (a) the identification of CBD targets among receptors in the same protein family and (b) the design of CBD derivatives and analogs. Given the close association between neuro-related diseases, such as epilepsy, psychosis, anxiety, movement disorder, and neurological pain, and the receptors GPR55, 5HT1A, and OPRD, this study provided evidence that CBD may act on these receptors to treat these medical conditions at the molecular level. Moreover, based on the proposed binding model and the CBD target mapping results, dopamine receptor 3 was further predicted to be a potential target for CBD. Negative controls for rhodopsin-like GPCRs were also introduced, revealing that the proposed CBD binding model was not universal but only for active targets.
References
Hall W, Degenhardt L. Adverse health effects of non-medical cannabis use. Lancet. 2009;374:1383–91.
D’Souza DC, Perry E, MacDougall L, Ammerman Y, Cooper T, Yu-te W, et al. The psychotomimetic effects of intravenous delta-9-tetrahydrocannabinol in healthy individuals: implications for psychosis. Neuropsychopharmacology. 2004;29:1558.
Ashton CH. Pharmacology and effects of cannabis: a brief review. Br J Psychiatry. 2001;178:101–6.
Zimmer A, Zimmer AM, Hohmann AG, Herkenham M, Bonner TI. Increased mortality, hypoactivity, and hypoalgesia in cannabinoid CB1 receptor knockout mice. Proc Natl Acad Sci U S A. 1999;96:5780–5.
Ramer R, Heinemann K, Merkord J, Rohde H, Salamon A, Linnebacher M, et al. COX-2 and PPAR-γ confer cannabidiol-induced apoptosis of human lung cancer cells. Mol Cancer Ther. 2013;12:69–82.
Bisogno T, Hanuš L, De Petrocellis L, Tchilibon S, Ponde DE, Brandi I, et al. Molecular targets for cannabidiol and its synthetic analogues: effect on vanilloid VR1 receptors and on the cellular uptake and enzymatic hydrolysis of anandamide. Br J Pharmacol. 2001;134:845–52.
Ryberg E, Larsson N, Sjögren S, Hjorth S, Hermansson NO, Leonova J, et al. The orphan receptor GPR55 is a novel cannabinoid receptor. Br J Pharmacol. 2007;152:1092–101.
Bih CI, Chen T, Nunn AV, Bazelot M, Dallas M, Whalley BJ. Molecular targets of cannabidiol in neurological disorders. Neurotherapeutics. 2015;12:699–730.
Aso E, Sánchez-Pla A, Vegas-Lozano E, Maldonado R, Ferrer I. Cannabis-based medicine reduces multiple pathological processes in AβPP/PS1 mice. J Alzheimers Dis. 2015;43:977–91.
Cheng D, Spiro AS, Jenner AM, Garner B, Karl T. Long-term cannabidiol treatment prevents the development of social recognition memory deficits in Alzheimer’s disease transgenic mice. J Alzheimers Dis. 2014;42:1383–96.
Devinsky O, Cilio MR, Cross H, Fernandez‐Ruiz J, French J, Hill C, et al. Cannabidiol: pharmacology and potential therapeutic role in epilepsy and other neuropsychiatric disorders. Epilepsia. 2014;55:791–802.
Costa B, Giagnoni G, Franke C, Trovato AE, Colleoni M. Vanilloid TRPV1 receptor mediates the antihyperalgesic effect of the nonpsychoactive cannabinoid, cannabidiol, in a rat model of acute inflammation. Br J Pharmacol. 2004;143:247–50.
McHugh D, Hu SS, Rimmerman N, Juknat A, Vogel Z, Walker JM, et al. N-arachidonoyl glycine, an abundant endogenous lipid, potently drives directed cellular migration through GPR18, the putative abnormal cannabidiol receptor. BMC Neurosci. 2010;11:44.
Jiang R, Yamaori S, Takeda S, Yamamoto I, Watanabe K. Identification of cytochrome P450 enzymes responsible for metabolism of cannabidiol by human liver microsomes. Life Sci. 2011;89:165–70.
Russo EB, Burnett A, Hall B, Parker KK. Agonistic properties of cannabidiol at 5-HT1a receptors. Neurochem Res. 2005;30:1037–43.
Kathmann M, Flau K, Redmer A, Tränkle C, Schlicker E. Cannabidiol is an allosteric modulator at mu-and delta-opioid receptors. Naunyn Schmiede Arch Pharmacol. 2006;372:354–61.
Liu H, Wang L, Lv M, Pei R, Li P, Pei Z, et al. AlzPlatform: an Alzheimer’s disease domain-specific chemogenomics knowledgebase for polypharmacology and target identification research. J Chem Inf Model. 2014;54:1050–60.
Xie XQ, Wang L, Liu H, Ouyang Q, Fang C, Su W. Chemogenomics knowledgebased polypharmacology analyses of drug abuse related G-protein coupled receptors and their ligands. Front Pharmacol. 2014;5:3.
Wang L, Ma C, Wipf P, Liu H, Su W, Xie XQ. TargetHunter: an in silico target identification tool for predicting therapeutic potential of small organic molecules based on chemogenomic database. AAPS J. 2013;15:395–406.
Rogers D, Hahn M. Extended-connectivity fingerprints. J Chem Inf Model. 2010;50:742–54.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–504.
Fiser A, Šali A. Modeller: generation and refinement of homology-based protein structure models. Methods Enzymol. 2003;374:461–91.
Webb B, Sali A. Protein structure modeling with MODELLER. Methods Mol Biol. 2014;1137:1–15.
Šali A, Potterton L, Yuan F, van Vlijmen H, Karplus M. Evaluation of comparative protein modeling by MODELLER. Proteins. 1995;23:318–26.
Shen My, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci. 2006;15:2507–24.
Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J Mol Biol. 1963;7:95–9.
Lovell SC, Davis IW, Arendall WB, de Bakker PI, Word JM, Prisant MG, et al. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins. 2003;50:437–50.
Consortium U. UniProt: a hub for protein information. Nucleic Acids Res. 2014;43:D204–12.
Fenalti G, Giguere PM, Katritch V, Huang XP, Thompson AA, Cherezov V, et al. Molecular control of [dgr]-opioid receptor signalling. Nature. 2014;506:191–6.
Chien EY, Liu W, Zhao Q, Katritch V, Han GW, Hanson MA, et al. Structure of the human dopamine D3 receptor in complex with a D2/D3 selective antagonist. Science. 2010;330:1091–5.
Segala E, Guo D, Cheng RK, Bortolato A, Deflorian F, Dore AS, et al. Controlling the dissociation of ligands from the adenosine A2A receptor through modulation of salt bridge strength. J Med Chem. 2016;59:6470–9.
Wu B, Chien EY, Mol CD, Fenalti G, Liu W, Katritch V, et al. Structures of the CXCR4 chemokine GPCR with small-molecule and cyclic peptide antagonists. Science. 2010;330:1066–71.
Bian YM, Feng ZW, Yang P, Xie XQ. Integrated in silico fragment-based drug design: case study with allosteric modulators on metabotropic glutamate receptor 5. AAPS J. 2017;19:1235–48.
Weiner SJ, Kollman PA, Nguyen DT, Case DA. An all atom force field for simulations of proteins and nucleic acids. J Comput Chem. 1986;7:230–52.
Meng EC, Shoichet BK, Kuntz ID. Automated docking with grid‐based energy evaluation. J Comput Chem. 1992;13:505–24.
Irwin JJ, Shoichet BK. ZINC: a free database of commercially available compounds for virtual screening. J Chem Inf Model. 2005;45:177–82.
Bento AP, Gaulton A, Hersey A, Bellis LJ, Chambers J, Davies M, et al. The ChEMBL bioactivity database: an update. Nucleic Acids Res. 2014;42:D1083–90.
Jo S, Kim T, Iyer VG, Im W. CHARMM‐GUI: a web‐based graphical user interface for CHARMM. J Comput Chem. 2008;29:1859–65.
Jo S, Lim JB, Klauda JB, Im W. CHARMM-GUI Membrane Builder for mixed bilayers and its application to yeast membranes. Biophys J. 2009;97:50–8.
Sadiq S, Guixa-Gonzalez R, Dainese E, Pastor M, De Fabritiis G, Selent J. Molecular modeling and simulation of membrane lipid-mediated effects on GPCRs. Curr Med Chem. 2013;20:22–38.
Grossfield A, Feller SE, Pitman MC. A role for direct interactions in the modulation of rhodopsin by ω-3 polyunsaturated lipids. Proc Natl Acad Sci U S A. 2006;103:4888–93.
Olausson BE, Grossfield A, Pitman MC, Brown MF, Feller SE, Vogel A. Molecular dynamics simulations reveal specific interactions of post-translational palmitoyl modifications with rhodopsin in membranes. J Am Chem Soc. 2012;134:4324–31.
Maier JA, Martinez C, Kasavajhala K, Wickstrom L, Hauser KE, Simmerling C. ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J Chem Theory Comput. 2015;11:3696–713.
Dickson CJ, Madej BD, Skjevik ÅA, Betz RM, Teigen K, Gould IR, et al. Lipid14: the amber lipid force field. J Chem Theory Comput. 2014;10:865–79.
Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, Klein ML. Comparison of simple potential functions for simulating liquid water. J Chem Phys. 1983;79:926–35.
Bayly CI, Cieplak P, Cornell W, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem. 1993;97:10269–80.
Frisch MJ, Trucks GW, Schlegel HB, Scuseria GE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Petersson GA, Nakatsuji H, Li X, Caricato M, Marenich AV, Bloino J, Janesko BG, Gomperts R, Mennucci B, Hratchian HP, Ortiz JV, Izmaylov AF, Sonnenberg JL, Williams-Young D, Ding F, Lipparini F, Egidi F, Goings J, Peng B, Petrone A, Henderson T, Ranasinghe D, Zakrzewski VG, Gao J, Rega N, Zheng G, Liang W, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, Vreven T, Throssell K, Montgomery JA Jr, Peralta JE, Ogliaro F, Bearpark MJ, Heyd JJ, Brothers EN, Kudin KN, Staroverov VN, Keith TA, Kobayashi R, Normand J, Raghavachari K, Rendell AP, Burant JC, Iyengar SS, Tomasi J, Cossi M, Millam JM, Klene M, Adamo C, Cammi R, Ochterski JW, Martin RL, Morokuma K, Farkas O, Foresman JB, Fox DJ. Gaussian 16, Revision B.01. Gaussian, Inc., Wallingford CT, 2016.
Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–74.
Wang J, Wang W, Kollman PA, Case DA. Automatic atom type and bond type perception in molecular mechanical calculations. J Mol Graph Model. 2006;25:247–60.
Case DA, Betz RM, Cerutti DS, Cheatham TE III, Darden TA, Duke RE, Giese TJ, Gohlke H, Goetz AW, Homeyer N, Izadi S, Janowski P, Kaus J, Kovalenko A, Lee TS, LeGrand S, Li P, Lin C, Luchko T, Luo R, Madej B, Mermelstein D, Merz KM, Monard G, Nguyen H, Nguyen HT, Omelyan I, Onufriev A, Roe DR, Roitberg A, Sagui C, Simmerling CL, Botello-Smith WM, Swails J, Walker RC, Wang J, Wolf RM, Wu X, Xiao L, Kollman PA. AMBER 2016, University of California, San Francisco, 2016.
Götz AW, Williamson MJ, Xu D, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 1. Generalized born. J Chem Theory Comput. 2012;8:1542–55.
Salomon-Ferrer R, Götz AW, Poole D, Le Grand S, Walker RC. Routine microsecond molecular dynamics simulations with AMBER on GPUs. 2. Explicit solvent particle mesh Ewald. J Chem Theory Comput. 2013;9:3878–88.
Loncharich RJ, Brooks BR, Pastor RW. Langevin dynamics of peptides: The frictional dependence of isomerization rates of N‐acetylalanyl‐N′‐methylamide. Biopolymers. 1992;32:523–35.
Izaguirre JA, Catarello DP, Wozniak JM, Skeel RD. Langevin stabilization of molecular dynamics. J Chem Phys. 2001;114:2090–8.
Darden T, York D, Pedersen L. Particle mesh Ewald: an N⋅log (N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–92.
Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–93.
Yamaori S, Ebisawa J, Okushima Y, Yamamoto I, Watanabe K. Potent inhibition of human cytochrome P450 3A isoforms by cannabidiol: role of phenolic hydroxyl groups in the resorcinol moiety. Life Sci. 2011;88:730–6.
Yamaori S, Koeda K, Kushihara M, Hada Y, Yamamoto I, Watanabe K. Comparison in the in vitro inhibitory effects of major phytocannabinoids and polycyclic aromatic hydrocarbons contained in marijuana smoke on cytochrome P450 2C9 activity. Drug Metab Pharmacokinet. 2012;27:294–300.
Yamaori S, Kushihara M, Yamamoto I, Watanabe K. Characterization of major phytocannabinoids, cannabidiol and cannabinol, as isoform-selective and potent inhibitors of human CYP1 enzymes. Biochem Pharmacol. 2010;79:1691–8.
Iannotti FA, Hill CL, Leo A, Alhusaini A, Soubrane C, Mazzarella E, et al. Nonpsychotropic plant cannabinoids, cannabidivarin (CBDV) and cannabidiol (CBD), activate and desensitize transient receptor potential vanilloid 1 (TRPV1) channels in vitro: potential for the treatment of neuronal hyperexcitability. ACS Chem Neurosci. 2014;5:1131–41.
Whyte LS, Ryberg E, Sims NA, Ridge SA, Mackie K, Greasley PJ, et al. The putative cannabinoid receptor GPR55 affects osteoclast function in vitro and bone mass in vivo. Proc Natl Acad Sci U S A. 2009;106:16511–6.
De Petrocellis L, Ligresti A, Moriello AS, Allarà M, Bisogno T, Petrosino S, et al. Effects of cannabinoids and cannabinoid‐enriched Cannabis extracts on TRP channels and endocannabinoid metabolic enzymes. Br J Pharmacol. 2011;163:1479–94.
Qin N, Neeper MP, Liu Y, Hutchinson TL, Lubin ML, Flores CM. TRPV2 is activated by cannabidiol and mediates CGRP release in cultured rat dorsal root ganglion neurons. J Neurosci. 2008;28:6231–8.
Huang H, McIntosh AL, Martin GG, Landrock D, Chung S, Landrock KK, et al. FABP1: a novel hepatic endocannabinoid and cannabinoid binding protein. Biochemistry. 2016;55:5243–55.
Acknowledgements
We acknowledge the funding support to the Xie laboratory from the NIH NIDA (P30 DA035778A1) and DOD (W81XWH-16-1-0490), and the funding support to the J. Wang laboratory from NIH R01-GM079383 & R21-GM097617. Computational support from the Center for Research Computing of University of Pittsburgh, and the Extreme Science and Engineering Discovery Environment (CHE090098), are also acknowledged.
Author contributions
X-QX designed the research; YB, XH, and YJ performed the research; X-QX, JW, and LW contributed the chemogenomics tools and methods; YB and XH wrote the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Electronic supplementary material
Rights and permissions
About this article

Cite this article
Bian, Ym., He, Xb., Jing, Yk. et al. Computational systems pharmacology analysis of cannabidiol: a combination of chemogenomics-knowledgebase network analysis and integrated in silico modeling and simulation. Acta Pharmacol Sin 40, 374–386 (2019). https://doi.org/10.1038/s41401-018-0071-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-018-0071-1
Keywords
This article is cited by
-
Altering biomolecular condensates as a potential mechanism that mediates cannabidiol effect on glioblastoma
Medical Oncology (2024)
-
Role of the endocannabinoid system in fragile X syndrome: potential mechanisms for benefit from cannabidiol treatment
Journal of Neurodevelopmental Disorders (2023)
-
Cannabidiol inhibits methamphetamine-induced dopamine release via modulation of the DRD1-MeCP2-BDNF-TrkB signaling pathway
Psychopharmacology (2022)
-
Generative chemistry: drug discovery with deep learning generative models
Journal of Molecular Modeling (2021)
-
Molecular dynamics simulations on RORγt: insights into its functional agonism and inverse agonism
Acta Pharmacologica Sinica (2019)
