
Abstract
Background
The impact of gonadotropin-releasing hormone (GnRH) antagonist and agonist (GnRHa) treatment on cardiovascular disease (CVD) risk in prostate cancer (PCa) remains inconclusive due to conflicting findings. We compared the effects of GnRH antagonist and GnRHa treatments on CVD risk in patients with PCa and pre-existing CVD, in a Taiwan population-based database.
Methods
We assessed the risk of major adverse CV events (MACE: ischemic heart disease [IHD], stroke, congestive heart failure [CHF] or all cause deaths) and composite CV events (IHD, stroke, CHF or CV deaths) occurring ≥90 days after androgen deprivation therapy (ADT) initiation in patients with PCa after 90 days of treatment with either GnRH antagonist (degarelix; n = 499) or GnRHa (goserelin, leuprolide, triptorelin; n = 15,127). Patients identified with pre-existing CVD had received cardiac therapy for IHD, reported a stroke or CHF within a year before ADT initiation. Adjusted hazard ratios (aHR) and 95% confidence interval (CI) were obtained for MACE and composite CV events risk after adjusting for age, baseline status of diabetes, hypertension and treatments received.
Results
All GnRH antagonist-treated patients showed lower risk of composite CV events than the GnRHa-treated patients. The lower composite CV events risk associated with GnRH antagonist was also observed in patients with metastasis at diagnosis (aHR 0.16; 95% CI, 0.04–0.38; p = 0.013) and those receiving ADT for more than six months (aHR 0.30; 95% CI, 0.16–0.54; p < 0.0001). In patients with pre-existing CVD, the MACE risk was 33% lower (aHR 0.67; 95% CI, 0.46–0.96; p = 0.0299) and composite CV events risk was 84% lower (aHR 0.16; 95% CI, 0.05–0.50; p = 0.0017) in GnRH antagonist-treated than the GnRHa-treated patients.
Conclusions
In patients with PCa and pre-existing CVD, GnRH antagonist use was associated with lower risks for composite CV events and MACE compared with GnRHa.
Similar content being viewed by others


Introduction
Prostate cancer (PCa) is globally the second most frequently diagnosed cancer in men, ranking after lung cancer [1]. In Taiwan, PCa increased in ranking from eighth to fifth among all cancers in men, with an incidence increase from 1.85% (1988–1992) to 8.96% (2013–2016) [2]. The PCa mortality rates in Taiwanese men increased from 8.3 to 11.5 per 100,000 people between 2006 and 2016 [3]. Among the non-cancer mortality causes in patients with PCa, cardiovascular disease (CVD) is the leading cause of death in the United States and many other countries [4,5,6]. The proportion of CVD fatalities increases over time after diagnosis, especially in those on long-term androgen deprivation therapy (ADT) [7].
ADT is the primary systemic therapy (standalone/concomitant) in PCa treatment for advanced disease. It reduces testosterone levels to those achieved by castration [8]. Approximately 50% of patients with PCa receive ADT during their treatment [9]. Androgen deprivation is achieved by gonadotropin-releasing hormone (GnRH) antagonists or agonists (GnRHa), which prevent luteinizing hormone (LH) secretion, and consequently inhibit testosterone production [10].
Reports of the relative associations between CVD and, GnRH antagonist/GnRHa are conflicting, especially in PCa patients with pre-existing CVD risk [11, 12]. Emerging literature suggests lower major adverse cardiovascular events (MACE) in patients treated with GnRH antagonists but remains inconclusive in patients with pre-existing CVD risk [12,13,14,15,16,17,18]. GnRHa has been associated with an increased CVD risk and related mortality, including in patients with PCa and prior CVD [13, 14, 16].
Clinical trials assessing the differential effect of GnRH antagonists and GnRHa on CVD remain inconclusive [15, 18,19,20]. Observational studies report a lower risk of CVD with GnRH antagonists than GnRHa [12, 14, 17]. However, the differential effect of GnRH antagonists or GnRHa monotherapy, without switching between drugs, is scarcely reported in Asian patients. Therefore, through this real-world data (RWD) analysis, we aim to compare the impacts of a GnRH antagonist (degarelix) and GnRHa treatments (goserelin, leuprolide, triptorelin), on the CVD risk in patients with PCa and pre-existing CVD, using Taiwan National Health Insurance Research Data (NHIRD).
Methods
Data source and study population
This population-based cohort study used NHIRD, which is linked by encrypted patient identifiers to the Taiwan Cancer Registry (TCR). The NHIRD includes medical claims data of demographics, enrollment profiles, disease diagnosis, procedures, and drug prescriptions; the TCR covers 97% of cancer cases in Taiwan and has a good level of data accuracy [13, 21, 22]. NHIRD are additionally linked to the Death Registry, to ascertain vital status and cause of death [23].
Patients, aged ≥20 years, with PCa (n = 18,835), who, according to prescription records, initiated ADT between January 1, 2015 and December 31, 2019, and sustained this for ≥3 months, were identified in the TCR, using the International Classification of Disease (ICD-O-3) code, C61.9. Patients who had a recorded orchiectomy (n = 146) or had received both GnRH antagonist and GnRHa for ≥3 months (n = 3063) were excluded.
Of eligible patients, GnRH antagonist exposure included patients treated with degarelix (n = 499; Anatomical Therapeutic Chemical Classification System [ATC] code: L02BX02), patients receiving GnRHa included goserelin, leuprolide and triptorelin treatment (n = 15,127; ATC codes: L02AE03, L02AE02, and L02AE04 respectively). Concomitant treatment data were extracted for: antiandrogens (bicalutamide, cyproterone, and flutamide; ATC codes: L02BB03, G03HA01 and L02BB01 respectively), estrogens (ATC code: L02AA), ketoconazole (ATC code: J02AB02), and androgen receptor (AR)-directed therapy for new-generation drugs (abiraterone, enzalutamide; ATC codes: L02BX03, L02BB04 respectively). Data were also extracted for chemotherapy and radiation therapy. The institutional review board of Taipei Medical University reviewed and approved this study (TMU-JIRB no. 201502042).
Covariates
For pre-existing CVD assessment, the following covariates were considered: hypertension (≥2 diagnoses within six months), cardiac therapy (≥2 prescriptions for drugs with ATC code: C01), acute myocardial infarction and other forms of ischemic heart disease (IHD), stroke, congestive heart failure (CHF), diabetes (≥2 diagnoses within six months), dyslipidemia (≥2 diagnoses within six months) and death (Supplementary Table 1).
The pre-existing CVD was assessed at baseline; patients receiving cardiac therapy, having a diagnosis of IHD, stroke or CHF 1 year before ADT initiation were categorized as having pre-existing CVD. Demographic and clinical characteristics were recorded at baseline. Additional covariates included age at diagnosis, clinical disease stage, Gleason score, prostate-specific antigen (PSA) level at diagnosis, and comorbidities. Patients’ ages were grouped as ≤54, 55–59, 60–69, 70–74, and ≥75 years.
The clinical stage was classified based on the TNM stage using the American Joint Committee on Cancer classification system (T scoring for area and size to give the extent of the main tumor: 1, 2, 3, 4 and Missing; N, for spread to lymph nodes, and M, for metastasis, both scoring: 0 or 1) [24]. Cancers were graded on differentiation using Gleason score (GS) and categorized based on score ranges 2–6, 7, and 8–10 [25]. The PSA (ng/mL) concentrations at diagnosis were also categorized as ≤50 ng/mL, >50 ng/mL and “missing”. To assess the burden of comorbidities and association with survival, the Charlson’s comorbidity index (CCI) was recorded at baseline, including assessment of claims during 1 year before the PCa diagnosis, excluding cancer diagnoses [26].
Patients were stratified based on assessed cancer risk, using the National Comprehensive Cancer Network (NCCN) staging, which considers: the clinical T stage, pre-treatment PSA level, and the GS (Table 1 footnote) [27]. The resulting risk groups allowed for comparison across patients with the same prognosis.
Outcome variables
Primary outcomes were MACE defined as IHD, stroke, CHF or all cause deaths occurring ≥90 days after ADT initiation, whichever came first. Secondary outcomes were composite CV events including IHD, stroke, CHF or CV deaths occurring ≥90 days after ADT initiation, whichever came first. Death Registry-recorded causes of death were used to determine CV deaths, defined by ICD-10 using I00–I99.
Statistical analysis
The baseline characteristics, MACE and composite CV events risk of the GnRH antagonist and GnRHa groups, also the pre-existing CVD and no pre-existing CVD subgroups were tested for significant difference by Chi-square test or Fisher’s exact test for categorical variables. The Cox proportional hazard model was used to estimate the differential of MACE associated with GnRH antagonist compared with GnRHa. The Fine and Gray hazard model, which considers non-CV deaths as competing risks in deriving the event probability over time, was employed to assess the risk of composite CV events [28, 29]. The hazard ratio (HR) with 95% confidence interval (CI) was calculated and adjusted for age, cancer stage, or receiving radiation therapy, chemotherapy, antiandrogen, abiraterone, and enzalutamide. The Kaplan–Meier survival curves were plotted for all patients as well as in subgroups of pre-existing CVD for MACE-free survival and composite CV event-free survival, for GnRH antagonist and GnRHa-treated patients. Log-rank tests were performed to test the differences in survival between treatment groups. Statistical analysis was performed using SAS version 9.4 (SAS Institute, Cary, NC, USA). Statistical significance is indicated by a p value of <0.05.
Results
Baseline characteristics
A total of 499 patients were included in the GnRH antagonist group and 15,127 patients in the GnRHa group. The mean (standard error) survival time for all patients was 2.62 (1.49) years. Patients from both groups were of similar ages (p = 0.4557), with the largest proportion of both groups being aged ≥75 years (Table 1).
A significantly higher proportion of patients had clinically advanced cancer stage in the GnRH antagonist treatment group (based on cancer stage, higher cancer grade, increased PSA and had “regional or metastatic” disease spread; Table 1). The proportion of patients with comorbidities was higher in the GnRH antagonist than the GnRHa group (Table 1). At baseline, a greater proportion of patients treated with GnRH antagonist had pre-existing CVD compared with the GnRHa-treated patients (33.5% vs 22.1% respectively, p < 0.0001; Table 1). A significantly lower proportion of patients from the GnRH antagonist than GnRHa group received baseline concomitant medications: antiandrogens (p < 0.0001), abiraterone (p = 0.0009), bicalutamide (p < 0.0001), cyproterone (p < 0.0001), and flutamide (p < 0.0001; Table 1).
Subgroup comparison
Cardiovascular outcomes
After 90 days of ADT, the proportions of patients receiving hyperlipidemia or cardiac treatments were significantly lower in the GnRH antagonist than GnRHa-treated group (hyperlipidemia treatment: 25.1% vs 34.3%, respectively; p < 0.0001; cardiac therapy: 24.6% vs 32.8% respectively; p < 0.0001). A similar trend was observed across both groups with pre-existing CVD (hyperlipidemia treatment: 36.5% vs 47.5%, respectively; p = 0.0057; cardiac therapy: 33.5% vs 41.3% respectively; p = 0.0478) and no pre-existing CVD (hyperlipidemia treatment: 19.3% vs 30.5% respectively; p < 0.0001; cardiac therapy: 20.2% vs 30.4% respectively; p < 0.0001; Table 2). Similarly, IHD, stroke, or CHF were observed in a significantly lower proportion of patients from the GnRH antagonist-treated group than the GnRHa-treated group (3.6% vs 11.6% respectively; p < 0.0001) and across both groups with pre-existing CVD (1.8% vs 12.3% respectively; p < 0.0001) and no pre-existing CVD (4.5% vs 11.4% respectively; p < 0.0001; Table 2). However, no significant difference in CV-related or other-cause deaths was observed among the groups.
MACE and composite CV events risk outcomes
MACE risk was not significantly different between the GnRH antagonist and GnRHa-treated patients. However, a significantly lower MACE risk was determined for patients with pre-existing CVD in the GnRH antagonist group than the GnRHa-treated group (adjusted hazard ratio [aHR] 0.67; 95% CI, 0.46–0.96; p = 0.0299; Table 3). In the no pre-existing CVD group, the MACE risk was not significantly different between the GnRH antagonist and GnRHa-treated groups (Table 3). The composite CV events risk was significantly lower in GnRH antagonist-treated patients than the GnRHa-treated patients (aHR 0.34; 95% CI, 0.21–0.55; p < 0.0001, Table 3). Similar lower risk of composite CV events was determined in GnRH antagonist-treated patients across the pre-existing (aHR 0.16; 95% CI, 0.05–0.50; p = 0.0017, Table 3) and no-pre-existing CVD (aHR 0.44; 95% CI, 0.26–0.74; p = 0.0019, Table 3) group than GnRHa-treated patients.
In patients with a pre-existing CVD and metastasis at diagnosis, the MACE risk was observed to be similar between patients treated with GnRH antagonist and GnRHa (aHR 0.98; 95% CI, 0.66–1.45; p = 0.9071; Table 4). Similarly, in patients with pre-existing CVD and receiving ADT for ≥6 months, no significant difference in MACE risk was observed between patients treated with GnRH antagonist and GnRHa (aHR 0.95; 95% CI, 0.74–1.22; p = 0.7023; Table 4). The risk of composite CV events was 84% lower in patients with pre-existing CVD and metastasis at diagnosis treated with GnRH antagonists than GnRHa (aHR 0.16; 95% CI, 0.04–0.38; p = 0.0130; Table 4). In patients with pre-existing CVD and receiving ADT for ≥6 months, a 88% lower risk of composite CV events was determined in GnRH antagonist treated patients than GnRHa-treated patients (aHR 0.12; 95% CI, 0.03–0.49; p < 0.0001; Table 4).
Survival analysis
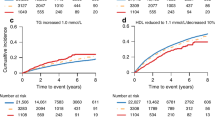
MACE-free survival probability was similar across GnRH antagonist and GnRHa-treated patients (p = 0.9569) as well as in patients across the pre-existing CVD (p = 0.1029), and no pre-existing CVD (p = 0.4228) group of patients (Fig. 1A–C). The composite CV event-free probability was significantly higher in GnRH antagonist-treated than GnRHa-treated patients (p < 0.0001, Fig. 1D). Similarly, the composite CV event-free survival probability was significantly higher in patients treated with GnRH antagonists with pre-existing CVD (p < 0.0001) as well as no pre-existing CVD (p = 0.0010) than GnRHa-treated patients (Fig. 1E, F).

Survival curves showing MACE-free survival probability (significance of difference tested by log rank test) in (A) all patients (p = 0.9569), (B) patients with pre-existing CVD (p = 0.1029), and (C) patients with no pre-existing CVD (p = 0.4228); composite CV event-free survival probability in (D) all patients (p < 0.0001), (E) patients with pre-existing CVD (p < 0.0001), and (F) patients with no pre-existing CVD (p = 0.0010). CV cardiovascular, CVD cardiovascular disease(s), GnRH gonadotropin-releasing hormone, MACE major adverse cardiovascular event (ischemic heart disease, stroke, congestive heart failure or all cause deaths), occurring ≥90 days after ADT initiation, whichever came first. Composite CV events: ischemic heart disease, stroke, congestive heart failure or CV deaths occurring ≥90 days after ADT initiation, whichever came first.
Discussion
This population-based study demonstrated that patients with PCa and pre-existing CVD treated with GnRH antagonist had a lower MACE risk (including IHD, stroke, CHF, or death), than those treated with GnRHa. A lower risk of composite CV events was observed in all GnRH antagonist-treated patients than GnRHa-treated patients, with a lowest risk determined in patients with pre-existing CVD. This is one of the few RWD studies, focusing on patients with pre-existing CVD, which demonstrates better CV outcomes in PCa treated with GnRH antagonist than GnRHa [13, 30,31,32]. This RWD provides important evidence for consideration in clinical practice, where patients receive ADT for long durations.
PRONOUNCE was the first global randomized trial with blinded adjudication of CV outcomes, in patients with PCa and recorded no difference in MACE risk in patients with known CVD, between degarelix (GnRH antagonist) and leuprolide (GnRHa). However, these results are inconclusive due to study limitations. Several studies have reported an association between GnRH antagonist and a lower risk of CV events compared with GnRHa treatment [13, 14, 16, 17, 33,34,35], but inclusion of baseline pre-existing CVD status was infrequently reported [13, 30,31,32].
We report lower MACE risk in patients treated with GnRH antagonist than GnRHa (aHR 0.67; 95% CI, 0.46–0.96; p = 0.0299) with pre-existing CVD. Our results are similar to those reported by Albertsen et al. and Margel et al. [14, 16]. We report 84% lower risk of composite CV events in patients treated with GnRH antagonist than GnRHa (aHR 0.16; 95% CI, 0.05–0.50; p = 0.0017) with pre-existing CVD. In a study using the same database that we used, but with a different study design, Chen et al. report a 52% lower risk of composite CV events (HR 0.48; 95% CI, 0.25–0.90; p = 0.0222) in patients with PCa treated with GnRH antagonist than GnRHa, at 12 months; the risk of CV events was not significantly lower in patients who had myocardial infarction, ischemic stroke, or CVD [13]. Differences in study design may explain the different findings (our study included patients who received ≥3 months GnRH antagonist or GnRHa monotherapy and had a longer treatment duration). Most, but not all RWD suggest fewer CVD events associated with GnRH antagonists than GnRHa [11, 13, 33, 34, 36, 37]. Conflicting results have been attributed to variation in baseline CVD, treatment duration, number of patients, methodological differences and CV outcomes not being the primary study outcome [17]. A Scottish Cancer Registry data analysis showed an increased CVD in patients treated with both GnRH antagonist (HR 1.5; 95% CI, 1.2–1.9) and GnRHa (HR 1.3; 95% CI, 1.2–1.4), compared with untreated patients with PCa; fewer patients were treated with GnRH antagonist than GnRHa, and there was limited adjustment for cancer stage [36]. In a multi-country (UK, Scotland, Belgium, Netherlands and France) RWD analysis and a French RWD analysis, no differences in CVD profiles were observed between the two treatment groups [11, 37]. Switching therapies during the study period, data limitations and small patient numbers on GnRH antagonist may explain these observations. Additionally, baseline CVD status was not considered in these previous studies.
Many conditions and risk factors are identified as contributing to CVD risks in patients with PCa. Therefore, several guidelines and statements have suggested a baseline CVD assessment before starting potentially cardiotoxic cancer treatments [9, 12, 38,39,40]. In our study, the definition of baseline CVD risk was based on review of codes used in previously published literature for claims-based studies [41]. However, cardiac biomarkers and lifestyle CVD risk factors measured before treatments may effectively manage patients during ADT. Patients with PCa reportedly present a burden of underassessed and undertreated CVD risk factors, including those receiving ADT [42]. Several CVD assessment methods are currently used for risk stratification, but that is not standardized [39, 43]. Intensive research to validate and refine risk stratification methods would mitigate treatment-related CVD risk and improve overall survival [44].
Both GnRH antagonist and GnRHa lead to castration-equivalent testosterone levels by different pathways [8, 33]. Antagonists bind directly to the GnRH receptors and in turn suppress LH and follicle-stimulating hormone (FSH) and reduce testosterone without causing any surge. GnRHa stimulate LH and FSH possibly causing a testosterone microsurge and subsequent receptor desensitization, resulting in castrate testosterone levels [14, 33]. The microsurges caused by GnRHa may have harmful CV effects, and the absence of these is advantageous with GnRH antagonist treatment [19, 45, 46].
Challa et al. hypothesize the possible reasons for GnRHa-related increased CV risks as: low FSH suppression (facilitates atheroma formation/progression), monocyte and T lymphocyte activation, and potentially testosterone microsurges [45]. Together, these actions may promote atherosclerotic plaque formation, disruption, and thrombosis [45]. In patients with pre-existing CVD, the differential effects of GnRH antagonist and GnRHa on MACE and composite CV events may be attributed to GnRHa destabilizing the preestablished atherosclerotic plaques [8, 14]. Proinflammatory T-helper 1 cells are dominant in atherosclerotic plaques; their lymphocytes express GnRH receptors and on activation by the GnRHa lead to T-cell proliferation [8, 14, 33, 44, 47, 48]. T lymphocytes may cause atherosclerotic plaque rupturing through release of proinflammatory cytokines and macrophage stimulation [44]. Macrophages maintain the local inflammatory response, increasing plaque development and thrombosis [49].
An increased invasive ability of macrophages has been reported in the presence of GnRHa, but not GnRH antagonist [13]. Lifshitz et al. hypothesize that in patients with pre-existing CVD, GnRH antagonist treatment has a direct protective effect on plaque stability in contrast to the plaque instability caused by GnRHa treatment. In patients with pre-existing CVD, GnRH antagonist treatment increased serum levels of five proteins associated with plaque stabilizing: human chitotriosidase, macrophage receptor with collagenous structure, cathepsin D, superoxide dismutase-2 and hydroxyacid oxidase-1 [50]. The microenvironment in patients with PCa with pre-existing CVD is altered with distinct lymphocyte, monocyte, and inflammatory modulation, enabling GnRH antagonist treatment to potentially have beneficial effects in patients with pre-existing CVD over GnRHa [50].
The prevalence of CVD is lower in Taiwan than in Western populations [51]. Therefore, treatment-related cardiotoxicity may be underestimated in patients with PCa. The decreased risk of composite CV events and MACE in patients with pre-existing CVD, treated with degarelix in this study population, provides important real-world evidence for physicians, highlighting relevant considerations regarding cardiotoxicity to cancer treatment.
Our study has a few limitations, including lack of body mass index and physical activity data-relevant for CVD baseline context. The number of patients treated with GnRH antagonist was lower than that of GnRHa. We have analyzed the RWD with distinct pre-existing CVD stratification and report the safety of degarelix (GnRH antagonist), including in patients with pre-existing CVD, adding to the limited data in Asian patients with PCa. Our study allows a true comparison between GnRH antagonist and GnRHa treatment, as patients received these single treatments, without overlap.
ADT is the main treatment in advanced stage PCa, and patients are potentially treated over a long duration, so our results provide additional valuable insight evaluating GnRH antagonist and GnRHa as treatment options. Given that PCa incidence rates increase with age, and CVD risk also becomes more likely, GnRH antagonist may provide a safer CV-risk profile for long-term PCa treatment.
Conclusion
GnRH antagonist reduced risk of MACE and composite CV events in patients with PCa and with pre-existing CVD, relative to GnRHa-treated patients. This effect was most pronounced in GnRH antagonist-treated patients with pre-existing CVD, metastasis at diagnosis and use of ADT ≥ 6 months. These real-world findings are relevant when considering the long-term treatment of PCa.
Data availability
This study was conducted using the Taiwan National Health Insurance Research Data (NHIRD) database. Due to legal restrictions imposed by the government of Taiwan in relation to the “Personal Information Protection Act”, data cannot be made publicly available. Requests for data can be sent as a formal proposal to the Health and Welfare Data Science Center, Taiwan.
Change history
10 October 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41391-022-00596-5
11 July 2022
A Correction to this paper has been published: https://doi.org/10.1038/s41391-022-00568-9
References
Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49.
Huang YC, Chen YH. Cancer incidence characteristic evolution based on the National Cancer Registry in Taiwan. J Oncol. 2020;2020:1408793.
Ministry of Health and Welfare ROCT. Taiwan health and welfare report 2017. Ministry of Health and Welfare, R.O.C. (Taiwan): Taipei City, Taiwan, 2018.
Sturgeon KM, Deng L, Bluethmann SM, Zhou S, Trifiletti DM, Jiang C, et al. A population-based study of cardiovascular disease mortality risk in US cancer patients. Eur Heart J. 2019;40:3889–97.
Elmehrath AO, Afifi AM, Al-Husseini MJ, Saad AM, Wilson N, Shohdy KS, et al. Causes of death among patients with metastatic prostate cancer in the US from 2000 to 2016. JAMA Netw Open. 2021;4:e2119568.
Chowdhury S, Robinson D, Cahill D, Rodriguez-Vida A, Holmberg L, Moller H. Causes of death in men with prostate cancer: an analysis of 50,000 men from the Thames Cancer Registry. BJU Int. 2013;112:182–9.
Stoltzfus KC, Zhang Y, Sturgeon K, Sinoway LI, Trifiletti DM, Chinchilli VM, et al. Fatal heart disease among cancer patients. Nat Commun. 2020;11:2011.
Muniyan S, Xi L, Datta K, Das A, Teply BA, Batra SK, et al. Cardiovascular risks and toxicity - The Achilles heel of androgen deprivation therapy in prostate cancer patients. Biochim Biophys Acta Rev Cancer. 2020;1874:188383.
Okwuosa TM, Morgans A, Rhee JW, Reding KW, Maliski S, Plana JC, et al. Impact of hormonal therapies for treatment of hormone-dependent cancers (breast and prostate) on the cardiovascular system: effects and modifications: a scientific statement from the American Heart Association. Circ Genom Precis Med. 2021;14:e000082.
Freedland SJ, Abrahamsson PA. Androgen deprivation therapy and side effects: are GnRH antagonists safer? Asian J Androl. 2021;23:3–10.
Scailteux LM, Vincendeau S, Balusson F, Leclercq C, Happe A, Le Nautout B, et al. Androgen deprivation therapy and cardiovascular risk: No meaningful difference between GnRH antagonist and agonists-a nationwide population-based cohort study based on 2010-2013 French Health Insurance data. Eur J Cancer. 2017;77:99–108.
Kokorovic A, So AI, Serag H, French C, Hamilton RJ, Izard JP, et al. Canadian Urological Association guideline on androgen deprivation therapy: Adverse events and management strategies. Can Urol Assoc J 2021;15:E307–E322.
Chen DY, Su PJ, See LC, Liu JR, Chuang CK, Pang ST, et al. Gonadotropin-releasing hormone antagonist associated with lower cardiovascular risk compared with gonadotropin-releasing hormone agonist in prostate cancer: a nationwide cohort and in vitro study. Prostate 2021;81:902–12.
Albertsen PC, Klotz L, Tombal B, Grady J, Olesen TK, Nilsson J. Cardiovascular morbidity associated with gonadotropin releasing hormone agonists and an antagonist. Eur Urol. 2014;65:565–73.
Leong DP, Fradet V, Shayegan B, Duceppe E, Siemens R, Niazi T, et al. Cardiovascular risk in men with prostate cancer: Insights from the RADICAL PC Study. J Urol. 2020;203:1109–16.
Margel D, Peer A, Ber Y, Shavit-Grievink L, Tabachnik T, Sela S, et al. Cardiovascular morbidity in a randomized trial comparing GnRH agonist and GnRH antagonist among patients with advanced prostate cancer and preexisting cardiovascular disease. J Urol. 2019;202:1199–208.
Abufaraj M, Iwata T, Kimura S, Haddad A, Al-Ani H, Abusubaih L, et al. Differential impact of gonadotropin-releasing hormone antagonist versus agonist on clinical safety and oncologic outcomes on patients with metastatic prostate cancer: a meta-analysis of randomized controlled trials. Eur Urol. 2021;79:44–53.
Lopes RD, Higano CS, Slovin SF, Nelson AJ, Bigelow R, Sorensen PS, et al. Cardiovascular safety of degarelix versus leuprolide in patients with prostate cancer: the primary results of the PRONOUNCE randomized trial. Circulation. 2021;144:1295–1307.
Klotz L, Boccon-Gibod L, Shore ND, Andreou C, Persson BE, Cantor P, et al. The efficacy and safety of degarelix: a 12-month, comparative, randomized, open-label, parallel-group phase III study in patients with prostate cancer. BJU Int. 2008;102:1531–8.
Sciarra A, Fasulo A, Ciardi A, Petrangeli E, Gentilucci A, Maggi M, et al. A meta-analysis and systematic review of randomized controlled trials with degarelix versus gonadotropin-releasing hormone agonists for advanced prostate cancer. Med (Baltim). 2016;95:e3845.
Chiang CJ, You SL, Chen CJ, Yang YW, Lo WC, Lai MS. Quality assessment and improvement of nationwide cancer registration system in Taiwan: a review. Jpn J Clin Oncol. 2015;45:291–6.
Wen CP, Tsai SP, Chung WS. A 10-year experience with universal health insurance in Taiwan: measuring changes in health and health disparity. Ann Intern Med. 2008;148:258–67.
Lu TH, Lee MC, Chou MC. Accuracy of cause-of-death coding in Taiwan: types of miscoding and effects on mortality statistics. Int J Epidemiol. 2000;29:336–43.
American Joint Committee on Cancer. Manual for Staging of Cancer, 5 edn. JB Lippincott: Philadelphia, 1997.
Kryvenko ON, Epstein JI. Changes in prostate cancer grading: Including a new patient-centric grading system. Prostate 2016;76:427–33.
Jespersen CG, Norgaard M, Bjerklund Johansen TE, Sogaard M, Borre M. The influence of cardiovascular morbidity on the prognosis in prostate cancer. Experience from a 12-year nationwide Danish population-based cohort study. BMC Cancer. 2011;11:519.
Mohler JL, Armstrong AJ, Bahnson RR, Boston B, Busby JE, D’Amico AV, et al. Prostate cancer, Version 3.2012: featured updates to the NCCN guidelines. J Natl Compr Canc Netw. 2012;10:1081–7.
Austin PC, Fine JP. Practical recommendations for reporting Fine-Gray model analyses for competing risk data. Stat Med. 2017;36:4391–4400.
Fine JP, Gray RJ. A proportional hazards model for the subdistribution of a competing risk. J Am Stat Assoc. 1999;94:496–509.
Kao HH, Kao LT, Li IH, Pan KT, Shih JH, Chou YC, et al. Androgen deprivation therapy use increases the risk of heart failure in patients with prostate cancer: A population-based cohort study. J Clin Pharm. 2019;59:335–43.
Wang LH, Liu CK, Chen CH, Kao LT, Lin HC, Huang CY. No increased risk of coronary heart disease for patients receiving androgen deprivation therapy for prostate cancer in Chinese/Taiwanese men. Andrology. 2016;4:128–32.
Wu SY, Fang SC, Hwang OR, Shih HJ, Shao YJ. Influence of baseline cardiovascular comorbidities on mortality after androgen deprivation therapy for metastatic prostate cancer. Cancers (Basel). 2020;12:189.
Davey P, Kirby MG. Cardiovascular risk profiles of GnRH agonists and antagonists: real-world analysis from UK general practice. World J Urol. 2021;39:307–15.
Perrone V, Degli Esposti L, Giacomini E, Veronesi C, Blini V, Oderda M. Cardiovascular risk profile in prostate cancer patients treated with GnRH agonists versus antagonists: an Italian real-world analysis. Ther Clin Risk Manag. 2020;16:393–401.
Shore ND, Saad F, Cookson MS, George DJ, Saltzstein DR, Tutrone R, et al. Oral relugolix for androgen-deprivation therapy in advanced prostate cancer. N. Engl J Med. 2020;382:2187–96.
Cardwell CR, O’Sullivan JM, Jain S, Harbinson MT, Cook MB, Hicks BM, et al. The risk of cardiovascular disease in prostate cancer patients receiving androgen deprivation therapies. Epidemiology 2020;31:432–40.
George G, Garmo H, Scailteux LM, Balusson F, De Coster G, De Schutter H, et al. Risk of cardiovascular disease following gonadotropin-releasing hormone agonists vs antagonists in prostate cancer: Real-world evidence from five databases. Int J Cancer. 2021;148:2203–11.
Armenian SH, Lacchetti C, Barac A, Carver J, Constine LS, Denduluri N, et al. Prevention and monitoring of cardiac dysfunction in survivors of adult cancers: American Society of Clinical Oncology clinical practice guideline. J Clin Oncol. 2017;35:893–911.
Lyon AR, Dent S, Stanway S, Earl H, Brezden-Masley C, Cohen-Solal A, et al. Baseline cardiovascular risk assessment in cancer patients scheduled to receive cardiotoxic cancer therapies: a position statement and new risk assessment tools from the Cardio-Oncology Study Group of the Heart Failure Association of the European Society of Cardiology in collaboration with the International Cardio-Oncology Society. Eur J Heart Fail 2020;22:1945–60.
Zamorano JL, Lancellotti P, Rodriguez Muñoz D, Aboyans V, Asteggiano R, Galderisi M, et al. 2016 ESC Position Paper on cancer treatments and cardiovascular toxicity developed under the auspices of the ESC Committee for Practice Guidelines: The Task Force for cancer treatments and cardiovascular toxicity of the European Society of Cardiology (ESC). Eur Heart J. 2016;37:2768–801.
George G, Scailteux LM, Garmo H, Balusson F, Cardwell C, Coster G, et al. Real-world insights into risk of developing cardiovascular disease following GnRH agonists versus antagonists for prostate cancer: a methodological protocol to a study using five European databases. Fundam Clin Pharm. 2019;33:479–99.
Sun L, Parikh RB, Hubbard RA, Cashy J, Takvorian SU, Vaughn DJ, et al. Assessment and management of cardiovascular risk factors among US veterans with prostate cancer. JAMA Netw Open. 2021;4:e210070.
Ng CF, Chiu PKF, Yee CH, Lau BSY, Leung SCH, Teoh JYC. Effect of androgen deprivation therapy on cardiovascular function in Chinese patients with advanced prostate cancer: a prospective cohort study. Sci Rep. 2020;10:18060.
Rosario DJ, Davey P, Green J, Greene D, Turner B, Payne H, et al. The role of gonadotrophin-releasing hormone antagonists in the treatment of patients with advanced hormone-dependent prostate cancer in the UK. World J Urol. 2016;34:1601–9.
Challa AA, Calaway AC, Cullen J, Garcia J, Desai N, Weintraub NL, et al. Cardiovascular toxicities of androgen deprivation therapy. Curr Treat Options Oncol. 2021;22:47.
Van Poppel H, Abrahamsson PA. Considerations for the use of gonadotropin-releasing hormone agonists and antagonists in patients with prostate cancer. Int J Urol. 2020;27:830–7.
Chen HF, Jeung EB, Stephenson M, Leung PC. Human peripheral blood mononuclear cells express gonadotropin-releasing hormone (GnRH), GnRH receptor, and interleukin-2 receptor gamma-chain messenger ribonucleic acids that are regulated by GnRH in vitro. J Clin Endocrinol Metab. 1999;84:743–50.
Tanriverdi F, Gonzalez-Martinez D, Hu Y, Kelestimur F, Bouloux PM. GnRH-I and GnRH-II have differential modulatory effects on human peripheral blood mononuclear cell proliferation and interleukin-2 receptor gamma-chain mRNA expression in healthy males. Clin Exp Immunol. 2005;142:103–10.
Barrett TJ. Macrophages in atherosclerosis regression. Arterioscler Thromb Vasc Biol. 2020;40:20–33.
Lifshitz K, Ber Y, Shenhar C, Nillson J, Peer A, Rosenbaum E, et al. Cardiovascular proteomics: A post hoc analysis from a phase II randomized clinical trial comparing GnRH antagonist vs GnRH agonist among men with advanced prostate cancer. J Urol. 2021;206:952–9.
Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990-2019: Update from the GBD 2019 Study. J Am Coll Cardiol 2020;76:2982–3021.
Acknowledgements
We would like to thank the Health and Welfare Data Science Center of Taiwan for providing data. Veena Ekbote, MIMS, provided editorial support, which was funded by Ferring Pharmaceuticals Ltd, in compliance with international guidelines for Good Publication Practice 3.
Funding
This research was funded by Ferring Pharmaceuticals. The funder had no role in design and conduct of the study or in collection, management, analysis, and interpretation of the data or in preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
YHJS designed the study, extracted the data and performed the statistical data analysis. All authors interpreted the data and contributed to the preparation of the manuscript. All authors critically reviewed drafts of manuscript and approved the final version.
Corresponding author
Ethics declarations
Competing interests
All authors declare no competing interests.
Ethics approval
The study was conducted according to the Declaration of Helsinki and the International Society for Pharmacoepidemiology Guidelines for Good Pharmacoepidemiology Practices. It was carried out retrospectively using an anonymized database. This study was reviewed and approved by the Institutional Review Board of Taipei Medical University (TMU-JIRB no. 201502042). All authors had full access to the data and take responsibility for data integrity and analysis.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised to correct the names of the authors, correct table 1 and 2 and the legend of figure 1.
The original online version of this article was revised: In Table 4 of this article, the data in the risk of MACE and composite CV events headed receiving more than 6 months of ADT were mistakenly listed under the headed preexisting CVD, receiving more than 6 months of ADT and vice versa.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shao, YH.J., Hong, JH., Chen, CK. et al. Cardiovascular risk of gonadotropin-releasing hormone antagonist versus agonist in men with prostate cancer: an observational study in Taiwan. Prostate Cancer Prostatic Dis 26, 722–729 (2023). https://doi.org/10.1038/s41391-022-00555-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41391-022-00555-0
This article is cited by
-
Cardiovascular disease risk assessment and multidisciplinary care in prostate cancer treatment with ADT: recommendations from the APMA PCCV expert network
World Journal of Urology (2024)
-
Cardiovascular adverse events-related to GnRH agonists and GnRH antagonists: analysis of real-life data from Eudra-Vigilance and Food and Drug Administration databases entries
Prostate Cancer and Prostatic Diseases (2023)
