
Abstract
Many bacteria and archaea harbor the adaptive CRISPR-Cas system, which stores small nucleotide fragments from previous invasions of nucleic acids via viruses or plasmids. This molecular archive blocks further invaders carrying identical or similar nucleotide sequences. However, few of these systems have been confirmed experimentally to be active in gut bacteria. Here, we demonstrate experimentally that the type I-C CRISPR-Cas system of the prevalent gut bacterium Eggerthella lenta can specifically target and cleave foreign DNA in vitro by using a plasmid transformation assay. We also show that the CRISPR-Cas system acquires new immunities (spacers) from the genome of a virulent E. lenta phage using traditional phage assays in vitro but also in vivo using gnotobiotic (GB) mice. Both high phage titer and an increased number of spacer acquisition events were observed when E. lenta was exposed to a low multiplicity of infection in vitro, and three phage genes were found to contain protospacer hotspots. Fewer new spacer acquisitions were detected in vivo than in vitro. Longitudinal analysis of phage-bacteria interactions showed sustained coexistence in the gut of GB mice, with phage abundance being approximately one log higher than the bacteria. Our findings show that while the type I-C CRISPR-Cas system is active in vitro and in vivo, a highly virulent phage in vitro was still able to co-exist with its bacterial host in vivo. Taken altogether, our results suggest that the CRISPR-Cas defense system of E. lenta provides only partial immunity in the gut.
Similar content being viewed by others


Introduction
Bacteria and archaea have the unique ability of acquiring resistance to various viruses (bacteriophages and archaeal viruses) and plasmids via CRISPR-Cas systems. This adaptive immunity is obtained by incorporating short fragments of DNA (spacers ~30 nucleotides) from the invading genetic elements within the CRISPR array of the host genome (the adaptation stage). This “memory” of past infections enables the cell to recognize and cleave the DNA/RNA from subsequent invaders with identical or similar sequences (the interference stage) [1]. The acquired spacers are located in a clustered regularly interspaced short palindromic repeat (CRISPR) array where each spacer is flanked by direct repeats. CRISPR-associated genes (Cas) are often flanking the CRISPR arrays and are coding for proteins needed for the above-mentioned stages. The CRISPR array is transcribed and processed into CRISPR RNAs (crRNA), which will, in the interference stage, guide Cas nucleases to search and cleave nucleic acids of the invader that match the spacer, and thereby ultimately prevent infection [2,3,4,5].
During the past decade, extensive analyses of Cas proteins have revealed highly diverse CRISPR-Cas systems, which are currently classified into two large classes (class 1 and class 2), six types (I-VI), and numerous subtypes [6]. For example, the class 1 type I-C CRISPR-Cas system is characterized by the following cas gene order cas3-cas5-cas8c-cas7-cas4-cas1-cas2, which are situated next to a CRISPR-array [7,8,9,10].
The microbes inhabiting the human gut (the gut microbiota, GM) play important roles in human health and disease [11]. It is therefore important to understand how bacteria defend themselves against phages in this ecosystem [12]. Most of the CRISPR-Cas research on gut-related bacteria is based on computational approaches [13,14,15], whereas experimental studies are sparse [16, 17]. However, Soto-Perez et al. demonstrated transcription and interference activity of a type I-C CRISPR-Cas system by constructing a Pseudomonas aeruginosa strain carrying Eggerthella lenta cas genes, that subsequently was infected by P. aeruginosa phages [16]. A type I-C CRISPR-Cas system previously found in Bifidobacterium spp. has recently also been described in the widespread human gut bacterium Eggerthella lenta [16, 18]. E. lenta is a common member of the human GM and seems to be more abundant in individuals suffering from type-2-diabetes mellitus (T2DM) [19, 20] and might play a role in disease etiology via its production of imidazole propionate that impairs insulin signaling [19].
Here we investigate the functionality (adaptation and interference activity) of the type I-C CRISPR-Cas system harbored by E. lenta DSM 15644 against the virulent E. lenta siphophage PMBT5 (genome size 30,930 bp) [21] in both in vitro and in vivo (in the gut) settings.
Methods
Bacterial strains, phage, and growth medium
Eggerthella lenta DSM 15644 (GCA_003340005.1), E. lenta DSM 2243T (GCF_000024265.1), and phage PMBT5 [21] (MH626557.1) were used in this study. Wilkins Chalgren Anaerobe medium (WCA, Sigma-Aldrich, St. Louis, Missouri, USA) was used for culturing as broth in Hungate tubes (Sciquip Limited, Newtown, UK), as solid media containing 1.5% (w/v) agar or as soft agar containing 0.5% (w/v) agar (Oxoid, Thermo Fisher Scientific, Waltham, Massachusetts, USA). Anoxic conditions were obtained as previously described [22]. Bacteria (cells from a single colony) were transferred from a WCA-plate to WCA-broth inside an anoxic chamber and the bacterial cultures were subsequently incubated at 37 °C for 1–3 days depending on the assay.
Phage propagation
For phage propagation, a culture of E. lenta DSM 15644 with an OD600nm ~0.25 (~5 × 108 colony forming units (CFU)/mL) was centrifuged for 10 min at 5000 × g and the supernatant was discarded. The bacterial pellet was resuspended in 200 µL of 40 mM anoxic CaCl2 and mixed with 100 µL phage PMBT5 lysate, followed by incubation for 10 min at room temperature to increase phage adsorption. The phage infected culture was subsequently added to either melted (52 °C) WCA soft agar for plaque assay or added to WCA-broth for phage amplification [21]. The inoculated WCA media were incubated under anoxic conditions for 17–20 h at 37 °C and OD600nm was measured with a Genesys30 Visible spectrophotometer (Thermo Fisher Scientific).
Culturing conditions for spacer acquisition in vitro
An E. lenta DSM 15644 culture with an OD600nm ~0.25 (~5 × 108 CFU/mL) was mixed with phage lysate of PMBT5 (~1 × 109 plaque forming units (PFU)/mL) to obtain multiplicity of infection (MOI, phage bacteria ratio) at 10, 1, 0.1, and 0.01. Bacteria (100 µL) and phage lysate (100 µL) were initially mixed in 40 mM anoxic CaCl2 (200 µL) in a 1.5 mL centrifuge tube, incubated for 10 min at room temperature, and subsequently added to a Hungate tube containing 9.6 mL WCA broth to reach a final volume of 10.0 mL. The bacteria-phage mixtures were prepared in triplicates and incubated at 37 °C for 144 h and OD600nm was measured at the following time points: 0, 4, 6, 24, 48, 72, 144 h. Differences in bacterial density were statistically evaluated with the ANOVA-based multiple pairwise comparison test and p values were adjusted with Tukey HSD test in the R version 4.2.1.
DNA extraction from cultures, lysates, and feces
DNA extraction from bacterial cultures, phage lysates and fecal pellets were performed using the Bead-Beat Micro AX gravity kit (A&A Biotechnology, Gdańsk, Poland), following the protocol of the manufacturer. Purified DNA was stored at −80 °C. A negative control representing E. lenta DSM 15644 with its native spacers along with a contamination control, consisting of autoclaved MilliQ water (Millipore corporation, Burlington, Massachusetts, USA), were included throughout all DNA extractions and PCR steps.
Primer design
Primers (Thermo Fisher Scientific) were designed with Geneious Prime v. 2019.0.4 and motif search was performed to ensure unique primer binding sites on the genome of E. lenta DSM 15644 and phage PMBT5 (Table S1). Primer specificity was tested in silico using NCBI primer-BLAST with strict parameters as described previously [23].
CRISPR-Cas interference assay
An interference assay was designed using plasmid pNZ123 [24] containing two different spacers originating from the native CRISPR array of E. lenta DSM 15644 (Spacer 2 (S2): 5ʹ-TCAGATTGTCGGGGTTGCCTGTCCCGCCTATCG-3ʹ, Spacer 1 (S1): 5ʹ-AATCGAATCTTCGCCCTTGCGGCCGAAAACCGG-3ʹ) which were flanked by different protospacer adjacent motifs (PAMs) (Table S1). Two native spacers were included in the construct to increase the interference activity. Based on literature investigating type I-C CRISPR-Cas systems [16, 25, 26], we tested the interference activity with two different PAMs (5ʹ-GGG, and 5ʹ-TTC), since no experimental data identifying a functional PAM for the type I-C CRISPR-Cas system in E. lenta DSM 15644 were available at that time. The pNZ123-derivatives were generated with the Gibson Assembly Cloning kit (NEB, Ipswich, Massachusetts, USA) and thereafter transformed into E. lenta DSM 15644 by electroporation as previously described [27], with the Gene Pulser electroporation system (Bio-Rad Laboratories, Hercules, California, USA) set at 25 μF, 200 Ω and 2.5 kV. Eggerthella lenta DSM 15644 competent cells were generated by incubation overnight at 37 °C in WCA broth (anoxic) containing 1% (w/v) glycine and 0.4 M D-sorbitol. Plasmid constructs were confirmed with PCR and Sanger sequencing (Macrogen, Amsterdam, Netherlands). A minimum inhibitory concentration test (Table S2) showed that E. lenta DSM 15644 was sensitive to chloramphenicol (Sigma-Aldrich) at concentrations above 1 µg/mL, thus 5 µg/mL chloramphenicol was used in media to select for cells that were transformed with a plasmid (pNZ123) carrying a chloramphenicol-resistance gene [24].
Detection of spacer acquisition
“CRISPR adaptation PCR technique using reamplification and electrophoresis” (CAPTURE) was applied to detect expanded CRISPR arrays in whole culture populations with increased sensitivity [28] in the type I-C CRISPR-Cas system harbored by E. lenta DSM 15644. The CAPTURE protocol is based on an initial PCR amplification followed by a reamplification (nested PCR) with primer sets representing different strategies (internal, degenerate, repeat) [28]. PCR was performed on a SureCycler8800 (Agilent Technologies, Santa Clara, California, USA) following the CAPTURE protocol [28] using the DreamTaq Green PCR Master Mix (Thermo Fisher Scientific), but annealing temperatures were adjusted to fit the designed primer sets (Table S1). After the initial PCR amplification, the PCR products were migrated on an 2% agarose gel suspended in 0.5X TBE buffer (45 mM Tris-Borate, 1 mM EDTA) at 110 V. The 1-kb plus DNA ladder (Thermo Fisher Scientific) was used as marker. Only every second lane was loaded with sample to minimize between-sample contamination. A sterile scalpel was used to cut out a fraction of the gel, with no visible band, that represented PCR-products with a DNA size ranging from 200–400 bp. The expected size for a single spacer acquisition in E. lenta DSM 15644 was 254 bp for the initial PCR (Table S3). The PCR-products were thereafter extracted from the gel with GeneJet Gel Extraction kit (Thermo Fisher Scientific) as recommended [28]. Reamplification was performed with the degenerate primer set according to the CAPTURE protocol [28] (Fig. S1). In a volume ratio of 2:1, AMPure XP bindings beads (Beckman Coulter, Brea, California, USA) were used to clean the extracted PCR products to remove DNA fragments (<100 bp) before library preparation.
Gnotobiotic mice study
Twelve germ-free outbred Swiss-Webster mice (Tac:SW, Taconic Biosciences A/S, Lille Skensved, Denmark) were bred at Section of Experimental Animal Models (University of Copenhagen) in an isolator where they were fed ad libitum chow diet (1314IRR, Brogaarden, Hørsholm, Denmark) that was sterilized by gamma irradiation. The mice represented 8 female and 4 male animals that were divided into 3 groups of 4 and were housed two-by-two according to the same sex (male-male, female-female, Table S4): E. lenta (EL) + PMBT5 (EL + Phage, n = 4), E. lenta + SM buffer (100 mM NaCl, 8 mM MgSO4, 50 mM Tris-HCl) (EL + Saline, n = 4), and a baseline (as control for the germ-free mice, n = 4) that were sacrificed at age 3 weeks (Fig. 1). The remaining 8 mice were transferred to the Department of Experimental Medicine (University of Copenhagen) in individual ventilated cages (IVCs) at age 5 weeks. Cage and housing conditions were as previously described [29]. The cages were sterilized and mounted to a sterile ventilation system. Animals were provided sterilized water and ad libitum chow diet (Safe D30, Scientific Diets, Rosenberg, Germany). After two weeks of acclimatization (i.e. 7 weeks of age), the mice were ear-tagged, weighed, and individual feces were sampled. The EL + Phage mice were orally administered with a mixture of bacterial host-phage cultures (E. lenta DSM 15644 and PMBT5) at a MOI of 1 (3 × 107 CFU and 3 × 107 PFU). The mice were considered as gnotobiotic (GB) after inoculation with bacteria and phages. With a volume of 40 µL, the bacteria and phages/saline were mixed in the ratio of 1:1 before being deposited on the tongue of the mice. This procedure was repeated after 6 h for a second inoculation. The bacterial cultures were in their exponential phase when orally administered to the mice and were grown under anoxic conditions prior to inoculation. Individual feces were then sampled (Fig. 1A) along with body weight measurements (Fig. S2) until the end of the experiment. Mouse feces were sampled at day 1 (before first inoculation), 1.5 (6 h after first culture inoculation), 2, 3, 4, 5, 12, 19, and 26. As controls, feces were also sampled when transferred from isolator to individual ventilated cages (arrival) and from baseline mice prior to inoculation. All samples were stored at −80 °C. The mice were euthanized by cervical dislocation at 10 weeks of age after anesthesia with a mixture of hypnorm (Apotek, Skanderborg, Denmark) and midazolam (Braun, Kronberg im Taunus, Germany) as described earlier [29]. Handling of mice during sampling was performed aseptically with the disinfectant VirkonS (Pharmaxim, Helsingborg, Sweden) as recommended by the manufacturer. The germ-free status was initially evaluated by the size of the cecum (enlarged) of the baseline mice and culture plating (no growth) confirming the germ-free status of the mice. For culturing by plating, blood agar plates were inoculated with feces from germ-free mice and dissolved in PBS buffer (NaCl 1.37 mM, KCl 27 mM, Na2HPO4 100 mM, and KH2PO4 18 mM) and incubated under anoxic conditions at 37 °C for 3 days. We also performed qPCR with universal primers targeting the 16 S rRNA gene and sequenced the full 16 S rRNA gene profile of both the mouse feed (provided in the breeding isolator and the IVCs) and fecal samples obtained at selected time points during the study (Fig. S3). Procedures were carried out in accordance with the Directive 2010/63/EU and the Danish Animal Experimentation Act (license-ID: 2017-15-0201-01262).

A Showing the lifespan of the mice included in the study. The mice were initially bred and housed in a germ-free isolator (light blue arrow) until age of 5 weeks when they were transferred to IVCs (dark blue arrow) for individual group caging followed by two weeks of acclimatization (gray arrow) prior intervention at age 7 weeks. Feces (brown cross) were sampled from each individual mouse before and after inoculation (yellow triangle) with phages and/or bacteria. The baseline mice were euthanized and sampled at age of 3 weeks. B Listing of the experimental groups, their abbreviation, and the inoculated bacterium and/or phage. SM buffer was used as saline solution.
Sequencing of PCR products
Sequencing was performed with NextSeq 550 (Illumina, San Diego, California, USA) using v2 MID output 2 × 150 cycles chemistry and barcodes as earlier described [30]. Illumina adaptors were designed specifically for E. lenta DSM 15644 (Table S1). To ensure the quality of the samples, additional cleaning with AMPure XP binding beads (Beckman Coulter), assessment of PCR-products size by gel electrophoresis, and DNA concentration measurements with Qubit HS (Thermo Fisher Scientific) were performed between each PCR step prior to sequencing. The average sequencing depth was 231,637 reads (minimum 54,123 reads and maximum 340,311 reads) for the in vitro samples, and 112,927 reads (minimum 15,138 reads and maximum 320,818 reads) for the in vivo samples (Accession: PRJEB47947, available at ENA). Full 16S rRNA gene sequencing was performed with the MinION platform (Oxford Nanopore Technologies, Oxford, UK), as previously described [31] (Accession: PRJEB52384, available at ENA). In brief, the 16S rRNA gene was amplified by PCR with primers [31] targeting the hypervariable regions V1-V8. The initial PCR (PCR1) reaction mixture included PCRBIO HiFi polymerase and PCRBIO buffer (PCR Biosystems Ltd., London, UK), primer mix, genomic DNA, and nuclease-free water. Gel electrophoresis was used to verify the size of the PCR products that subsequently were barcoded by an additional PCR (PCR2) reaction using the same reagents, but with barcoded primers. The final PCR products were purified using AMPure XP beads (Beckman Coulter) and pooled in equimolar concentrations. The pooled barcoded amplicons were ligated according to 1D genomic DNA using a ligation protocol (SQK-LSK109) to complete library preparation for sequencing on a R9.4.1 flow cell.
Processing of raw sequencing data
Paired ends of raw sequencing reads were merged with Usearch 11.0.667 [32] (-fastq_mergepairs) with default settings to ensure overlapping sequences of the forward and reverse reads. Subsequently, redundant sequences of primers and adaptors were removed with cutadapt 2.6 [33] (Fig. S4).
Bioinformatic analysis of sequencing and genomic data
The alignment package BWA [34], which is based on Burrow-Wheeler transformation, was used for alignment of short reads against the phage PMBT5 genome and visually interpreted with the use of Tablet 1.21.02.08 [35]. Samples with ≤30 reads that could be assigned to the PMBT5 phage genome were not considered, due to the numerous PCR cycles [28] and the cut of gel fragments that might had introduced minor contaminations. Local BLASTn [36] was used to match spacers originating from the type I-C CRISPR array of E. lenta DSM 15644 to viral genomes in the HuVirDB [16]. WebLogo [37] was used to visualize PAM sequences. CRISPRDetect [38] was used to identify CRISPR-Cas systems in genome sequences. The database of potential anti-CRISPR (Acr) protein [39] was used to screen for Acr proteins encoded by phage PMBT5 by the “-pblast” option with default settings in the alignment tool DIAMOND [40], and visualized in CLC Sequence viewer 8.0. The requirements of potential Acr protein candidates were set to a minimum 40% of the amino acid (AA) identity sequence, length at minimum 100 AA, and for the alignment to contain shared domains with contiguous sequences.
High-throughput qPCR (HT-qPCR) assays
The BioMark HD system was used for qPCR analysis with a Flex Six IFC chip (Fluidigm, San Francisco, California, USA) as previously described [23]. For bacteria and phage quantification, strain-specific primers (Table S1) were designed to target the cas1c gene (DSM15644-Cas1, NCBI GeneID: 69511386) in E. lenta DSM 15644 and a tail-associated lysin encoding gene (PMBT5-Tail, NCBI GeneID: 54998184) in PMBT5. A universal 16S rRNA primers targeting the V3-region was used as a control (Table S1). The quality of the primers was evaluated with AriaMX Real-time and Brilliant III Ultra-Fast SYBR Green Low ROX qPCR Master Mix (Agilent Technologies) prior to HT qPCR analysis as earlier described [23]. Bacterial culture of E. lenta DSM 15644 (~5 × 108 CFU/mL–OD600nm = ~0.25) was mixed with feces from germ-free mice prior DNA extraction to ensure that the genomic DNA used for the standard curve was treated as the investigated samples. The criteria for including a primer set for qPCR analysis was absence of primer dimers, no additional PCR fragments (evaluated by the melting curve), and a standard curve with efficiency between 98 and 102%, R2 > 0.991, slope ~ −3.2, and intercept around 38. Samples with less than 10 gene copies were discarded from the analysis.
Results
In this study we investigated the activity of the type I-C CRISPR-Cas system (Fig. 2) harbored by Eggerthella lenta DSM 15644, when the bacterial cells were infected with the virulent phage PMBT5 during either in vitro or in vivo settings. To investigate if the type I-C CRISPR-Cas system had previously acquired spacers from other phages, we aligned the 25 native spacers in the native CRISPR array with the HuVirDB (Human virome database) [16]. Only three spacers (S18, S9, and S7) were assigned to 7 viral contigs in the HuVirDB (Table S5). These matches were further supported by the spacers matching two recently assembled phage genomes [41]; S18 matched a Siphoviridae isolate (GenBank ID: BK046045.1) and S9 and S7 an unknown phage (GenBank ID: BK052885.1) [41]. None of the native spacers of E. lenta DSM 15644 matched the phage PMBT5 genome (Table S6).

R repeat, S spacer, TR terminal repeat.
Co-existence of E. lenta and phages in vitro
The infection of E. lenta DSM 15644 with the virulent phage PMBT5 was assayed at four different MOI for 144 h (Fig. 3A). The bacterial cultures infected at MOI 10 and 1 grew to a significantly (q < 5 × 10−7) higher cell density (OD600nm = 0.16–0.17, 48 h after infection) compared to MOI 0.1 and 0.01 (OD600nm = 0.04–0.05) (Fig. 3A). The cell density of the phage-infected cultures at MOI 10 and 1 was still markedly decreased (q < 5 × 10−6) compared to the bacterial cultures with no phages. The bacterial abundance was similar after 48 h of infection and decreased 0.3–0.8 log after 4 h after infection for all four MOI (Fig. 3B). The phage abundance in the cultures with MOI 0.01, 0.1, and 1 increased by 4.6, 3.3, and 2.0 log at 48 h, respectively. The MOI 10 culture on the other hand only had an increase of phage abundance with 1 log. This indicates that the phage titer was MOI dependent (Fig. 3C). Considering the MOI-dependent outcome of cell density (Fig. 3A) and phage abundance (Fig. 3C), the difference in the basic principles of measuring cell density (intact cells) and gene abundance (both intact and lysed cells) likely to explain why the bacterial gene abundance was similar for all MOI, while cell density was significantly different 48 h after infection.

A Growth curve of E. lenta DSM 15644 during infection with phage PMBT5 at four different multiplicities of infections (MOI) and a control with no phages added were performed with biological triplicates (n = 3). Bacterial growth was measured (absorbance at OD600nm) at several time points for 144 h. The bacterial (B) and phage abundance (C) was measured by qPCR (technical duplicates of the biological triplicates, n = 6). Primers designed to specifically target the genomes of E. lenta DSM 15644 (cas1 gene) and phage PMBT5 (gene coding for a tail-associated lysin) were used to measure total gene copies found in the cultures. A minimum threshold of 10 gene copies was applied. The error bars show the standard deviation within each MOI.
Type I-C CRISPR-Cas system of E. lenta can acquire new immunities in vitro and the new spacers preferentially target three hotspots in the phage PMBT5 genome
Different in vitro assays (Supplementary Methods) were performed to try to isolate CRISPR-protected bacteriophage insensitive mutants as well as plasmid interfering mutants, but to no avail (Fig. S5). However, sequencing of PCR-amplified CRISPR-arrays from whole populations of E. lenta DSM 15644 revealed 13 newly acquired spacers that matched phage PMBT5 genome in cultures with all four MOI (Figs. 4 and S6). The size of the acquired phage-associated spacers varied from 29–37 bp. The matching 13 unique protospacers were located in the genes coding for a phage terminase large subunit, portal-, type I restriction-modification-, arnA-like, SHOCT domain-containing-, replication initiator-, or four uncharacterized proteins (Fig. 4B, C, & Table S7). Based on these 13 protospacers, the PAM was predicted as 5ʹ-TTC, but no clear motifs could be detected in the flanking sequences on both sides of the protospacer (Fig. S7 and Table S7). Three out of the thirteen phage protospacers appeared as hotspots since they together represented 91.7% of the reads (174 637 reads out of 190 317 reads) matching the phage genome in all four MOI (Fig. 4B, C). These three protospacer hotspots were found within the coding sequences of a portal protein (gp04), SHOCT domain-containing protein (gp12), and a replication initiator protein (gp39) (Table S7). The ratio of spacer acquisitions from the hotspots varied between the MOI, e.g. the fraction of spacer acquisitions targeting a SHOCT domain-containing protein (gp12++) were 11.48%, 92.63%, 92.60%, and 14.43% for MOI 10, 1, 0.1, and 0.01, respectively (Table S7).

A Expanded CRISPR arrays generated by PCR on whole culture populations in selected samples (Fig. S6 for all samples) representing two replicates of all four MOI after 48 h and 24 h of incubation of E. lenta DSM 15644 exposed to phage PMBT5. DNA ladder is a 100-bp scale. With the degenerate primers, the expanded CRISPR array with one spacer “+1” was expected to yield a PCR product at ~110 bp (Fig. S1). No expanded CRISPR arrays were observed in samples with no added phages (after 48 h incubation) or with MilliQ water added. The PCR-product at ~40 bp likely represented primer dimers. B The annotated genome of phage PMBT5 highlights the genes that are presented in (C) with a bar plot showing the number of reads/spacers that matched to phage genes with biological triplicate (n = 3) at MOI 10, 1, 0.01, and 0.01. Three genes appeared as hotspots of spacer acquisitions and were coding for a portal protein (gp04+), a SHOCT domain-containing protein (gp12++), a replication initiator protein (gp39+++). These are marked by boxes with red dashed lines. A few genes were targeted at different positions within the same gene. D Graph illustrating a tendency of an inverse relation between MOI and cell density (OD600nm) of the average number of reads/spacer acquisitions in E. lenta DSM 15644 exposed to phage PMBT5 (n = 3). The error bars show the standard deviation within each MOI.
A relatively low number of reads matched the phage PMBT5 genome at MOI 10 and 1 (MOI 10: 2648 reads (0.09%) of total 2,792,090 reads, MOI 1: 15 832 reads (0.56%) of total 2,808,635 reads). The bacterial cultures infected at MOI 0.1 and 0.01 grew only to a low cell density, yet a relatively high fraction of cells acquired new spacers that matched the genome of phage PMBT5 (MOI 0.1: 72 397 reads (2.35%) of total 3,082,415 reads, MOI 0.01: 99 440 reads (3.66%) of total 2,719,689 reads). The number of spacer acquisitions matching the phage PMBT5 genome was almost linear from MOI 10 to 0.01, while bacterial biomass as determined by OD600nm had an inverse tendency with a decreased growth from MOI 10 to 0.01 (Fig. 4D). This suggested that a low MOI may favor the adaptation activity of the type I-C CRISPR-Cas system. Taken altogether, the type I-C CRISPR-Cas system of E. lenta DSM 15644 can acquire new spacers from an invading phage genome.
Sequencing of all samples yielded a total of 12,276,803 reads of which 1.55% (190 317 reads) contained spacer acquisition events that could be assigned to phage PMBT5 genome, but each read only contained a single phage-associated spacer acquisition. The remaining reads (98.45%) could be assigned to PCR products with no spacer acquisitions (primer dimers, 74%) and chromosomal DNA from E. lenta DSM 15644 (24.45%). The reads assigned to the chromosomal DNA covered the native CRISPR array (positions 1,572,740 to 1,574,444 bp) and showed a 100% nucleotide identity to 24 out of 25 spacers (Fig. S8). No reads matched other parts of the bacterial DNA. This phenomenon was observed at all four MOI as well as with the control without phage, suggesting that it did not depend on the presence of phages.
Efficient interference activity of the type I-C CRISPR-Cas system
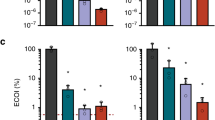
A plasmid interference assay was also conducted to further evaluate the functionality of the type I-C CRISPR-Cas system of E. lenta DSM 15644. Two protospacers, matching S1 and S2 from the native CRISPR-array of E. lenta, were cloned into vector pNZ123 with one of two PAMs (5ʹ-TTC or 5ʹ-GGG) and introduced into E. lenta. This yielded three transformants (15644-pNZ123::GGG-S2-GGG-S1, 15644-pNZ123::TTC-S2-TTC-S1, 15644-pNZ123::WT). While the 5ʹ-TTC motif was identified in our above phage assays, other studies [16, 25, 26] suggested that 5ʹ-GGG may be the PAM of the type I-C CRISPR-Cas system of E. lenta. Note that plasmid pNZ123 provides chloramphenicol resistance to the bacterial transformants. If the interference complexes of the type I-C CRISPR-Cas system recognize and cleave the two protospacers (S2 and S1), cloned into the pNZ123 vector, the chloramphenicol resistance will be lost and these bacterial transformants will be sensitive to the antibiotic. The efficiency of transformation (CFU/µg DNA) was clearly reduced (>5 logs) with the two recombinant plasmids pNZ123::GGG-S2-GGG-S1 and 15644 pNZ123::TTC-S2-TTC-S1 compared to the control pNZ123::WT (Fig. 5). These data indicate that the type I-C CRISPR-Cas system of E. lenta is also functional against plasmid invasion and has PAM flexibility.

E. lenta DSM 15644 was transformed with pNZ123 (WT) and two derivatives containing each the same two protospacers but a different PAM (pNZ123::GGG-S2-GGG-S1, pNZ123::TTC-S2-TTC-S1, pNZ123::WT). Absence of plasmid transformation indicates interference activity of the type I-C CRISPR-Cas system. Transformation assays were, respectively, replicated 2, 2, 4, and 4 times with 3 technical replicates. The error bars show the standard deviation.
Co-existence of E. lenta and phages in the gut of gnotobiotic mice
While spacer acquisition events could be noted when E. lenta DSM 15644 was infected with phage PMBT5 in vitro, this study also aimed to explore CRISPR-Cas activities in vivo. In total, 12 gnotobiotic (GB) mice were used to (i) investigate the coexistence of E. lenta DSM 15644 and phage PMBT5, and to (ii) see if the type I-C CRISPR-Cas system contribute to phage resistance. The mice received either a mixture of E. lenta (EL) and phages (EL + Phage) or EL and saline (EL + Saline) (Fig. 1). EL + Phage mice showed sustained co-existence of bacteria and phages throughout the study (Fig. 6); however, at day 26 both bacteria and phages simultaneously decreased in abundance. Phages appeared consistently to be 1 log higher compared to its bacterial host until day 19. E. lenta could co-exist with its antagonist virulent phage, as the bacterial abundance detected in the EL + Phage was comparable to the EL + Saline mice (Fig. 6).

Where day –1 were feces samples from GB mice sacrificed at the age of 3 weeks, day 0 were feces samples from GB mice when transferred from isolator to individual ventilated cages at another housing facility, and day 1 were just before culture inoculation. Primers designed to specifically target the genomes of E. lenta DSM 15644 (cas1 gene) and phage PMBT5 (gene coding for a tail-associated lysin) were used to measure total gene copies found in the feces samples. A minimum threshold of 10 gene copies was applied. The error bars show the standard deviation within each treatment group at the given day.
Temporary and limited CRISPR-Cas adaptation activity in the gut of gnotobiotic mice
The CAPTURE protocol [28] followed by sequencing was used again to investigate if the CRISPR array of E. lenta DSM 156444 had expanded during colonization in the gut of GB mice. In contrary to the in vitro settings, only one newly acquired spacer (with 75,742 reads out of 76,846 total phage-associated reads, 98.6%) targeted a phage gene coding for a DNA gyrase inhibitor protein (gp34, Fig. 7) and was detected in the EL + Phage mice at day 12 until day 26. The PAM for this single protospacer was 5ʹ-TTC (Fig. S7). The sequencing yielded a total of 10,716,969 reads of which only 0.7% contained spacer acquisitions that could be assigned to the phage PMBT5 genome. The remaining reads (99.3%) were assigned to PCR products with no spacer acquisition (primer dimers, 89.2%), and to the E. lenta genome (10.1%) as also observed in the in vitro experiment. The size of the DNA fragments on an agarose gel (Fig. 7) suggested expanded CRISPR arrays containing even multiple spacer acquisitions, but the sequences of these spacers matched the native CRISPR array in the E. lenta genome (Fig. S8) and not the phage genome. These CRISPR arrays that were expanded with multiple spacers were observed both in samples from EL + Phage mice (Fig. 7), EL + Saline mice, and pure bacterial cultures of E. lenta DSM 15644 (Fig.S9), suggesting that the observation was independent of the presence of phages. Overall, the results indicated a temporary and limited CRISPR-Cas mediated adaptation activity when exposed to phage PMBT5 in a GB mouse model.

A An agarose gel showing expanded CRISPR arrays generated by PCR on whole populations in selected samples representing the four EL + Phage mice (Mouse ID; 9, 10, 11, 12) from day 5, 12, 19, and 26, as well as from controls at arrival (Day 1) and baseline mice (Mouse ID: 1, 2, 3, 4). A 100-bp DNA ladder was used to estimate PCR product size. With the degenerate primers, the acquisition of one spacer “+1” was expected to yield a PCR product at ~110 bp (Fig. S1) and then ~70 bp for additional spacers. The PCR-product at ~40 bp likely represented primer dimers. B The annotated phage genome of PMBT5 highlights the one gene coding for a DNA gyrase inhibitor (gp34) that is presented in (C) with a line plot showing average number of reads/spacers over time (n = 4) that matched the phage genome. The error bars show the standard deviation within each treatment group at the given day.
Discussion
Here we report the activity of a type I-C CRISPR-Cas system harbored by the prevalent human gut bacterium E. lenta [42] when exposed to virulent phages in both in vitro and in vivo settings. With a highly sensitive PCR-based protocol [28] and sequencing, we detected MOI-dependent CRISPR-Cas adaptation activity against phage PMBT5 when infecting E. lenta DSM 15644. The bacterial cultures infected at MOI of 0.1 and 0.01 had a relatively higher number of new spacers matching the phage genome as compared to cultures infected at MOI 10 and 1 (Fig. 4). The determination of bacterial and phage abundance by qPCR revealed that phage abundance was MOI-dependent as it increased by 4.6 log at an initial MOI of 0.01 compared to a 1 log increase at an MOI of 10 after 48 h of incubation. On the other hand, the bacterial abundance was similar at all four MOI. In contrast, the cell density (measured by absorbance) at MOI 10 was significantly higher than at a MOI of 0.01 (Fig. 3A). This suggested that at a MOI of 10, the host-phage dynamics favored sub-populations of bacteria with other phage resistance strategies [43,44,45] than CRISPR-Cas immunity [46]. At the lower MOI of 0.01, the host-phage dynamics favored diverse CRISPR-Cas immunity as well as constant production of phage particles until 48 h of incubation (Fig. 3C). This indicated that cells with CRISPR-Cas immunity against phage PMBT5 were not sufficient to hamper the replication of phage particles and/or that a substantial population did not acquire CRISPR-immunity and hence remained susceptible to infection. It was not possible to detect anti-CRISPR phage (Acr) proteins with high confidence (Table S8 and Fig. S10). Similar host-phage dynamics have previously been suggested between Pseudomonas aeruginosa PA14 and its phage DMS3vir, where high risk of infection (high titers) was associated with frequent mutations in phage receptors, whereas CRISPR-Cas immunity was less frequent [47].
Using in vitro settings, 13 protospacers of phage PMBT5 were targeted at all four MOI, of which 3 appeared at preferred targets while only 1 protospacer was targeted in the in vivo settings of the GB mice. These 3 hotspots of spacer acquisition were located within genes coding for a portal protein (involved in virion assembly, DNA packaging, and DNA delivery [48, 49]), SHOCT domain-containing protein (suggested to be involved in oligomerization and nucleic acid binding [50]), replication initiator protein (essential for precise initiation and termination of replication [51]), and a DNA gyrase inhibitor protein (inhibits the replication of DNA and transcription process [52, 53]) (Figs. 4 and 7, and Table S7). Considering that only 1 new spacer acquisition was detected in GB mice, it suggests that the type I-C CRISPR-Cas in E. lenta DSM 15644 does not constitute the main phage resistance strategy under the investigated conditions. Our findings may be supported by another study using plants as a model, showing that phage resistance evolution in vitro is not reflected in vivo [54], and that both biotic and abiotic parameters affect evolution of phage resistance, including CRISPR-Cas immunity [55]. The distinctly different environmental conditions for host-phage interactions in test tubes versus the spatial heterogeneity found in real gastrointestinal conditions in GB mice [56] may explain this clear difference in the number of unique acquired spacers between the in vitro and in vivo settings. Based on the 14 protospacers (both from the in vitro and in vivo experiments), the adaptation PAM was predicted as 5′-TTC (Figure S7), which is in agreement with another study that predicted similar adaptation PAM for type I-C CRISPR-Cas systems in 15 different E. lenta strains using computational approaches [16].
The numbers of reads representing the acquired spacers can only be considered as relative arbitrary values due to the basic principles of the CAPTURE protocol [28], which prevent quantitative analyses. The differences in the number of reads between protospacers should therefore be interpreted in the light of their relative abundance nature and that the method cannot differentiate between free DNA, non-viable cells, or viable cells. Considering the low relative abundance of reads with spacer acquisition after numerous PCR cycles and the sensitivity limit of degenerate primers (1 spacer acquisition per 105 cells) [28], it appears that spacer acquisition may be relatively rare in E. lenta DSM 15644 when exposed to phage PMBT5 in both in vitro and in vivo settings. This would also be in accordance with other studies investigating spacer acquisitions under laboratory conditions [57, 58]. A hypothetical protein (gp31) and a type I restriction-modification protein (gp05) encoded by phage PMBT5 had identical (E-value <10−23) AA domains as four computational predicted Acr protein clusters [39] (gp31: cluster 2517 + 20298 and gp05: cluster 12618 + 59,526) (Fig. S10). The phage protein sequences of PMBT5 were also aligned with 108 confirmed Acr proteins which showed low confidence matches (E-value <10−2) between three PMBT5 proteins and four Acr proteins associated to CRISPR-Cas system type II-A and VI-A (Table S8). If these phage proteins contain Acr features, it might have challenged the detection of spacer acquisitions in E. lenta and thereby limited CRISPR-Cas immunity. However, experimental studies need to be conducted to validate the presence of Acr proteins associated with phage PMBT5.
In both the in vitro and in vivo settings, less than 2% of the total sequenced reads could be assigned to the phage PMBT5 genome, while up to ~25% were assigned to the genome of E. lenta, and the remaining reads were primer dimers. The reads that matched to the chromosomal DNA of E. lenta framed almost the entire native CRISPR array (Fig. S8) and no other bacterial genes. The associated expanded CRISPR arrays even appeared with multiple spacer acquisitions (Fig. S9). This phenomenon was detected in all samples independent of the presence of phages in the in vivo samples. Whether this observation has biological relevance or is just PCR-generated artefacts is not known. It does not seem likely that these observations are “real” spacer acquisition since PAMs are necessary for spacer acquisition and PAMs are not present in the CRISPR array [59]. Other potential biological explanations may be homologous recombination (driven by the repeats) or a mechanism where the spacers are shuffled to increase expression as spacers are more expressed at the leader end of the CRISPR array. Self-targeting immune memories of CRISPR-Cas have previously been demonstrated [60,61,62], but does not seem to explain our observation of spacers matching the CRISPR array of the host, since no chromosomal genes were targeted.
Using a plasmid system in which we cloned two spacers (S2 and S1) originating from the native CRISPR array of E. lenta DSM 15644, we showed clear interference activity of the type I-C CRISPR-Cas system, including the necessity of the PAM 5ʹ-GGG and 5ʹ-TTC (Fig. 5). Considering that the adaptation and interference stages consist of different protein complexes being formed, the PAM requirements may be different for both stages [63]. Thereby explaining why both PAM 5ʹ-GGG and 5ʹ-TTC showed high interference efficiency, while 5ʹ-TTC appeared to be the preferred PAM that is involved in spacer acquisition. However, one protospacer was detected with 5ʹ-GGG as PAM (Table S7). The observed interference activity of the type I-C CRISPR-Cas system in E. lenta DSM 15644 is in line with another study that reported transcription and interference activity of a type I-C CRISPR-Cas system from the closely related strain E. lenta DSM 2243T [16]. Soto-Perez et al. conducted an experimental design using the evolutionary distinct (from E. lenta) bacterium P. aeruginosa and its associated P. aeruginosa phages [16]. Whereas we investigated CRISPR-Cas immunity of E. lenta using natural host-phage relations. Despite the high genomic similarity between E. lenta DSM 2243T and DSM 15644 [16], the E. lenta DSM 2243T showed no susceptibility against phage PMBT5 (Fig. S11).
The phage PMBT5 was highly virulent in vitro since the bacterial culture was completely cleared after phage amplification (Fig. S11). It is therefore intriguing, why the bacterial abundance was stable and similar with and without the presence of this seemingly highly virulent phage (Fig. 6) during the 26 days in GB mice. The contribution of physical parameters should not be neglected, since small cavities in the intestinal lumen, mucus production (from host cells) [64, 65], protection by numerous bacterial cell layers in microcolony structures [66,67,68], and the overall spatial distribution in the gut may have protected the bacteria from the phages. This would be in accordance with a study showing that the spatial heterogeneity of the gut limits predation and favors the coexistence of phages and bacteria [56]. Other studies have also shown host-phage coexistence in different experimental and theoretical settings using the bacterium Streptococcus thermophilus and its phage 2972 [69, 70]. Avoiding infections would mean less phage resistance, and perhaps even avoiding impaired fitness that is sometimes associated with phage resistance [43,44,45].
Both qPCR and sequencing targeting the 16S rRNA gene of bacteria revealed high numbers of gene copies (>109 copies pr. gram feces) and sequence analysis revealed additional organisms in addition to E. lenta DSM 15,644 (Fig. S3) in the GB mice. The high number of 16S rRNA gene copies may partly be explained by the primers being able to bind to the 16S rRNA region of chloroplast found in plants [71, 72]. The 16S rRNA gene amplicon sequencing of the mouse feed showed up to 88% relative abundance of reads of plant origin (Fig. S3). Given the enlarged cecum and the absence of growth after plating, we speculated that these observations reflect dead bacteria killed by sterilization of the feed. The majority of the bacterial taxa detected by sequencing was associated to spore-forming taxa [73,74,75,76,77,78,79]. For example, spore-forming Clostridium spp. was detected in both feces and feed. It is possible that spore DNA may still be detectable after sterilization by gamma irradiation [80, 81]. The remaining spore forming taxa was not detected in the mouse feed. The E. lenta host-phage pair co-existence remained stable despite the detection of other bacterial species by 16S rRNA gene sequencing, but we cannot rule out that it can have affected the animal model.
Although, the CRISPR-Cas system only provided limited phage immunity, this study showed the activity of the type I-C CRISPR-Cas system in E. lenta targeting its antagonist phage in both in vitro and in vivo settings.
Data availability
Most of the datasets generated during and/or analyzed during the current study are available in the OSF repository (https://osf.io/n5dqj), while the remaining datasets are available from the corresponding author on reasonable request. Sequencing data are available through the European nucleotide archive (ENA) with the accession numbers PRJEB47947 and PRJEB52384.
References
McGinn J, Marraffini LA. Molecular mechanisms of CRISPR-Cas spacer acquisition. Nat Rev Microbiol. 2019;17:7–12.
Brouns SJJ, Jore MM, Lundgren M, Westra ER, Slijkhuis RJH, Snijders APL, et al. Small CRISPR RNAs guide antiviral defense in prokaryotes. Science. 2008;321:960–4.
Barrangou R, Fremaux C, Deveau H, Richards M, Boyaval P, Moineau S, et al. CRISPR provides acquired resistance against viruses in prokaryotes. Science. 2007;315:1709–12.
Ishino Y, Shinagawa H, Makino K, Amemura M, Nakatura A. Nucleotide sequence of the iap gene, responsible for alkaline phosphatase isoenzyme conversion in Escherichia coli, and identification of the gene product. J Bacteriol. 1987;169:5429–33.
Bolotin A, Quinquis B, Sorokin A, Ehrlich SD. Clustered regularly interspaced short palindrome repeats (CRISPRs) have spacers of extrachromosomal origin. Microbiol (Read). 2005;151:2551–61.
Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, et al. Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol. 2019;18:67–83.
Makarova KS, Wolf YI, Alkhnbashi OS, Costa F, Shah SA, Saunders SJ, et al. An updated evolutionary classification of CRISPR-Cas systems. Nat Rev Microbiol. 2015;13:722–36.
Hochstrasser ML, Taylor DW, Kornfeld JE, Nogales E, Doudna JA. DNA targeting by a minimal CRISPR RNA-guided cascade. Mol Cell. 2016;63:840–51.
Lee H, Zhou Y, Taylor DW, Sashital DG. Cas4-dependent prespacer processing ensures high-fidelity programming of CRISPR arrays. Mol Cell. 2018;70:48–59.e5.
Lee H, Dhingra Y, Sashital DG. The Cas4-Cas1-Cas2 complex mediates precise prespacer processing during CRISPR adaptation. eLife. 2019;8:1–26.
Rasmussen TS, Koefoed AK, Jakobsen RR, Deng L, Castro-Mejía JL, Brunse A, et al. Bacteriophage-mediated manipulation of the gut microbiome - promises and presents limitations. FEMS Microbiol Rev. 2020;44:507–21.
Bernheim A, Sorek R. The pan-immune system of bacteria: antiviral defence as a community resource. Nat Rev Microbiol. 2020;18:113–9.
Louwen R, Staals RHJ, Endtz HP, van Baarlen P, van der Oost J. The role of CRISPR-Cas systems in virulence of pathogenic bacteria. Microbiol Mol Biol Rev. 2014;78:74–88.
Grissa I, Vergnaud G, Pourcel C. CRISPRcompar: a website to compare clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2008;36:W145–8.
Tajkarimi M, Wexler HM. CRISPR-Cas systems in Bacteroides fragilis, an important pathobiont in the human gut microbiome. Front Microbiol. 2017;8:2234.
Soto-Perez P, Bisanz JE, Berry JD, Lam KN, Bondy-Denomy J, Turnbaugh PJ. CRISPR-Cas system of a prevalent human gut bacterium reveals hyper-targeting against phages in a human virome catalog. Cell Host Microbe. 2019;26:325–35.e5.
Cornuault JK, Moncaut E, Loux V, Mathieu A, Sokol H, Petit M-A, et al. The enemy from within: a prophage of Roseburia intestinalis systematically turns lytic in the mouse gut, driving bacterial adaptation by CRISPR spacer acquisition. ISME J. 2020;14:771–87.
Pan M, Nethery MA, Hidalgo-Cantabrana C, Barrangou R. Comprehensive mining and characterization of CRISPR-Cas systems in. Bifidobacterium Microorg. 2020;8:720.
Koh A, Molinaro A, Ståhlman M, Khan MT, Schmidt C, Mannerås-Holm L, et al. Microbially produced imidazole propionate impairs insulin signaling through mTORC1. Cell. 2018;175:947–61.e17.
Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature. 2012;490:55–60.
Sprotte S, Rasmussen TS, Cho G, Brinks E, Lametsch R, Neve H, et al. Morphological and genetic characterization of Eggerthella lenta bacteriophage PMBT5. Viruses. 2022;14:1–16.
Rasmussen TS, Streidl T, Hitch TCAA, Wortmann E, Deptula P, Kofoed MVW, et al. Sporofaciens musculi gen. nov., sp. nov., a novel bacterium isolated from the caecum of an obese mouse. Int J Syst Evol Microbiol. 2021;71:004673.
Muhammed MK, Krych L, Nielsen DS, Vogensen FK. A high-throughput qPCR system for simultaneous quantitative detection of dairy Lactococcus lactis and Leuconostoc bacteriophages. PLoS One. 2017;12:e0174223.
De Vos W. Gene cloning and expression in lactic streptococci. FEMS Microbiol Lett. 1987;46:281–95.
Rao C, Chin D, Ensminger AW. Priming in a permissive type I-C CRISPR-Cas system reveals distinct dynamics of spacer acquisition and loss. RNA. 2017;23:1525–38.
Burmistrz M, Rodriguez Martinez JI, Krochmal D, Staniec D, Pyrc K. Clustered regularly interspaced short palindromic repeat (CRISPR) RNAs in the Porphyromonas gingivalis CRISPR-Cas I-C system. J Bacteriol. 2017;199:e00275–17.
Lemay M-L, Tremblay DM, Moineau S. Genome engineering of virulent lactococcal phages using CRISPR-Cas9. ACS Synth Biol. 2017;6:1351–8.
McKenzie RE, Almendros C, Vink JNA, Brouns SJJ. Using CAPTURE to detect spacer acquisition in native CRISPR arrays. Nat Protoc. 2019;14:976–90.
Rasmussen TS, de Vries L, Kot W, Hansen LH, Castro-Mejía JL, Vogensen FK, et al. Mouse vendor influence on the bacterial and viral gut composition exceeds the effect of diet. Viruses. 2019;11:435.
Krych Ł, Kot W, Bendtsen KMB, Hansen AK, Vogensen FK, Nielsen DS. Have you tried spermine? A rapid and cost-effective method to eliminate dextran sodium sulfate inhibition of PCR and RT-PCR. J Microbiol Methods. 2018;144:1–7.
Hui Y, Tamez-Hidalgo P, Cieplak T, Satessa GD, Kot W, Kjærulff S, et al. Supplementation of a lacto-fermented rapeseed-seaweed blend promotes gut microbial- and gut immune-modulation in weaner piglets. J Anim Sci Biotechnol. 2021;12:1–14.
Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–1.
Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17:10.
Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–60.
Milne I, Stephen G, Bayer M, Cock PJAA, Pritchard L, Cardle L, et al. Using Tablet for visual exploration of second-generation sequencing data. Brief Bioinform. 2013;14:193–202.
Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. NCBI BLAST: a better web interface. Nucleic Acids Res. 2008;36:W5–9.
Crooks GE, Hon G, Chandonia J-M, Brenner SE. WebLogo: a sequence logo generator. Genome Res. 2004;14:1188–90.
Biswas A, Staals RHJ, Morales SE, Fineran PC, Brown CM. CRISPRDetect: A flexible algorithm to define CRISPR arrays. BMC Genomics. 2016;17:356.
Gussow AB, Park AE, Borges AL, Shmakov SA, Makarova KS, Wolf YI, et al. Machine-learning approach expands the repertoire of anti-CRISPR protein families. Nat Commun. 2020;11:3784.
Buchfink B, Xie C, Huson DH. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 2015;12:59–60.
Tisza MJ, Buck CB. A catalog of tens of thousands of viruses from human metagenomes reveals hidden associations with chronic diseases. Proc Natl Acad Sci USA. 2021;118:1–11.
Koppel N, Bisanz JE, Pandelia M-E, Turnbaugh PJ, Balskus EP. Discovery and characterization of a prevalent human gut bacterial enzyme sufficient for the inactivation of a family of plant toxins. eLife. 2018;7:33953.
Koskella B, Lin DM, Buckling A, Thompson JN. The costs of evolving resistance in heterogeneous parasite environments. Proc Biol Sci. 2012;279:1896–903.
Harcombe WR, Bull JJ. Impact of phages on two-species bacterial communities. Appl Environ Microbiol. 2005;71:5254–9.
Seed KD, Yen M, Shapiro BJ, Hilaire IJ, Charles RC, Teng JE, et al. Evolutionary consequences of intra-patient phage predation on microbial populations. eLife. 2014;3:e03497.
Hampton HG, Watson BNJ, Fineran PC. The arms race between bacteria and their phage foes. Nature. 2020;577:327–36.
Meaden S, Capria L, Alseth E, Gandon S, Biswas A, Lenzi L, et al. Phage gene expression and host responses lead to infection-dependent costs of CRISPR immunity. ISME J. 2021;15:534–44.
Lokareddy RK, Sankhala RS, Roy A, Afonine PV, Motwani T, Teschke CM, et al. Portal protein functions akin to a DNA-sensor that couples genome-packaging to icosahedral capsid maturation. Nat Commun. 2017;8:1–11.
Prevelige PE, Cortines JR. Phage assembly and the special role of the portal protein. Curr Opin Virol. 2018;31:66–73.
Eberhardt RY, Bartholdson SJ, Punta M, Bateman A. The SHOCT domain: a widespread domain under-represented in model organisms. PLoS One. 2013;8:1–7.
Campos-Olivas R, Louis JM, Clérot D, Gronenborn B, Gronenborn AM. The structure of a replication initiator unites diverse aspects of nucleic acid metabolism. Proc Natl Acad Sci USA. 2002;99:10310–5.
Reece RJ, Maxwell A, Wang JC. DNA gyrase: structure and function. Crit Rev Biochem Mol Biol. 1991;26:335–75.
Chitra SR, Ramalakshmi N, Arunkumar S, Manimegalai P. A comprehensive review on DNA gyrase inhibitors. Infect Disord Drug Targets. 2020;20:765–77.
Hernandez CA, Koskella B. Phage resistance evolution in vitro is not reflective of in vivo outcome in a plant-bacteria-phage system. Evolution (N. Y). 2019;73:2461–75.
Alseth EO, Pursey E, Luján AM, McLeod I, Rollie C, Westra ER. Bacterial biodiversity drives the evolution of CRISPR-based phage resistance. Nature. 2019;574:549–52.
Lourenço M, Chaffringeon L, Lamy-Besnier Q, Pédron T, Campagne P, Eberl C, et al. The spatial heterogeneity of the gut limits predation and fosters coexistence of bacteria and bacteriophages. Cell Host Microbe. 2020;28:390–401.e5.
McGinn J, Marraffini LA. CRISPR-Cas systems optimize their immune response by specifying the site of spacer integration. Mol Cell. 2016;64:616–23.
Heler R, Samai P, Modell JW, Weiner C, Goldberg GW, Bikard D, et al. Cas9 specifies functional viral targets during CRISPR-Cas adaptation. Nature. 2015;519:199–202.
Kieper SN, Almendros C, Behler J, McKenzie RE, Nobrega FL, Haagsma AC, et al. Cas4 facilitates PAM-compatible spacer selection during CRISPR adaptation. Cell Rep. 2018;22:3377–84.
Weissman JL, Stoltzfus A, Westra ER, Johnson PLF. Avoidance of self during CRISPR immunization. Trends Microbiol. 2020;28:543–53.
Stern A, Keren L, Wurtzel O, Amitai G, Sorek R. Self-targeting by CRISPR: gene regulation or autoimmunity? Trends Genet. 2010;26:335–40.
Stachler A-E, Turgeman-Grott I, Shtifman-Segal E, Allers T, Marchfelder A, Gophna U. High tolerance to self-targeting of the genome by the endogenous CRISPR-Cas system in an archaeon. Nucleic Acids Res. 2017;45:5208–16.
Shah SA, Erdmann S, Mojica FJM, Garrett RA. Protospacer recognition motifs: mixed identities and functional diversity. RNA Biol. 2013;10:891–9.
Sausset R, Petit MA, Gaboriau-Routhiau V, de Paepe M. New insights into intestinal phages. Mucosal Immunol. 2020;13:205–15.
Yang C, Ottemann KM. Control of bacterial colonization in the glands and crypts. Curr Opin Microbiol. 2019;47:38–44.
Eriksen RS, Mitarai N, Sneppen K. Sustainability of spatially distributed bacteria-phage systems. Sci Rep. 2020;10:3154.
Eriksen RS, Svenningsen SL, Sneppen K, Mitarai N. A growing microcolony can survive and support persistent propagation of virulent phages. Proc Natl Acad Sci USA. 2017;115:337–42.
Abedon ST. Phage ‘delay’ towards enhancing bacterial escape from biofilms: a more comprehensive way of viewing resistance to bacteriophages. AIMS Microbiol. 2017;3:186–226.
Weissman JL, Holmes R, Barrangou R, Moineau S, Fagan WF, Levin B, et al. Immune loss as a driver of coexistence during host-phage coevolution. ISME J. 2018;12:585–97.
Common J, Morley D, Westra ER, van Houte S. CRISPR-Cas immunity leads to a coevolutionary arms race between Streptococcus thermophilus and lytic phage. Philos Trans R Soc Lond B Biol Sci. 2019;374:20180098.
Hanshew AS, Mason CJ, Raffa KF, Currie CR. Minimization of chloroplast contamination in 16S rRNA gene pyrosequencing of insect herbivore bacterial communities. J Microbiol Methods. 2013;95:149–55.
Dron M, Rahire M, Rochaix JD. Sequence of the chloroplast 16S rRNA gene and its surrounding regions of Chlamydomonas reinhardii. Nucleic Acids Res. 1982;10:7609–20.
Gupta RS, Patel S. Robust demarcation of the family Caryophanaceae (Planococcaceae) and its different genera including three novel genera based on phylogenomics and highly specific molecular signatures. Front Microbiol. 2019;10:2821.
Tanaka M, Onizuka S, Mishima R, Nakayama J. Cultural isolation of spore-forming bacteria in human feces using bile acids. Sci Rep. 2020;10:15041.
Browne HP, Almeida A, Kumar N, Vervier K, Adoum AT, Viciani E, et al. Host adaptation in gut Firmicutes is associated with sporulation loss and altered transmission cycle. Genome Biol. 2021;22:204.
Beno SM, Cheng RA, Orsi RH, Duncan DR, Guo X, Kovac J, et al. Paenibacillus odorifer, the predominant Paenibacillus species isolated from milk in the United States, demonstrates genetic and phenotypic conservation of psychrotolerance but clade-asociated differences in nitrogen metabolic pathways. mSphere. 2020;5:e00739–19.
Paredes-Sabja D, Torres JA, Setlow P, Sarker MR. Clostridium perfringens spore germination: characterization of germinants and their receptors. J Bacteriol. 2008;190:1190–201.
Shen A, Edwards AN, Sarker MR, Paredes-Sabja D. Sporulation and germination in clostridial pathogens. Microbiol Spectr. 2019;7:903–26.
Setlow P. Germination of spores of Bacillus species: what we know and do not know. J Bacteriol. 2014;196:1297–305.
Setlow P, Christie G. What’s new and notable in bacterial spore killing! World J Microbiol Biotechnol. 2021;37:144.
Setlow P. Spores of Bacillus subtilis: Their resistance to and killing by radiation, heat and chemicals. J Appl Microbiol. 2006;101:514–25.
Acknowledgements
We thank Professor Alejandro Reyes (Universidad de los Andes, Colombia) for initial discussions of the design of the gnotobiotic mouse (GB) model and we also thank Dr. Frank Hille (Max Rubner-Institut, Germany) for suggestions that improved the initial draft of the manuscript. In addition, we are grateful to Helene Farlov and Mette Nelander at Section of Experimental Animal Models (University of Copenhagen, Denmark) for handling GB mice in the isolator, as well as the staff at Department of Experimental Medicine (University of Copenhagen, Denmark) for their cooperation in housing the GB mice. We also thank lab technicians Pernille Lærke Jensen and Emilie Gaal at Section of Microbiology and Fermentations (University of Copenhagen, Denmark) for helping with the interference activity assay.
Funding
Funding was provided by the Danish Council for Independent Research with grant ID: DFF-6111-00316 (PhageGut). This work is supported by the Joint Programming Initiative “Healthy Diet for a Healthy Life”, specifically here, the Danish Agency for Science and Higher Education. S.M. acknowledges funding from the Canadian Institutes of Health Research (Team grant on Intestinal Microbiomics, Institute of Nutrition, Metabolism, and Diabetes). S.M. holds a Tier 1 Canada Research Chair in Bacteriophages.
Author information
Authors and Affiliations
Contributions
TSR, DSN, AKH, and SM conceived the research idea and designed the study; TSR and AKK performed the experiments; TSR, AKK, DSN, SM, GR, WK, LD, MKM, SS, HN, CMAPF, and FKV performed laboratory and data analysis; TSR wrote the first draft of the manuscript. All authors critically revised and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Rasmussen, T.S., Koefoed, A.K., Deng, L. et al. CRISPR-Cas provides limited phage immunity to a prevalent gut bacterium in gnotobiotic mice. ISME J 17, 432–442 (2023). https://doi.org/10.1038/s41396-023-01358-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-023-01358-4
