
Abstract
Several abundant but yet uncultivated bacterial groups exist in extreme iron- and sulfur-rich environments, and the physiology, biodiversity, and ecological roles of these bacteria remain a mystery. Here we retrieved four metagenome-assembled genomes (MAGs) from an artificial acid mine drainage (AMD) system, and propose they belong to a new deltaproteobacterial order, Candidatus Acidulodesulfobacterales. The distribution pattern of Ca. Acidulodesulfobacterales in AMDs across Southeast China correlated strongly with ferrous iron. Reconstructed metabolic pathways and gene expression profiles showed that they were likely facultatively anaerobic autotrophs capable of nitrogen fixation. In addition to dissimilatory sulfate reduction, encoded by dsrAB, dsrD, dsrL, and dsrEFH genes, these microorganisms might also oxidize sulfide, depending on oxygen concentration and/or oxidation reduction potential. Several genes with homology to those involved in iron metabolism were also identified, suggesting their potential role in iron cycling. In addition, the expression of abundant resistance genes revealed the mechanisms of adaptation and response to the extreme environmental stresses endured by these organisms in the AMD environment. These findings shed light on the distribution, diversity, and potential ecological role of the new order Ca. Acidulodesulfobacterales in nature.
Similar content being viewed by others


Introduction
Iron and sulfur are biologically important elements that are cycled dynamically between the geosphere and biosphere. Microorganisms capable of catalyzing dissimilatory redox transformations of sulfur and iron had a profound impact on Earth evolution [1, 2], and thus have drawn extensive attention from different research fields. Both elements participate in the formation of many minerals, particularly pyrite, which is ubiquitous in nature [3]. When pyrite and other sulfide minerals are exposed to air and water at Earth’s surface, microbially catalyzed oxidative dissolution occurs, generating acid mine drainage (AMD) [4]. AMD, typically characterized by extreme acidity and elevated concentrations of metals and sulfate, represents an extreme habitat to life, as well as a major global environmental challenge [5]. Iron and sulfur oxidations are the primary biochemical transformations occurring in AMD [6], and hence numerous studies have revealed microbial diversity, metabolic functions, and ecological roles of iron- and/or sulfur-oxidizing microbes affiliated with Acidithiobacillus, Leptospirillum, “Ferrovum”, Ferroplasma, Thermoplasmatales, and the ARMAN (archaeal Richmond Mine acidophilic nanoorganisms) in AMD ecosystems [7,8,9,10]. Moreover, the acidophiles involved in the subsequent reduction of oxidized sulfur and iron species (sulfate and ferric iron reduction) in AMD ecosystems cannot be neglected because they are thought to retard AMD generation and contribute to AMD bioremediation [11,12,13]. So far, a few studies have surveyed the taxonomic diversity of acidophilic sulfate-reducing microorganisms (aSRMs) in AMD ecosystems [12, 14, 15], and several pure cultures of aSRMs have been studied, such as Desulfosporosinus acidiphilus, Desulfosporosinus acididurans, and Desulfovibrio sp. TomC [16,17,18]. Even so, some abundant microorganisms in AMD ecosystems remain elusive.
In the 2000s, uncultured bacteria BA71 and BA18, affiliated with the deltaproteobacterial Candidate Sva0485 clade were first reported and speculated as potential sulfate/iron reducers, accounting for 4.30 and 1.08%, respectively, of the 16S ribosomal RNA (rRNA) gene clone library of a slime biofilm from the Richmond Mine [19]. Since then, numerous studies have affirmed the presence of the Sva0485 clade (relative abundance ≥ 5%) in AMD ecosystems (including water, sediments, and biofilms) [20,21,22,23,24], including Xiang Mountain, China (Fig. 1c), where they make up 51.1% of a 16S rRNA gene clone library in an AMD sediment [25]. This clade was also found in diverse environments, such as Fe nodules from Quaternary sediments (relative abundance, 9.4%), an inactive hydrothermal sulfide chimney in the deep sea (relative abundances, 12.3–13.8%), and a deep ferruginous sediment from Lake Towuti (relative abundances, ~2–20%) [26,27,28]. The clade is considered to be composed of potential sulfate-reducing bacteria (SRB) as it is commonly found in active sulfate-reducing consortia [29,30,31]. However, because of the lack of microbial isolates or genomes, the physiology and ecological role of this taxon remains unknown.

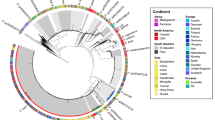
The reconstructed genomes of Ca. Acidulodesulfobacterales. a Geographical location of an artificial AMD system from which AMD samples were collected, with ESOM image highlighting the four genomes within metagenomic data. b Relative metagenomic abundance and metatranscriptomic abundance of the four Ca. Acidulodesulfobacterales genomes. S1 through S5 represent July 2016, August 2016, December 2016, February 2017, and August 2017 samples, respectively. c A maximum-likelihood phylogenetic tree of 16S rRNA genes from four genomic bins and representatives of the class Deltaproteobacteria, and the abundance distribution pattern of Ca. Acidulodesulfobacterales in natural environments. d Phylogenetic placement of the four bins and their orthoANI and 16S rRNA similarity values. The maximum-likelihood phylogenetic tree was constructed using the concatenated alignment of 16 ribosomal proteins. Bootstrap values were based on 100 replicates, and percentages ≥50% are shown with black circles
In this study, four draft genomes belonging to the Sva0485 clade were retrieved from AMD metagenomes and the name “Candidatus Acidulodesulfobacterales” (Ca. Acidulodesulfobacterales) is proposed for this taxon. Then, we surveyed the distribution patterns of these species in AMDs across Southeast China and one artificial AMD system, and evaluated the deterministic factors explaining the spatial distribution pattern of Ca. Acidulodesulfobacterales. In addition, the potential roles of these new species in nature were probed by reconstructing their metabolic pathways and examining in situ gene expression by metatranscriptomics. This study expands our understanding of the biogeography, taxonomic diversity, and ecological role of this new order Ca. Acidulodesulfobacterales.
Materials and methods
Sampling, physicochemical analyses, DNA and RNA extraction and sequencing
An artificial system for simulating and tracking the oxidation of natural pyrite was established with material collected from Yunfu Pyrite Mine, Guangdong Province, on 26 June 2009 in a ventilated greenhouse on the Sun Yat-sen University campus (Fig. 1a) [32]. The details of this artificial system and the pyrite oxidation experiment were described in our previous study [32]. In July 2016 (S1), August 2016 (S2), December 2016 (S3), February 2017 (S4), and August 2017 (S5), representing a seasonal cycle of summer–winter–summer, approximately 50 L of acidic water was pre-filtered using 3.0 and 0.8 μm filters (149 mm diameter; Whatman) to remove coarse particles and eukaryotes [33], and then the filtrate was concentrated to ~500 mL by tangential flow filtration system (PES membrane, 1000 kDa pore size; Merck Millipore, Germany). Each concentrate was divided equally into two parts: one for DNA extraction and the other for RNA extraction, and then was centrifuged (10,000 × g, 10 min, 4 °C) to obtain a cell pellet. The samples for RNA extraction were preserved in 1 mL of RNAlater (Ambion) and all samples were stored at −80 °C until nucleic acid extraction. Physicochemical characteristics were measured as previously reported [34], and summarized in Supplementary Table S1. The detailed methods are provided in the Supplementary Information, including details of genomic DNA and RNA isolation and sequencing.
Genome-resolved metagenomic analysis
Of raw reads generated by Illumina HiSeq and MiSeq sequencers, duplicates were removed as artifacts using an in-house perl script, and unique reads were filtered to remove low quality bases/reads using Sickle (version 1.33) with the parameters “-q 20 -l 50” [35]. After that, all high-quality datasets were co-assembled using SPAdes (version 3.11.0) with the parameters “-k 21, 33, 55, 77, 99, 127 --meta” [36]. To calculate scaffold coverage, all high-quality reads from metagenomic datasets were mapped to the assembled scaffolds (length ≥ 2000 bp) using BBMap with the parameters “minid = 0.97, local = t”. These scaffolds were binned using MetaBAT (version 0.32.4) with the parameters “-m 2000 --unbinned” [37], which considers both tetranucleotide frequencies and the coverage of these scaffolds. The retrieved bins from MetaBAT were evaluated for taxonomic assignment, genome completeness, potential contamination, and strain heterogeneity, using CheckM [38], and were visualized using ESOM [39]. Afterwards, a cluster of four bins belonging to the Sva0485 group was further optimized to obtain high-quality genomes as previously described [8].
Analyses of genome bins
Four high-quality genomes were submitted to the JGI IMG/MER system for gene calling and annotation [40]. Subsequently, predicted gene functions were manually curated and revised by comparisons with the databases including NCBI-nr, KEGG, and eggNOG. Based on gene annotation, metabolic pathways were constructed for these bins. The 16S rRNA gene sequences were identified using RNAmmer [41] and were then used to search for the closely related 16S rRNA gene sequences in NCBI GenBank using BLASTn. Hits with an alignment coverage ≥ 85% and sequence identity ≥ 85% were downloaded for subsequent analysis. Calculation of relative abundance and transcript abundance was described in the Supplementary Information.
Phylogenetic analyses
Datasets consisting of 16 ribosomal proteins [42] from the four bins, along with 65 genomes belonging to the Delta- and Epsilon-proteobacteria and one genome from the phylum Euryarchaeota were individually aligned using MUSCLE (version 3.8.31) [43], and then were trimmed to remove columns composed of ≥ 95% gaps and the taxa with < 50% of the expected alignment columns using TrimAL with the parameters (-gt 0.95 -cons 50) [44]. The curated alignments were concatenated for phylogenetic analyses, and a maximum-likelihood tree was constructed using RAxML (version 8.1.24) [45], with the parameters set as “-f a -n boot -m PROTGAMMALG -c 4 -e 0.001 -# 100”. In addition, a total of 79 16S rRNA gene sequences belonging to the Deltaproteobacteria were aligned using MUSCLE (version 3.8.31) [43], and then the alignment was filtered through TrimAL to remove columns comprised of ≥ 95% gaps, generating a final alignment containing 79 taxa and 1861 alignment positions. The 16S rRNA gene tree was constructed using RAxML (version 8.1.24) [45], with the parameters set as “-f a -m GTRGAMMAI -n boot -c 4 -e 0.001 -# 100”. In addition, combined with the reference DsrAB data set from a previous study [46], a total of 506 DsrAB sequences were used for tree construction using RAxML with the parameters set as “-f a -m PROTGAMMAIJTT -n boot -c 4 -e 0.001 -# 100”. The newick files with the best tree topology were uploaded to iTOL [47] for visualization and formatting.
Data collection for meta-analysis
To reveal broader patterns in the distribution of Ca. Acidulodesulfobacterales in the AMD environment, we collected microbial data and site properties of 59 AMD samples across Southeast China [34] for meta-analysis. A total of 131,720 quality reads were downloaded from the European Nucleotide Archive database (accession no. PRJEB9908) [48]. These reads were combined with the V4 region of 16S rRNA gene sequences from Ca. Acidulodesulfobacterales and then were clustered into operational taxonomic units (OTUs) at the 97% similarity level with the UPARSE pipeline [49]. Relative abundance of each OTU was calculated as previously reported [50].
Statistical analyses
All statistical analyses were implemented using SPSS 18.0, SigmaPlot 10.0, and various R packages (http://www.r-project.org). The relationships between relative abundances/transcript abundances of the retrieved bins and physiochemical properties were assessed using the Spearman’s Rho/Pearson correlation. Redundancy analysis was carried out to identify environmental parameters that could explain the variation in relative abundance of the OTUs related to Ca. Acidulodesulfobacterales. Multiple linear regression (MLR) with stepwise method were conducted to test the significance between the relative abundance of Ca. Acidulodesulfobacterales and environmental properties. To quantify the contributions of the selected environmental variables and geographical distance to the Ca. Acidulodesulfobacterales abundance, we used the Lindeman–Merenda–Gold method in the relaimpo package [51]. In addition, orthologous average nucleotide identity (orthoANI) [52] was calculated among genomes from Ca. Acidulodesulfobacterales.
Accession numbers
The genomes reported in this study are available at the JGI IMG/MER under the Study ID Gs0128962 (accessions: Ga0325886–Ga0325889) and also at the NCBI GenBank under the BioProjectID PRJNA517999 (accessions: SGBB00000000–SGBD00000000 and SHMQ00000000).
Results and discussion
A new order Ca. Acidulodesulfobacterales in the class Deltaproteobacteria
In this study, a total of approximately 434 GB metagenomic raw data were generated for five samples (Supplementary Table S2). The Sva0485 clade was abundant (4.7–61.5%) in all samples (Fig. 1b) and comprised four distinct metagenomic bins, designated AP1-4. AP1-4 formed a distinct clade within the Deltaproteobacteria in a concatenated ribosomal protein tree (Fig. 1d). Therefore, we propose that AP1-4 represent a new order and designate it as Candidatus Acidulodesulfobacterales (Ca. Acidulodesulfobacterales). The 16S rRNA gene sequence identities and OrthoANI values between four bins were estimated, ranging from 91 to 100 and 70 to 80%, respectively (Fig. 1d). 16S rRNA genes have been widely used for taxonomic assignment and phylogenetic relationship determination, as the “gold standard” in both microbial phylogeny and ecology studies for several decades [53,54,55]. However, some counter examples have been reported that different species shared ≥ 97% of 16S rRNA sequence similarity, such as Bacillus psychrophilus and Bacillus globisporus (99.8%), and Serpula hyodysenteriae B78 and Serpula innocens B256 (99.5%) [55, 56]. This phenomenon was also observed in our research, where AP1 and AP2 and AP3 and AP4 shared 97 and 100% 16S rRNA sequence similarity, despite relatively low ANI values of 80 and 73%. As their OrthoANI values were much lower than proposed species cutoff values of 95% [57], they were considered to be four new species affiliated with two new genera, Candidatus Acididesulfobacter (Ca. Acididesulfobacter, including AP1 and AP2) and Candidatus Acidulodesulfobacterium (Ca. Acidulodesulfobacterium, including AP3 and AP4) of Ca. Acidulodesulfobacterales. The details of their taxonomic epithets are provided in the Supplementary Information.
The biogeography of Ca. Acidulodesulfobacterales in AMD environments
To reveal the spatial distribution pattern of Ca. Acidulodesulfobacterales in AMD environments, we reanalyzed microbial data in 59 AMD samples across Southeast China generated by 454 pyrosequencing [34]. As shown in Fig. 2a, members of Ca. Acidulodesulfobacterales were nearly ubiquitous, being present in 49 samples, with large differences in their total relative abundances from 0.03 to 45.4%. The genera Ca. Acididesulfobacter (AP1 and AP2) and Ca. Acidulodesulfobacterium (AP3 and AP4) were similarly widespread and abundant. Subsequently, MLR analysis with multivariate models indicated that ferrous iron was a major factor correlating with the abundance of Ca. Acididesulfobacter, Ca. Acidulodesulfobacterium, and their order Ca. Acidulodesulfobacterales, accounting for 22.6, 30.6 and 28.8% of the relative influence, respectively, followed by latitude, sulfate, and pH (Fig. 2b). For Ca. Acididesulfobacter, Cu and Zn were also important factors.

Environmental distribution of Ca. Acidulodesulfobacterales. a The relative abundance of Ca. Acidulodesulfobacterales in AMDs across Southeast China. b Relative influence (%) of environmental properties and geographical distance on the relative abundance of Ca. Acidulodesulfobacterales
General genomic features of Ca. Acidulodesulfobacterales and their dynamics in an artificial AMD system
The basic genomic characteristics of the four Ca. Acidulodesulfobacterales genomes are summarized in Table 1. Their genome sizes were between 1.9 and 2.5 Mb with estimated completeness between 92 and 94% and <1% contamination. In comparison with Ca. Acidulodesulfobacterium (AP3 and AP4), Ca. Acididesulfobacter (AP1 and AP2) had larger genomes and lower GC contents. The number of predicted genes ranged from 1805 to 2208, and the gene annotation rate ranged from 79 to 86, 79 to 86, and 66 to 78% in the NCBI-nr, KEGG, and eggNOG databases, respectively. Interestingly, the AP1 genome had more CRISPR loci and higher diversity of spacers than the others, suggesting more interactions present between AP1 and phages.
As mentioned above, relatively low OrthoANI values among these Ca. Acidulodesulfobacterales genomes might translate to differing metabolic potentials. Comparative analysis based on KEGG Orthology (KO) showed that 870 core KOs were shared by the four species, with 939 and 922 shared KOs in Ca. Acididesulfobacter (AP1 and AP2) and Ca. Acidulodesulfobacterium (AP3 and AP4), respectively (Supplementary Fig. S1 and Supplementary Table S3) (Materials and methods in the Supplementary Information). Notably, the number of species-specific KOs (59 in AP1, 21 in AP2, 41 in AP3, and 38 in AP4) was much lower than the core KOs, suggesting that genomic differences among these species were small. In spite of this, we found that for Ca. Acidulodesulfobacterales, urea metabolism was found only in AP1, and nitric oxide reduction occurred only in AP3. Metabolic comparisons among these species are also discussed in the subsequent section. These species-specific genes, and the functions they confer, likely enable these species to avoid competitive exclusion and thus coexist.
A total of 57 MAGs (with estimated completeness > 50 and <2% contamination) belonging to Proteobacteria, Euryarchaeota, Nitrospirae, Micrarchaeota, Parvarchaeota, Firmicutes, and Actinobacteria were also retrieved from the metagenomes of the artificial AMD system. The drastic change in relative abundances of these phyla over a 13-month period implied the potentially important roles of different dominant taxa at different times and that this ecosystem was in an unstable state (Supplementary Fig. S2). Subsequently, the relative abundances of the four species affiliated with Ca. Acidulodesulfobacterales and their transcripts were examined in the artificial AMD system over the 13-month period (Fig. 1b). The relative abundance of Ca. Acidulodesulfobacterales fluctuated between 4.7 and 61.5%. AP4 was the dominant species with relative abundance 4.2–42.0%, except in S5 when AP2 became the dominant species with relative abundance 9.4%. A similar trend was reflected in transcript abundance (Fig. 1b). The transcript abundance of Ca. Acidulodesulfobacterales ranged greatly from 7.5 to 70.6%, and AP4 had the highest transcript abundance (6.4−67.1%), except in S5, when AP2 showed the highest transcript abundance (~4.0%). There was a significant relationship (all P < 0.05) between the relative abundance of genomes (recruited from metagenomes) and transcript abundance for Ca. Acidulodesulfobacterales AP3, and AP4 (Supplementary Fig. S3), suggesting these lineages may not only be abundant, but also highly active.
Metabolic potential of Ca. Acidulodesulfobacterales
Metabolic potentials of these species were constructed from the genomes (Fig. 3 and Supplementary Table S4), and further supported by expression of the genes, as evaluated through metatranscriptomics (Supplementary Tables S5 and S6). Overall, the 20 transcripts with the highest abundance (Reads Per Kilobase per Million mapped reads, RPKM) and the highest relative transcriptional activity (RTA) were characterized, among which 10 transcripts related to stress response (rubredoxin, hsp20, and dnaK), flagellar assembly (fliC and fliD), transport (pal), transcriptional regulation (fmdB), nitrogen fixation (nifU and nifU family maturases), and iron oxidation (cyt572) were shared by the two expression profiles of RPKM and RTA (Fig. 4). The unshared transcripts were associated with thiamine metabolism (thiM and iscS), methane metabolism (hdrB2 and hdrC2), glyoxylate and dicarboxylate metabolism (gcvH), sulfur metabolism (sqr), sulfur relay system (sirA and sirA-like genes), metal-sulfur cluster assembly (suf), lipopolysaccharide biosynthesis (lpxC), pilus assembly (pilA), flagellar assembly (flgE), protein export (tatB), RNA degradation (groEL), and stress response (rpoD, rpoZ, cspA, xseB, and hupB) (Fig. 4). One-way analysis of variance (ANOVA) analysis with a Duncan test illustrated that RPKM levels for the top 20 transcripts of AP4 were always higher than the other species (all P < 0.05). But, at the RTA levels, this trend was not found. Supplementary Tables S5 and S6 provide additional details on key transcripts involved in carbon, nitrogen, sulfur, and iron metabolisms, oxidative phosphorylation and fermentation, and response to environmental stresses, which are individually described below.

Metabolic capabilities of Ca. Acidulodesulfobacterales. Pathways associated with carbon, nitrogen, sulfur, and iron metabolism are shown, along with oxidative phosphorylation, fermentation, and stress response. Genes appearing in all four genomes, two or three genomes and one genome are shown in green, orange, and purple, respectively. Genes absent in all bins are gray. The copy numbers of each gene in the four bins are provided in Supplementary Table S4

Expression profiles for 20 genes with the highest abundance (RPKM, left) and the highest relative transcriptional activity (RTA, right). Genes within the red dashed frame were shared by the two expression profiles. Asterisks indicate that a given transcript is significantly different in special bins comparing with in the other bins using one-way ANOVA analysis
Carbon fixation
As shown in Fig. 3, the reductive tricarboxylic acid (rTCA) cycle was identified in all four genomes, implying that Ca. Acidulodesulfobacterales have the genetic potential for carbon fixation via the rTCA cycle, in agreement with the fact that the rTCA cycle is often utilized by anaerobic or microaerobic species of Proteobacteria, especially Deltaproteobacteria [58]. Meanwhile, the transcriptional analysis confirmed the expression of genes in the rTCA cycle. Importantly, the key genes ccsAB, encoding citryl-CoA synthetase, were expressed at higher levels (RPKM) in AP4 than in the other three genomes (ANOVA, P = 0.004), suggesting Ca. Acidulodesulfobacterales (especially AP4) may be important autotrophs for the AMD community.
Nitrogen metabolism
The expression of the nifDKH genes, encoding nitrogenase, was confirmed in all of the four bins, illustrating that Ca. Acidulodesulfobacterales have the potential to fix nitrogen to synthesize organic nitrogen for growth. Among the four species, gene expression analysis revealed the higher expression level (RPKM) of nitrogenase in AP4 (ANOVA, P = 0.02), and no significant difference in the RPKM values of the nifDKH genes in Ca. Acidulodesulfobacterales across time. Also, the nitrogenase RTA in Ca. Acidulodesulfobacterales as a whole increased significantly (ANOVA, P = 0.004) across time, illustrating their increasing activity for nitrogen fixation. Meanwhile, we found the high expression of the genes nifS and nifU (as two of the top 20 transcripts), which are maturases required for the formation of the iron–sulfur cluster in nitrogenase [59], and the highest RPKM and RTA of the nifU gene were present in AP4. These findings provide evidence for an important role of Ca. Acidulodesulfobacterales, particularly AP4, in supplying nitrogen for the AMD community. Furthermore, statistical analyses demonstrated that RTA of the nifDKH genes in Ca. Acidulodesulfobacterales increased significantly (both P < 0.05) with the decrease of environmental pH and the increase of total iron (Supplementary Fig. S4a, b).
In addition to nitrogen fixation, Ca. Acidulodesulfobacterales could potentially reduce hydroxylamine to generate ammonium as evidenced by the expression of the hcp gene in AP1, AP2, and AP4. RTA analysis revealed the highest activity of the hcp gene in AP2. It is also noteworthy that AP1 harbored genes encoding urea transport system (urtABCDE) and urease (ureDABCEFG), indicating urea could potentially serve as an alternative nitrogen resource for AP1, similar to other known microorganisms (e.g., Ferrovum spp.) in AMD [60]. For ammonium transport and assimilation, the genes amt, glnA, and gltD, encoding ammonium transporter, glutamine synthetase, and glutamate synthase, respectively, were expressed in all four species, suggesting that Ca. Acidulodesulfobacterales likely gain organic nitrogen by ammonium assimilation for growth, like other microorganisms in AMD [60]. Interestingly, the amt gene showed the same trends in RPKM and RTA as the nifDKH genes, implying the enhanced demand and capacity of each member of the Ca. Acidulodesulfobacterales (especially AP4) to gain nitrogen for anabolism. Moreover, AP3 harbored a potential for reducing nitric oxide to nitrogen due to the detected expression of the norB and nosZ genes, which encode nitric oxide reductase subunit B and nitrous-oxide reductase, respectively. In summary, multiple strategies for the utilization of nitrogen resources (nitrogen, ammonium, urea etc.) were identified in Ca. Acidulodesulfobacterales, similar to Leptospirillum and a few other AMD taxa, as reported in previous studies [61,62,63].
Sulfur metabolism
One crucial characteristic of AMD is the high concentration of sulfate [7]. A complete dissimilatory sulfate reduction pathway was found in the genomes of AP1, AP3, and AP4, whereas the concatenated DsrAB protein tree demonstrated that the dsrAB genes from Ca. Acidulodesulfobacterales were the reductive type (Fig. 5a), showing that Ca. Acidulodesulfobacterales are potential acidophilic SRB (aSRB). The concatenated DsrAB protein tree also provides evidence for the fact that organisms of Deltaproteobacteria likely acquired the dsrAB genes in multiple lateral gene transfer events [64]. AP1, AP3, and AP4 were further confirmed as potential aSRB due to the dsrD gene present in their dsr operons, which was found widely in SRB but not in sulfur-oxidizing bacteria [65,66,67]. However, it is surprising to simultaneously detect the dsrL and especially dsrEFH genes (with well-known roles in sulfur oxidation) in the dsr operons of AP1, AP3, and AP4 (Fig. 5b). Previous studies have shown that the DsrL is a homolog of the small subunit of bacterial glutamate synthase and is essential for sulfur oxidation [68, 69]. The dsrEFH genes, which encode sulfur trafficking enzymes, are ubiquitous in sulfur oxidizers but absent in SRB [70, 71]. Given the occurrence of the dsrL and dsrEFH genes, these new species may be capable of oxidizing sulfide to sulfate via the reverse sulfate reduction pathway, analogous to the pathway model in Desulfurivibrio alkaliphilus [72]. To date, similar dsr operons have been found in several organisms from Actinobacteria (four genomes), Ca. Lambdaproteobacteria (four genomes), Nitrospirae (one genome), and Deltaproteobacteria (one genome), as shown in Fig. 5 [64]. Significant correlations (both P < 0.05) between the RTA values of the dsrAB and dsrEFH genes and between the RTA values of the dsrAB genes and the sulfate concentration were observed (Supplementary Fig. S4c, d). Considering the low concentration of oxygen in AMD bulk water (0.65–2.93 mg L−1), it is possible that planktonic Ca. Acidulodesulfobacterales or those on biofilm surfaces may be performing sulfide oxidation, whereas those within the biofilm matrix or anaerobic sediments may be able to simultaneously reduce sulfate. The latter process may be dominant in the environment, as evidenced by the much higher RTA of the dsrD gene than that of the dsrEFH genes. Therefore, we suggest that Ca. Acidulodesulfobacterales may not only reduce sulfate, but also oxidize sulfide, depending on oxygen concentrations and/or oxidation reduction potential in the environment. In addition, transcriptional results provided evidence that AP4 may play a more important role in sulfur metabolism than AP1 and AP3 due to its much higher expression level (RPKM) and RTA of the dsrAB genes (ANOVA, both P ≤ 0.05).

The dsr operon of Ca. Acidulodesulfobacterales. a Phylogenic analysis of the concatenated DsrAB proteins. Bootstrap values were based on 100 replicates, and only bootstrap values higher than 50% are shown with black circles. b The dsr operon structure in Ca. Acidulodesulfobacterales and previously reported genomes
Interestingly, Ca. Acidulodesulfobacterales also harbored a partial pathway for assimilatory sulfate reduction due to their absence of the cysJI and sir genes, encoding sulfite reductase (Fig. 3). In addition, the sqr gene was expressed, with its highest RPKM and RTA in AP4 (ANOVA, both P < 0.05), suggesting that Ca. Acidulodesulfobacterales (especially AP4) have the potential to produce polysulfide, which plays an integral role in a wide variety of geochemical processes (e.g., pyrite formation, organic matter sulfidization, and isotope exchange among reduced sulfur species, and metal chelation) and thus contributes to detoxification and elemental burial and sequestration [73]. Furthermore, the presence of the soxA, soxB, soxY, and soxZ genes in AP1, AP2, and AP3 suggests that Ca. Acidulodesulfobacterales harbor the potential to oxidize S2O32- to produce S(0) via the partial SOX (sulfur oxidation) system without the soxCD genes [74]. The expression analysis of the soxB gene suggests that AP3 was highly active in oxidizing thiosulfate compared with the others (t-test, all P < 0.05). In addition, the cysE and cysM genes, encoding serine O-acetyltransferase and cysteine synthase B, respectively, were identified in all four genomes, illustrating that Ca. Acidulodesulfobacterales may utilize the generated sulfide to synthesize the l-cysteine and acetate for growth.
Iron metabolism
Considering the high availability of iron in AMD environments, genes involved in iron oxidation were searched for in the new genomes [60, 75]. Results revealed that all four genomes had cyt572- and cyt1-like genes, and only AP1 and AP4 had the cyt579-like gene (Supplementary Table S7). These cytochromes have been shown to participate in iron oxidation in Leptospirillum Group II and III [76], which are common in AMD environments [77]. As such, it is inferred that Ca. Acidulodesulfobacterales are likely able to oxidize iron and transfer electrons via the cyt572–cyt579–cyc1 pathway. The key cyt572-like gene, one of the top 20 transcripts, showed a high expression level (RPKM) and a RTA in AP4. Moreover, an iro-like gene was identified in AP4 (Supplementary Table S7), which encodes the iron oxidase in Acidithiobacillus ferrivorans [75], demonstrating AP4 may be a potential Fe(II) oxidizer.
In addition to the above potential iron oxidation genes, genes encoding iron transport and storage proteins and the related regulators were also detected in Ca. Acidulodesulfobacterales. For Fe(II) transport, the double-copy gene mntH was confirmed in all four bins (Supplementary Table S5). Previous studies have demonstrated that the mntH gene encodes a secondary Fe(II) transporter with the primary function of Mn(II) uptake, as well as Fe(II) in certain circumstances [78]. Gene expression analysis uncovered that across time Ca. Acidulodesulfobacterales showed an increasing RTA of the mntH gene but no significant RPKM difference of the mntH gene was detected. AP4 was always the main contributor to the mntH RPKM, suggesting Ca. Acidulodesulfobacterales (especially AP4) as an important participant in iron metabolism. Also, four different TonB-dependent outer membrane Fe(III) siderophore transporter groups belonging to the CirA-linear and FepA-cyclic catecholate types were found in Ca. Acidulodesulfobacterales (Supplementary Table S8). These different types of siderophore receptors may provide a metabolic versatility for the growth of Ca. Acidulodesulfobacterales in AMD environments where iron co-precipitation with phosphates or sulfate during iron and sulfur biooxidation might compromise their ability to scavenge iron, as suggested for Acidithiobacillus spp. [78]. In most prokaryotes, diverse ferric uptake regulators (Fur) are responsible for iron regulation, and the iron storage proteins play a central role in maintaining iron homeostasis [78]. The present study revealed that only AP4 harbored the fur gene, but the bfr gene encoding bacterioferritin was expressed in all four bins with its highest RPKM in AP4. Ortholog analysis demonstrated that the bacterioferritin of Ca. Acidulodesulfobacterales was the classical iron storage protein due to the residue Met52, different from the Leu52 in the bacterioferritin of Acidithiobacillus spp. (Supplementary Fig. S5) [78]. Many studies have reported that the classical bacterioferritin can catalyze the oxidation and hydrolysis of iron at specific sites inside the protein shell, resulting in the formation of a mineral core of hydrated ferric oxide within the protein cavity, whereas it also has the capacity to re-utilize the iron stored inside its cavity and then release iron [79]. Thus, Ca. Acidulodesulfobacterales, particularly AP4, are likely to be important regulators of iron metabolism through complex iron management mechanisms.
Oxidative phosphorylation
Nearly complete pathways for oxidative phosphorylation along with high-affinity O2 terminal oxidases were found in the four bins (Fig. 4), indicating Ca. Acidulodesulfobacterales likely utilize oxygen as a terminal electron acceptor. All of the four bins harbored the cydA and cydB genes encoding cytochrome bd ubiquinol oxidase, which is a high-affinity terminal oxygen reductase capable of functioning under low oxygen concentrations [80, 81]. The RTA for cydAB genes was higher in AP4 and AP2 than in AP1 and AP3 (t-test, all P < 0.05), with the highest expression level (RPKM) in AP4. Furthermore, these species contained nearly complete NADH:quinone oxidoreductase (nuo) complexes (except the nuoE, nuoF, and nuoG genes) necessary for NADH-mediated oxidative phosphorylation and complete F-type ATPase, which is ubiquitous and phylogenetically conserved among Bacteria and considered to be the ancestral bacterial ATPase [82]. Thus, we propose that Ca. Acidulodesulfobacterales can respire oxygen in situ.
Fermentation
The present study revealed that Ca. Acidulodesulfobacterales are likely able to ferment, producing lactate (the LDH gene) and ethanol (the adhP and ALDH genes). Metatranscriptomic analysis revealed that among these four bins, AP4 had the lowest RTA of the LDH gene and the highest RTA of the adhP gene (ANOVA, all P < 0.05). The RPKM and RTA of the adhP gene was always higher than those of the LDH gene in AP4, implying that AP4 may prefer ethanol fermentation. AP1 and AP2 might perform acetate fermentation via the Pta-Ack pathway (the pta-ackA genes) that is widely distributed in bacteria [83], suggesting some Ca. Acidulodesulfobacterales are able to grow as acetogens. Thus, Ca. Acidulodesulfobacterales are likely to be facultative anaerobes.
Stress response
AMD is an extreme environment due to the low pH and high concentrations of dissolved metals [5, 84], where indigenous microbes must thus adapt to the extreme environmental stresses (including acid, heavy metals, and oxidation) for survival [60, 85]. For acid stress, the expression of the kdpABC and kup genes, encoding a complete potassium-transporting ATPase system and potassium uptake protein, respectively, indicated that Ca. Acidulodesulfobacterales may generate an inside-positive membrane potential by taking up K+ to partially deflect the inward flow of protons. Meanwhile, Ca. Acidulodesulfobacterales likely maintain a near-neutral cytoplasm by the metabolism of proton buffer molecules, such as phosphate (pstSCAB) and arginine (pdaD). These strategies respond to acid stress and have been widely used by AMD microbes [60, 85].
In response to heavy metal stress, diverse heavy metal transporters were expressed, such as the Cu(I)/Ag(I) efflux system (cusAB) and the divalent metal cation (Fe/Co/Zn/Cd) transporter (such as copB and czcD) (Supplementary Tables S5 and S6), suggesting that efflux of metal ions is an important strategy to resist against heavy metal stress for Ca. Acidulodesulfobacterales, as occurs in other known AMD microorganisms [60, 85]. Moreover, another strategy may be to reduce metal ions or metalloids (such as mercury) to less toxic reduced forms, which may then be exported.
For oxidative stress, genes encoding peroxiredoxin and thioredoxin reductase were expressed in Ca. Acidulodesulfobacterales, which are widely used to remit harm of oxidative stress by AMD microorganisms [86]. Besides, a novel cytoplasmic oxidative stress protection system composed of rubrerythrin (rbr) and rubredoxin (rbo, one of the top 20 transcripts) in anaerobic microbes [87] was found in Ca. Acidulodesulfobacterales, especially AP4, fully demonstrating that they were facing severe oxidative stress. Moreover, other proteins (such as cytochrome c551 peroxidase and thiol peroxidase) for the response of oxidative stress were also expressed in Ca. Acidulodesulfobacterales. In conclusion, all above findings explain the mechanisms how Ca. Acidulodesulfobacterales can survive in the extreme AMD environment.
Concluding remarks
As abundant taxa in the extreme iron- and sulfur-bearing environments (including AMD ecosystems, Quaternary sediments, and deep-sea vents), Ca. Acidulodesulfobacterales was first detected through the 16S rRNA gene clone library 18 years ago [19]. We now describe the first four genomes affiliated with Ca. Acidulodesulfobacterales. This study demonstrated that ferrous iron was a main factor controlling the distribution pattern of Ca. Acidulodesulfobacterales in AMDs across Southeast China. The reconstruction of metabolic pathways with metatranscriptomics shed light on the metabolic versatility of Ca. Acidulodesulfobacterales, revealing that they are important participants in biogeochemical cycles of carbon, nitrogen, sulfur, and iron. Our work also provides insights into the mechanisms of adaptation and response to the extreme environmental stresses. As facultatively anaerobic autotrophs, Ca. Acidulodesulfobacterales likely use oxygen as terminal electron acceptor and thus couple Fe(II) and sulfide oxidation to aerobic respiration under aerobic conditions in AMD water and on biofilm surface, while they may perform dissimilatory sulfate reduction, Fe(III) respiration and fermentations under anaerobic conditions in the interior of biofilms and sediments. To further understand the physiological and genetic properties of Ca. Acidulodesulfobacterales, future studies should strive to obtain pure cultures or defined mixed cultures of these species to confirm these ecological functions. Meanwhile, future research should expand the number of genomes belonging to Ca. Acidulodesulfobacterales to further address questions related to biogeography, comparative genomics, and evolution. Such efforts will further expand our understanding of the taxonomic diversity and ecological role of the new order Ca. Acidulodesulfobacterales in nature.
References
Osorio H, Mangold S, Denis Y, Ñancucheo I, Esparza M, Johnson DB, et al. Anaerobic sulfur metabolism coupled to dissimilatory iron reduction in the extremophile Acidithiobacillus ferrooxidans. Appl Environ Microbiol. 2013;79:2172–81.
Colman DR, Poudel S, Hamilton TL, Havig JR, Selensky MJ, Shock EL, et al. Geobiological feedbacks and the evolution of thermoacidophiles. ISME J. 2018;12:225–36.
Johnson DB, Kanao T, Hedrich S. Redox transformations of iron at extremely low pH: fundamental and applied aspects. Front Microbiol. 2012;3:96–108.
Nordstrom DK, Alpers CN. Negative pH, efflorescent mineralogy, and consequences for environmental restoration at the Iron Mountain Superfund site, California. Proc Natl Acad Sci USA. 1999;96:3455–62.
Rothschild LJ, Mancinelli RL. Life in extreme environments. Nature. 2001;409:1092–101.
Méndez-García C, Peláez AI, Mesa V, Sánchez J, Golyshina OV, Ferrer M. Microbial diversity and metabolic networks in acid mine drainage habitats. Front Microbiol. 2015;6:475–92.
Chen LX, Huang LN, Méndezgarcía C, Kuang JL, Hua ZS, Liu J, et al. Microbial communities, processes and functions in acid mine drainage ecosystems. Curr Opin Biotechnol. 2016;38:150–8.
Chen LX, Méndez-García C, Dombrowski N, Servín-Garcidueñas LE, Eloe-Fadrosh EA, Fang BZ, et al. Metabolic versatility of small archaea Micrarchaeota and Parvarchaeota. ISME J. 2017;12:756–75.
Golyshina OV, Kublanov IV, Tran H, Korzhenkov AA, Lünsdorf H, Nechitaylo TY, et al. Biology of archaea from a novel family Cuniculiplasmataceae (Thermoplasmata) ubiquitous in hyperacidic environments. Sci Rep. 2016;6:39034–45.
Golyshina OV, Lünsdorf H, Kublanov IV, Goldenstein NI, Hinrichs KU, Golyshin PN. The novel, extremely acidophilic, cell wall-deficient archaeon Cuniculiplasma divulgatum gen. nov., sp. nov. represents a new family of Cuniculiplasmataceae fam. nov., order Thermoplasmatales. Int J Syst Evol Microbiol. 2016;66:332–40.
Neculita CM, Zagury GJ, Bussière B. Passive treatment of acid mine drainage in bioreactors using sulfate-reducing bacteria. J Environ Qual. 2007;36:1–16.
Sánchez-Andrea I, Stams AJM, Amils R, Sanz JL. Enrichment and isolation of acidophilic sulfate-reducing bacteria from Tinto River sediments. Environ Microbiol Rep. 2013;5:672–8.
Hedrich S, Johnson DB. Remediation and selective recovery of metals from acidic mine waters using novel modular bioreactors. Environ Sci Technol. 2014;48:12206–12.
Karnachuk OV, Gerasimchuk AL, Banks D, Frengstad B, Stykon GA, Tikhonova ZL, et al. Bacteria of the sulfur cycle in the sediments of gold mine tailings, Kuznetsk Basin, Russia. Microbiology. 2009;78:483–91.
Sánchez-Andrea I, Rojas-Ojeda P, Amils R, Sanz JL. Screening of anaerobic activities in sediments of an acidic environment: Tinto River. Extremophiles. 2012;16:829–39.
Abicht HK, Mancini S, Karnachuk OV, Solioz M. Genome sequence of Desulfosporosinus sp. OT, an acidophilic sulfate-reducing bacterium from copper mining waste in Norilsk, Northern Siberia. J Bacteriol. 2011;193:6104–5.
Karnachuk OV, Mardanov AV, Avakyan MR, Kadnikov VV, Vlasova M, Beletsky AV, et al. Draft genome sequence of the first acid-tolerant sulfate-reducing deltaproteobacterium Desulfovibrio sp. TomC having potential for mine water treatment. FEMS Microbiol Lett. 2015;362:1–3.
Sánchez-Andrea I, Stams AJ, Hedrich S, Ňancucheo I, Johnson DB. Desulfosporosinus acididurans sp. nov.: an acidophilic sulfate-reducing bacterium isolated from acidic sediments. Extremophiles. 2015;19:39–47.
Bond PL, Smriga SP, Banfield JF. Phylogeny of microorganisms populating a thick, subaerial, predominantly lithotrophic biofilm at an extreme acid mine drainage site. Appl Environ Microbiol. 2000;66:3842–9.
Brofft JE, McArthur JV, Shimkets LJ. Recovery of novel bacterial diversity from a forested wetland impacted by reject coal. Environ Microbiol. 2002;4:764–9.
Druschel GK, Baker BJ, Gihring TH, Banfield JF. Acid mine drainage biogeochemistry at Iron Mountain, California. Geochem Trans. 2004;5:13–32.
Sánchez-Andrea I, Rodriguez N, Amils R, Sanz JL. Microbial diversity in anaerobic sediments at Rio Tinto, a naturally acidic environment with a high heavy metal content. Appl Environ Microbiol. 2011;77:6085–93.
Yang Y, Yang LI, Sun QY. Archaeal and bacterial communities in acid mine drainage from metal-rich abandoned tailing ponds, Tongling, China. Tran Nonferr Metal Soc. 2014;24:3332–42.
Grettenberger CL, Pearce AR, Bibby KJ, Jones DS, Burgos WD, Macalady JL. Efficient low-pH iron removal by a microbial iron oxide mound ecosystem at Scalp Level Run. Appl Environ Microbiol. 2017;83:15–17.
Hao C, Zhang H, Haas R, Bai Z, Zhang B. A novel community of acidophiles in an acid mine drainage sediment. World J Microbiol Biotechnol. 2007;23:15–21.
Yoshida H, Yamamoto K, Murakami Y, Katsuta N, Hayashi T, Naganuma T. The development of Fe-nodules surrounding biological material mediated by microorganisms. Environ Geol. 2008;55:1363–74.
Sylvan JB, Toner BM, Edwards KJ. Life and death of deep-sea vents: bacterial diversity and ecosystem succession on inactive hydrothermal sulfides. MBio. 2012;3:e00279–11.
Vuillemin A, Horn F, Friese A, Winkel M, Alawi M, Wagner D, et al. Metabolic potential of microbial communities from ferruginous sediments. Environ Microbiol. https://doi.org/10.1111/1462-2920.14343
Kleindienst S, Herbst F-A, Stagars M, von Netzer F, von Bergen M, Seifert J, et al. Diverse sulfate-reducing bacteria of the Desulfosarcina/Desulfococcus clade are the key alkane degraders at marine seeps. ISME J. 2014;8:2029–44.
Bar-Or I, Ben-Dov E, Kushmaro A, Eckert W, Sivan O. Methane-related changes in prokaryotes along geochemical profiles in sediments of Lake Kinneret (Israel). Biogeosciences. 2015;12:2847–60.
Concheri G, Stevanato P, Zaccone C, Shotyk W, D’Orazio V, Miano T, et al. Rapid peat accumulation favours the occurrence of both fen and bog microbial communities within a Mediterranean, free-floating peat island. Sci Rep. 2017;7:8511.
Chen YT, Li JT, Chen LX, Hua ZS, Huang LN, Liu J, et al. Biogeochemical processes governing natural pyrite oxidation and release of acid metalliferous drainage. Environ Sci Technol. 2014;48:5537–45.
Carradec Q, Pelletier E, Da Silva C, Alberti A, Seeleuthner Y, Blanc-Mathieu R, et al. A global ocean atlas of eukaryotic genes. Nat Commun. 2018;9:373.
Kuang JL, Huang LN, Chen LX, Hua ZS, Li SJ, Hu M, et al. Contemporary environmental variation determines microbial diversity patterns in acid mine drainage. ISME J. 2013;7:1038–50.
Joshi NA, Fass JN. Sickle: a sliding-window, adaptive, quality-based trimming tool for FastQ files. 2011. https://githubcom/najoshi/sickle.
Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol. 2012;19:455–77.
Kang DD, Froula J, Egan R, Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 2015;3:e1165.
Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015;25:1043–56.
Ultsch A, Mörchen F. ESOM-Maps: tools for clustering, visualization, and classification with Emergent SOM. Tech Rep Dept Math Comp Sci, University of Marburg, Germany. 2005;46:1–7.
Markowitz VM, Chen IMA, Palaniappan K, Chu K, Szeto E, Grechkin Y, et al. IMG: the integrated microbial genomes database and comparative analysis system. Nucleic Acids Res. 2011;40:D115–22.
Lagesen K, Hallin P, Rødland EA, Stærfeldt H-H, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–8.
Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle CJ, et al. A new view of the tree of life. Nat Microbiol. 2016;1:16048–53.
Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–7.
Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009;25:1972–3.
Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–3.
Müller AL, Kjeldsen KU, Rattei T, Pester M, Loy A. Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi)sulfite reductases. ISME J. 2015;9:1152–65.
Letunic I, Bork P. Interactive Tree Of Life (iTOL): an online tool for phylogenetic tree display and annotation. Bioinformatics. 2006;23:127–8.
Kuang JL, Huang LN, He ZL, Chen LX, Hua ZS, Jia P, et al. Predicting taxonomic and functional structure of microbial communities in acid mine drainage. ISME J. 2016;10:1527–39.
Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods. 2013;10:996–1000.
Liu J, He XX, Lin XR, Chen WC, Zhou QX, Shu WS, et al. Ecological effects of combined pollution associated with e-waste recycling on the composition and diversity of soil microbial communities. Environ Sci Technol. 2015;49:6438–47.
Grömping U. Relative importance for linear regression in R: the package relaimpo. J Stat Softw. 2006;17:1–27.
Lee IC, Kim YO, Park SC, Chun JS. OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int J Syst Evol Microbiol. 2016;66:1100–3.
Woese CR. Bacterial evolution. Microbiol Rev. 1987;51:221–71.
Woese CR, Kandler O, Wheelis ML. Towards a natural system of organisms: proposal for the domains Archaea, Bacteria, and Eucarya. Proc Natl Acad Sci USA. 1990;87:4576–9.
Sun DL, Jiang X, Wu QL, Zhou NY. Intragenomic heterogeneity in 16S rRNA genes causes overestimation of prokaryotic diversity. Appl Environ Microbiol. 2013;79:5962–9.
Fox GE, Wisotzkey JD, Jurtshuk JRP. How close is close: 16S rRNA sequence identity may not be sufficient to guarantee species identity. Int J Syst Evol Microbiol. 1992;42:166–70.
Jain C, Rodriguez-R LM, Phillippy AM, Konstantinidis KT, Aluru S. High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun. 2018;9:5114.
Berg IA. Ecological aspects of the distribution of different autotrophic CO2 fixation pathways. Appl Environ Microbiol. 2011;77:1925–36.
Jacobson MR, Cash VL, Weiss MC, Laird NF, Newton WE, Dean DR. Biochemical and genetic analysis of the nifUSVWZM cluster from Azotobacter vinelandii. Mol Gen Genet. 1989;219:49–57.
Hua ZS, Han YJ, Chen LX, Liu J, Hu M, Li SJ, et al. Ecological roles of dominant and rare prokaryotes in acid mine drainage revealed by metagenomics and metatranscriptomics. ISME J. 2015;9:1280–94.
Ram RJ, VerBerkmoes NC, Thelen MP, Tyson GW, Baker BJ, Blake RC, et al. Community proteomics of a natural microbial biofilm. Science. 2005;308:1915–20.
Bertin PN, Heinrich-Salmeron A, Pelletier E, Goulhen-Chollet F, Arsène-Ploetze F, Gallien S, et al. Metabolic diversity among main microorganisms inside an arsenic-rich ecosystem revealed by meta-and proteo-genomics. ISME J. 2011;5:1735–47.
Tyson GW, Lo I, Baker BJ, Allen EE, Hugenholtz P, Banfield JF. Genome-directed isolation of the key nitrogen fixer Leptospirillum ferrodiazotrophum sp. nov. from an acidophilic microbial community. Appl Environ Microbiol. 2005;71:6319–24.
Anantharaman K, Hausmann B, Jungbluth SP, Kantor RS, Lavy A, Warren LA, et al. Expanded diversity of microbial groups that shape the dissimilatory sulfur cycle. ISME J. 2018;12:1715–28.
Hittel DS, Voordouw G. Overexpression, purification and immunodetection of DsrD from Desulfovibrio vulgaris Hildenborough. Antonie Van Leeuwenhoek. 2000;77:271–80.
Mizuno N, Voordouw G, Miki K, Sarai A, Higuchi Y. Crystal structure of dissimilatory sulfite reductase D (DsrD) protein-possible interaction with B- and Z-DNA by its winged-helix motif. Structure. 2003;11:1133–40.
Rabus R, Venceslau SS, Wöhlbrand L, Voordouw G, Wall JD, Pereira IA. A post-genomic view of the ecophysiology, catabolism and biotechnological relevance of sulphate-reducing prokaryotes. Adv Microb Physiol. 2015;66:55–21.
Lübbe YJ, Youn HS, Timkovich R, Dahl C. Siro(haem)amide in Allochromatium vinosum and relevance of DsrL and DsrN, a homolog of cobyrinic acid a,c-diamide synthase, for sulphur oxidation. FEMS Microbiol Lett. 2006;261:194–202.
Weissgerber T, Sylvester M, Kröninger L, Dahl C. A comparative quantitative proteomic study identifies new proteins relevant for sulfur oxidation in the purple sulfur bacterium Allochromatium vinosum. Appl Environ Microbiol. 2014;80:2279–92.
Grimm F, Franz B, Dahl C. Regulation of dissimilatory sulfur oxidation in the purple sulfur bacterium Allochromatium Vinosum. Front Microbiol. 2011;2:51–62.
Yvonne S, Venceslau SS, Michaele J, Hans-Georg S, Pereira IAC, Christiane D. Cytoplasmic sulfurtransferases in the purple sulfur bacterium Allochromatium vinosum: evidence for sulfur transfer from DsrEFH to DsrC. PLoS ONE. 2012;7:e40785.
Thorup C, Schramm A, Findlay AJ, Kai WF, Schreiber L. Disguised as a sulfate reducer: growth of the deltaproteobacterium Desulfurivibrio alkaliphilus by sulfide oxidation with nitrate. MBio. 2017;8:e00671–711.
Findlay AJ. Microbial impact on polysulfide dynamics in the environment. FEMS Microbiol Lett. 2016;363:fnw103.
Hensen D, Sperling D, Trüper HG, Brune DC, Dahl C. Thiosulphate oxidation in the phototrophic sulphur bacterium Allochromatium vinosum. Mol Microbiol. 2006;62:794–810.
Bonnefoy V, Holmes DS. Genomic insights into microbial iron oxidation and iron uptake strategies in extremely acidic environments. Environ Microbiol. 2012;14:1597–11.
Ilbert M, Bonnefoy V. Insight into the evolution of the iron oxidation pathways. Biochim Biophys Acta. 2013;1827:161–75.
Hao C, Wang L, Gao Y, Zhang L, Dong H. Microbial diversity in acid mine drainage of Xiang Mountain sulfide mine, Anhui Province, China. Extremophiles. 2010;14:465–74.
Osorio H, Martínez V, Nieto PA, Holmes DS, Quatrini R. Microbial iron management mechanisms in extremely acidic environments: comparative genomics evidence for diversity and versatility. BMC Microbiol. 2008;8:203–21.
Yang X, Le Brun NE, Thomson AJ, Moore GR, Chasteen ND. The iron oxidation and hydrolysis chemistry of Escherichia coli bacterioferritin. Biochemistry. 2000;39:4915–23.
Pitcher RS, Watmough NJ. The bacterial cytochrome cbb3 oxidases. Biochim Biophys Acta. 2004;1655:388–99.
Borisov VB, Gennis RB, Hemp J, Verkhovsky MI. The cytochrome bd respiratory oxygen reductases. Biochim Biophys Acta. 2011;1807:1398–413.
Mulkidjanian AY, Makarova KS, Galperin MY, Koonin EV. Inventing the dynamo machine: the evolution of the F-type and V-type ATPases. Nat Rev Microbiol. 2007;5:892–9.
He Y, Li M, Perumal V, Feng X, Fang J, Xie J, et al. Genomic and enzymatic evidence for acetogenesis among multiple lineages of the archaeal phylum Bathyarchaeota widespread in marine sediments. Nat Microbiol. 2016;1:16035–43.
Huang LN, Kuang JL, Shu WS. Microbial ecology and evolution in the acid mine drainage model system. Trends Microbiol. 2016;24:581–93.
Chen LX, Hu M, Huang LN, Hua ZS, Kuang JL, Li SJ, et al. Comparative metagenomic and metatranscriptomic analyses of microbial communities in acid mine drainage. ISME J. 2015;9:1579–92.
Ferrer A, Orellana O, Levicán G. Oxidative stress and metal tolerance in extreme acidophiles. In: Johnson DB, Quatrini R, editors. Acidophiles: life in extremely acidic environments. Norfolk, UK: Caister Academic Press; 2016. p. 63–76.
Lumppio HL, Shenvi NV, Summers AO, Voordouw G, Kurtz DM. Rubrerythrin and Rubredoxin oxidoreductase in Desulfovibrio vulgaris: a novel oxidative stress protection system. J Bacteriol. 2001;183:101–8.
Acknowledgements
We thank the three anonymous reviewers for their valuable comments. We thank Aharon Oren from the Hebrew University of Jerusalem for providing suggestions for the Latin name of this new order. This work was financially supported by the National Natural Science Foundation of China (grant no. U1501232, 41622106, 31570500, 31570506, 41603074, 31600077, 41773132, and 41630103), the Natural Science Foundation of Guangdong Province, China (grant no. 2016A030312003), and U.S. National Science Foundation (grant no. DEB-1557042).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tan, S., Liu, J., Fang, Y. et al. Insights into ecological role of a new deltaproteobacterial order Candidatus Acidulodesulfobacterales by metagenomics and metatranscriptomics. ISME J 13, 2044–2057 (2019). https://doi.org/10.1038/s41396-019-0415-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41396-019-0415-y
This article is cited by
-
Soil microbial ecology through the lens of metatranscriptomics
Soil Ecology Letters (2024)
-
Microbe-driven elemental cycling enables microbial adaptation to deep-sea ferromanganese nodule sediment fields
Microbiome (2023)
-
Microbial community dynamics and cycling of plutonium and iron in a seasonally stratified and radiologically contaminated pond
Scientific Reports (2023)
-
Stepwise pathway for early evolutionary assembly of dissimilatory sulfite and sulfate reduction
The ISME Journal (2023)
-
Effects of metal contamination with physicochemical properties on the sediment microbial communities in a tropical eutrophic-hypereutrophic urban reservoir in Brazil
Environmental Science and Pollution Research (2023)
