
Abstract
Background
Neonates have high levels of cold-shock proteins (CSPs) in the normothermic brain for a limited period following birth. Hypoxic–ischemic (HI) insults in term infants produce neonatal encephalopathy (NE), and it remains unclear whether HI-induced pathology alters baseline CSP expression in the normothermic brain.
Methods
Here we established a version of the Rice–Vannucci model in PND 10 mice that incorporates rigorous temperature control.
Results
Common carotid artery (CCA)-ligation plus 25 min hypoxia (8% O2) in pups with targeted normothermia resulted in classic histopathological changes including increased hippocampal degeneration, astrogliosis, microgliosis, white matter changes, and cell signaling perturbations. Serial assessment of cortical, thalamic, and hippocampal RNA-binding motif 3 (RBM3), cold-inducible RNA binding protein (CIRBP), and reticulon-3 (RTN3) revealed a rapid age-dependent decrease in levels in sham and injured pups. CSPs were minimally affected by HI and the age point of lowest expression (PND 18) coincided with the timing at which heat-generating mechanisms mature in mice.
Conclusions
The findings suggest the need to determine whether optimized therapeutic hypothermia (depth and duration) can prevent the age-related decline in neuroprotective CSPs like RBM3 in the brain, and improve outcomes during critical phases of secondary injury and recovery after NE.
Impact
-
The rapid decrease in endogenous neuroprotective cold-shock proteins (CSPs) in the normothermic cortex, thalamus, and hippocampus from postnatal day (PND) 11–18, coincides with the timing of thermogenesis maturation in neonatal mice.
-
Hypoxia–ischemia (HI) has a minor impact on the normal age-dependent decline in brain CSP levels in neonates maintained normothermic post-injury.
-
HI robustly disrupts the expected correlation in RNA-binding motif 3 (RBM3) and reticulon-3 (RTN3).
-
The potent neuroprotectant RBM3 is not increased 1–4 days after HI in a mouse model of neonatal encephalopathy (NE) in the term newborn and in which rigorous temperature control prevents the manifestation of endogenous post-insult hypothermia.
Similar content being viewed by others


Introduction
Mammalian cold-shock proteins (CSPs) are critical mediators of the cellular adaptive response to cold stress.1,2 While the translational efficiencies of most proteins are coupled to the activity of temperature-sensitive enzymes involved in cap-dependent synthesis, CSPs evolved unique molecular mechanisms allowing them to escape translational repression at cooler temperatures.3,4,5 Together they modulate critical biological processes including inflammation,6,7,8 fever,9 neuroprotection,5,10,11,12 neurotransmission,13,14 bone homeostasis,15,16 and directly counteract the inhibitory effect of hypothermia on cap-dependent expression thereby promoting global protein synthesis (GPS).17
The brain is highly vulnerable to changes in external temperature during infancy.18,19 Both the fetus in utero and neonates early postpartum, depend on a caregiver for temperature maintenance. Life’s unpredictability precludes a guarantee that a mother can sustain the thermoneutrality of the fetus or newborn unabated.20,21 Evidence suggests that CSPs are temporarily increased during early neurodevelopment as molecular safeguards to protect the neonatal brain from changes in external temperature until endogenous mechanisms of thermogenesis mature.22,23 Consequently, baseline brain levels of the major CSPs, RNA-binding motif 3 (RBM3) and cold-inducible RNA-binding protein (CIRBP), are inversely associated with postnatal age in mice, rats, and in humans.22,24,25,26 Similarly, brain levels of small reticulon-3 (RTN3) isoforms, some of which may also be novel CSPs, are inversely associated with postnatal age in mice and in humans.5,25,27 Moreover, brain morphology is normal in newborn RBM3 KO mice if maintained at normothermia, but pups subjected to hypothermia in utero by exposing the dam to external cold stress, are born with severe central nervous system (CNS) abnormalities.23
Hypoxia–ischemia (HI) from a sentinel event produces devastating damage to the newborn brain resulting in neonatal encephalopathy (NE).28 Therapeutic hypothermia (TH) to 33 °C for 72 h, initiated within 6 h of birth, is the standard of care to improve long-term neurological outcomes in term newborns with moderate-to-severe NE.29,30,31 It is unknown whether TH increases neuroprotective CSPs in this and related settings.31,32 Furthermore, it is unknown whether HI disturbs the normal neurodevelopmental time course of transient CSP expression in post-resuscitated infants. Here we address this latter question. We first characterized neuropathology in a version of the Rice–Vannucci (RV) model modified to include rigorous temperature monitoring/control in PND 10 pups to model perinatal asphyxia in the normothermic term infant, and comprehensively assessed CSPs (RBM3, CIRBP, RTN3) in the normothermic cortex, thalamus, and hippocampus at 1, 4, and 8 days post-injury, to test the hypothesis that HI alters the neurodevelopmental time course of CSP expression. We provide a foundational dataset using rigorous temperature control to inform the design of future studies on methods to induce and/or modulate neuroprotective CSPs in HI-induced NE.
Materials and methods
Additional details are provided in the Supplementary Methods.
Animals
Studies were approved by the IACUC of the University of Pittsburgh. Tissues were analyzed at the University of South Florida via a Material Transfer Agreement. C57BL/6 foster dams with litters consisting of 7–8 male pups were purchased from Charles River. Mice on a 12 h light/dark cycle had ad libitum access to standard chow.
HI injury model
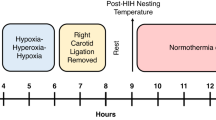
PND 10 pups were isoflurane anesthetized (3% induction and 1.5–3% maintenance) in a 2:1 mixture of N2O/O2 and weighed. The core temperature during sham or occlusion surgery was monitored via a neonatal rectal probe (Physitemp). Body temperature (Tb) was maintained to ~36 °C. Post-surgical temperature was measured with a Braun no-touch infra-red pediatric thermometer (Braun Healthcare US). Pilots in naive PND 10 pups (n = 6) showed high concordance between temperatures measured by a rectal thermometer (Tb of 36.5 ± 0.73°C) vs. the Braun IR thermometer (Braun IRT) (Tb of 36.6 ± 0.30 °C). Normothermia was ~36 °C based on pilots in cage-nesting naive PND 10 pups in which Tb was found to be 36.1 ± 0.34 °C (n = 6). The right common carotid artery (CCA) was accessed by a 0.2–0.3 cm neck incision, sutured, and cut. The incision was closed with 3M Vetbond Tissue Adhesive (Maplewood) and Bupivacaine (0.25%) applied to the wound. Mice advanced in blocks of four. A second technician maintained pups at ~36 °C in the post-surgical phase by applying heat as needed and by regular Tb monitoring with the Braun IRT. The four-pup block was then returned to the foster dam for 1 h. Next, pups advanced to the hypoxia phase and were placed inside a specialized acrylic hypoxia chamber glove box (Coy; Grass Lake, MI). In pilots, we observed a high rate of mortality when hypoxia (8% O2/92% N2) was maintained for >30 min. Thus, we titrated hypoxia to a duration that produced reproducible brain damage but no mortality, resulting in the 25 min insult level used in this study. An ambient hypoxia chamber temperature setting of 38–40 °C maintained neonatal Tb to ~36 °C. Shams receive identical procedures without CCA ligation and were placed inside the chamber for 25 min at normoxia (21% O2/79% N2). After hypoxia or normoxia, Tb was managed to ~36 °C in room air for 2 h:5 min on a benchtop with lamps and heating blankets, and the temperature was recorded every 15 min. Pups were then returned to dams until weaning or euthanasia. Cages were supplemented with multiple types of bedding, including Enviro-dri®, to promote high-quality nest construction and decrease thermal stress caused by standard housing.33,34 Details of our approach are provided (Supplementary Fig. 1).
Histology staining and neuropathology assessment
Anesthetized pups were transcardially perfused with heparinized cold saline, and 4% paraformaldehyde in 4% sucrose/0.1 M phosphate buffer/pH 7.2 (Electron Microscopy Sciences). Isolated brains were post-fixed 24 h and shipped to Neuroscience Associates (NSA, Knoxville, TN). Brains were embedded in a gelatin matrix (MultiBrain® Technology, NSA). The block was frozen by immersion in chilled 2-methylbutane and mounted on a freezing stage of an AO 860 sliding microtome. The MultiBrain® block was sectioned coronally through the entire specimen length and collected into a series of 24 cups containing antigen preservation solution (50% phosphate-buffered saline pH 7.0, 50% ethylene glycol, 1% polyvinyl pyrrolidone). Free-floating sections of an entire cup—each cup having section representation across the full brain rostral to caudal (i.e., a total of 29 “levels”)—were stained with either Fluro-Jade (FJ), GFAP, Iba-1, Weil, or Amino Cupric Silver/Neutral Red Counter. Stained sections were mounted on glass slides. Based on preliminary observations, whole brains at levels 15 and 18, across all brains and stains, were digitally scanned by NSA at ×20 magnification on a TissueScope LE120 (Huron Digital Pathology). Data were uploaded to Concentriq (Proscia; Philadelphia, PA) for the manual acquisition of regions of interest at ×2, ×4, ×10, and ×20 magnification for figure compilation. Slides of Amino Cupric Silver/Neutral Red Counter, Iba1, and GFAP were analyzed by an expert veterinary neuropathologist (R.H.G.) to construct a comprehensive neuroanatomical assessment table of microscopic pathology that spanned all brain levels (1–29). The diagnostic category of “neuropil argyrophilia” was used when a pattern of finely grained silver deposition was present in the absence (or in addition to) distinct neuronal cell body staining by the Amino Cupric Silver stain.35 Neuron argyrophilia scores are based on estimates of the percentage of degenerating (silver positive) vs. normal (unstained) neurons counted in each neuroanatomical region and take into account the size of the brain region (lesions may be focal, multifocal, or diffuse). Normal (0) = no stained neurons observed. Minimal (−) = very few neurons judged to be degenerative. Mild (+) = <~25% of neurons affected. Moderate (++) = ~25–50% of neurons affected. Marked (+++) = ~50–75% of neurons affected. Severe (###) = >75% of neurons affected. All regions characterized by neuron argyrophilia also exhibited reactive microglia and astrocytes characterized microscopically by increased staining of microglial cells (Iba1) or of astrocytes (GFAP). In both processes—designated respectively as microgliosis and astrocytosis—there was also altered morphology of the reactive microglia and astrocytes characterized by thickened more prominent cytologic processes. Foci with severe loss of cellularity (shown with all stains) were classified as “necrosis.” Examples of staining at a single brain level are shown (Supplementary Figures).
Western blot
We used our standard protocol25 (see Supplementary Methods).
Lesion volume analysis
Whole fixed brains were embedded in paraffin. Sections were taken every 0.5 mm from posterior to anterior and stained with hematoxylin–eosin. The hemispheric area was measured on each slide and summed for total volume (MCID software, St. Catherines, Ontario). The hippocampal area was measured on every slide with a visible hippocampus and the volume was calculated.
Statistical analysis
Western blot densitometry of total protein stain (TPS treated membranes) and CSPs, MBP, and cell death targets was measured with the UN-SCAN-IT software (Silk Scientific). Each target of interest (densitometry value) was standardized by dividing by the densitometry value of TPS corresponding to the target’s lane. Standardized densitometric values within each blot were then normalized. For CSP/MBP analysis (48/brain region), samples were split evenly across two 26-well criterion gels to ensure equal injury representation (Sham vs. HI) and postnatal age on each blot. Data were analyzed using a two-way analysis of variance and Tukey’s multiple comparison post hoc test. Molecular analysis of cell death pathways at 3 days post-injury in hippocampal homogenates was analyzed by an unpaired two-tailed t test. Data were significant at p < 0.05.
Results
Male PND 10 pups were subjected to HI or sham manipulations (Supplementary Fig. 1). Neuronal death was assessed by FJ staining at 3, 5, and 7 days post-injury. Increased FJ staining was detected in the ipsilateral hippocampus at 3 days post-injury (Fig. 1a). FJ staining was generally absent in HI-injured brains 5–7 days later (Fig. 1a). Next, cell signaling changes were investigated. The levels of the p75 neurotrophin receptor (P75NTR) and cyclophilin C (CYPC) decreased 3 days post-injury in the hippocampus (Figs. 1b, d and Supplementary Fig. 2A, C). The levels of the pro-apoptotic protein Bax increased (Fig. 1c and Supplementary Fig. 2B). There was no change in the levels of cyclophilin A (CYPA) (Fig. 1e and Supplementary Fig. 2D). Finally, 14 days post-injury lesion volume analysis confirmed robust tissue loss in both the ipsilateral hemisphere (Fig. 1f) and ipsilateral hippocampus (Fig. 1g) of injured pups, vs. either the contralateral hemisphere/hippocampus of injured pups or vs. sham ipsilateral hemisphere/hippocampus.

a Fluro jade (FJ) staining in the ipsilateral hippocampus 3 days post-injury (n = 2 representative mice per time point). b–e Densitometric analysis (n = 4/group) showed p57NTR and CYPC levels decreased 3 days post-injury in the ipsilateral hippocampus. No change in CYPA was detected. Bax was increased 3 days post-injury. f, g Chronic brain tissue loss was seen in the cortical hemisphere and hippocampus ipsilateral to the CCA ligation (n = 11/group sham and n = 12/group HI). Data were significant at p < 0.05. Box plots show minimum, maximum, interquartile range (IQR), and median. Asterisks in the graphs indicate post hoc significance. p75NTA P75 neurotrophin receptor, CYPC cyclophilin C, CYPA cyclophilin A, CCA common carotid artery.

a–c Iba-1 staining (purple) and GFAP staining (brown) in the contralateral/ipsilateral hippocampus in a representative mouse at 3, 5, and 7 days, post-injury. d–g ×20 magnification of Iba-1 and GFAP staining in the contralateral vs. ipsilateral hippocampus at 3 days post-injury in a representative mouse. Iba-1 ionized calcium-binding adapter molecule 1, GFAP glial fibrillary acidic protein.
A comprehensive neuropathological assessment of microgliosis, astrocytosis, and neuronal degeneration was also performed. Microgliosis and astrocytosis were most severe in the injured hippocampus and were detected 3–7 days post-injury (Fig. 2, Supplementary Figs. 3 and 4, and Table 1). All hippocampal sectors (CA1, CA2, CA3, CA4) had activated microglia and astrocytes. In contrast, in the dentate gyrus (DG), microgliosis and astrocytosis were robustly increased 3 days post-injury, but began to decrease thereafter and were minimal by 7 days (Fig. 2a–g and Table 1). Additional brain regions in some pups showed mild persistent microgliosis and astrocytosis up to 7 days post-injury, including in the temporal, occipital, and parietal cortex and thalamus (Table 1). Neuronal degeneration was generally accompanied by microgliosis and astrocytosis. Neuron argyrophilia was most severe, persisted the longest, and was seen in all HI-injured pups in the hippocampus (Fig. 3, Supplementary Fig. 5, and Table 1). Curiously, the magnitude and duration of ongoing neuronal degeneration were greatest in the CA2, followed by the CA3 and CA4. In contrast, neuronal degeneration in the CA1 began to resolve by 7 days post-injury.

a, c, e Amino Cupric Silver staining (black) and Congo red counterstaining (pink) in brain sections from two representative mice at 3, 5, and 7 days, post-injury. b ×4 magnification of silver staining in the hippocampus and corpus callosum 3 days post-injury. d ×10 magnification of CA1 hippocampal neuron degeneration 5 days post-injury. g ×4 magnification of silver staining in the thalamus and cortices 3 days post-injury. f ×10 magnification of CA3 hippocampal neuron degeneration at 7 days post-injury.
Next, we assessed thalamic white matter injury. Myelin-basic protein (MBP) levels were measured in protein extracts from the isolated thalamus at 24 h, 4 days, and 8 days post-injury. MBP levels were below detection limits in PND 11 sham and in injured pups (Supplementary Fig. 6A, B). In contrast, MBP levels were robustly detected in the thalamus by PND 18 (Supplementary Fig. 6). Injured pups showed a nonsignificant (p = 0.06) trend for decreased thalamic MBP levels. Weil’s myelin stain further supported myelin sheath damage in the thalamus in some HI-injured pups. Myelin staining (deep blue/black) developmentally increased in the thalamus from PND 13 to PND 17 (Supplementary Figs. 6C–E and 7), but two of three HI-injured pups at the 7 days post-injury time point showed decreased Weil staining in the ipsilateral thalamus and cerebral peduncles (Supplementary Fig. 6E–G).
We next tested the hypothesis that HI alters the neurodevelopmental expression of RBM3, CIRBP, and RTN3. All three CSPs robustly decreased in the cortex with advancing age in shams (Fig. 4 and Supplementary Figs. 8 and 10A). Cortical RBM3 levels were not affected by HI injury, nor was there interaction with age (Fig. 4a, b and Supplementary Fig. 8A, B). Cortical CIRBP levels showed a significant interaction with age and injury (Fig. 4c, d and Supplementary Fig. 8C, D). Cortical RTN3 levels were significantly affected by injury and showed a non-significant trend toward a potential interaction with age (Fig. 4e, f and Supplementary Fig. 8E, F). Specifically, a post hoc significant increase in RTN3c levels was detected 24 h post-injury (Fig. 4f).

a, c, e Representative western blot (n = 4/group) of cortical RBM3, CIRBP, and RTN3 levels in shams vs. HI-injured pups 24 h, 4 days, and 8 days post-injury b, d, f Densitometric analysis of cortical RBM3, CIRBP, and RTN3 levels (n = 8/group). Total protein stains are available in the supplementary. Data were significant at p < 0.05. Box plots show minimum, maximum, interquartile range (IQR), and median. Asterisks in the graphs indicate post hoc significance. RBM3 RNA-binding motif 3, CIRBP cold-inducible RNA-binding protein, RTN3 reticulon 3.
In the thalamus, all three CSPs decreased with advancing age in shams but were unaffected by HI injury (Fig. 5 and Supplementary Figs. 9 and 10B). Thalamic RBM3 levels were not affected by HI injury, nor was there interaction with age (Fig. 5a, b and Supplementary Fig. 9A, B). Thalamic CIRBP levels were not affected by HI injury, nor was there interaction with age (Fig. 5c, d and Supplementary Fig. 9C, D). Thalamic RTN3 levels were not affected by HI injury, nor was there an interaction with age (Fig. 5e, f and Supplementary Figs. 9E, F and 10B).

a, c, e Representative western blot (n = 4/group) of thalamic RBM3, CIRBP, and RTN3 levels in shams vs. HI-injured pups 24 h, 4 days, and 8 days post-injury. b, d, f Densitometric analysis of thalamic RBM3, CIRBP, and RTN3 levels (n = 8/group). Total protein stains are available in the supplementary. Data were significant at p < 0.05. Box plots show minimum, maximum, interquartile range (IQR), and median. Asterisks in graphs indicate post hoc significance. RBM3 RNA-binding motif 3, CIRBP cold-inducible RNA-binding protein, RTN3 reticulon 3.
In the hippocampus, there were marked differences in CSP detection (Fig. 6 and Supplementary Figs. 11 and 12). Hippocampal RBM3 levels were readily detectable, decreased in shams with advancing age, and showed a significant interaction with injury (Fig. 6a, b and Supplementary Fig. 11A, B). RBM3 levels increased 8 days post-injury in HI-injured pups vs. shams but did not reach significance on post hoc analysis. (Fig. 6b). CIRBP was readily detectable, decreased in shams with advancing age, and had a significant interaction with injury manifested by a slight increase at 8 days post-injury, but which was not significant on post hoc analysis. (Fig. 6c, d and Supplementary Fig. 11C, D). Hippocampal RTN3 and RTN3c were faintly detected (Supplementary Fig. 12). The absence of RTN3 in most samples precluded meaningful densitometric analysis. RBM3 has been shown to regulate RTN3 expression, thus, we performed a Spearman correlation on CIRBP or RTN3 vs. RBM3 levels, across all brain tissues/ages. In the cortex, RBM3/CIRBP was highly correlated in shams (Fig. 6e). and maintained after injury (Fig. 6f). RBM3/RTN3 were also correlated in shams, but not after HI injury (Fig. 6e, f). The same phenomenon was observed in the thalamus (Fig. 6g, h). Lack of RTN3 expression in the hippocampus precluded a correlation analysis, but hippocampal RBM3/CIRBP levels were correlated in shams and after injury (Fig. 6i, j).

a, c Representative western blot (n = 4/group) of hippocampal RBM3 and CIRBP levels in shams vs. HI-injured pups 24 h, 4 days, and 8 days post-injury. b, d Densitometric analysis of hippocampal RBM3 and CIRBP levels (n = 8/group). Total protein stains are available in the supplementary. e–j Spearman correlation analysis of RBM3 vs. CIRBP and RTN3 levels in shams and HI injury pups across all time points in the cortex, thalamus, and hippocampus. Data were significant at p < 0.05. Box plots show minimum, maximum, interquartile range (IQR), and median. Asterisks in graphs indicate post hoc significance. RBM3 RNA-binding motif 3, CIRBP cold-inducible RNA-binding protein, RTN3 reticulon 3.
Finally, we investigated whether inter-subject differences in insult severity correlated with changes in RBM3 and CIRBP levels in the injured hippocampus. Here, GFAP and Iba1 protein levels were used as surrogate markers of insult severity (Supplementary Figs. 13–15). GFAP and Iba1 levels were significantly increased at 4 and 8 days post-HI but showed high inter-subject variability (Supplementary Figs. 13C and 14C). Hippocampal RBM3 levels did not correlate with GFAP levels (Supplementary Fig. 13D, E) or Iba1 levels (Supplementary Fig. 14D, E) on 4 or 8 days post-injury. In contrast, hippocampal CIRBP levels at 4 days post-injury, but not at 8 days, showed a significant negative correlation with GFAP levels (Supplementary Fig. 13F, G) and Iba1 levels (Supplementary Fig. 14F, G).
Discussion
The RV model of unilateral ischemia is the gold standard to study preclinical HI-mediated neuropathology in neonatal rodents. Based on neurodevelopmental metrics, a PND 7–10 pup approximates a 36–40-week term newborn.36 Here we established a modified RV model in PND 10 mice which incorporates non-invasive temperature surveillance to rigorously maintain target temperature across the surgical, hypoxic, and post-resuscitation phases. This ensured that brief acute endogenous neuroprotective hypothermia did not manifest immediately after injury as reported.37
Neuronal death and brain injury markers in temperature-managed PND 10 HI-injured mice
The RV model results in neuronal apoptosis and necrosis.38 FJ staining increased in the HI-injured hippocampus 3 days post-injury but was sporadic-to-absent 5–7 days later. FJ signal positivity is linked to eosinophilic necrosis or “red neurons”.39 The limited FJ staining here mimics reports that fewer neurons show evidence of eosinophilic-positive necrotic death in the neonatal RV model (although other types of necrosis are readily detected), vs. adult models of brain HI.38 However, the detection of FJ signals at 3 days post-injury in our model, suggests that it has utility in assessing the efficacy of neuroprotective therapies at that time point.
Next, we screened for potential HI injury markers at 3 days post-injury in hippocampal homogenates. P75NTR robustly decreased after HI. Studies show that neuronal KO of p75NTR disrupted neurodevelopment in mice and led to a profound decrease in cortical and hippocampal volumes by ~40% and ~35%, respectively, vs. WTs.40 The pro-apoptotic protein Bax modestly increased 3 days post-injury, consistent with reports by others showing that Bax inhibiting peptides protect in the RV model.41 Finally, we observed a robust decrease in CYPC. This was specific as we did not observe a similar change in CYPA post-HI.
Lastly, we measured 14 days hemispheric and hippocampal lesion volume.42 Significant brain tissue volume loss was detected ipsilateral to CCA-ligation. Together the findings confirm robust neuronal death in the acute and sub-acute phases after HI in our modified RV model.
Gliosis in temperature-managed PND 10 HI-injured mice
Astrocytosis and microgliosis are secondary injury mechanisms in perinatal asphyxia, and molecular signals released by these processes have clinical prognostic value for assessing HI severity.43,44 GFAP and Iba-1 robustly increased in ipsilateral hippocampus 3–7 days post-injury. Temporal changes in microglial staining intensity agree with other studies in the RV model.45 The small sample here (n = 3/group for injured time points) is a limitation. However, we sought to confirm the expected patterns of neuropathology studied by others, now with highly rigorous temperature control, before studying CSPs.
White matter injury in temperature-managed PND 10 HI-injured mice
White matter injury is common on neuroimaging in infants with moderate/severe HI and is associated with damage to the basal ganglia and thalamus.46 Studies in the RV model suggest that thalamic neurodegeneration blossoms in the first week.38,47 We characterized white matter damage with Weil stain and MBP immunoblotting. MBP robustly increased in shams by PND 18, at which time HI-injured pups began to manifest a trend toward decreased levels 8 days post-injury (p = 0.06). Weil stain of the myelin sheath in ipsilateral thalamus and cerebral peduncles also showed a decrease in staining 7 days post-injury.
Tissue degeneration in temperature-managed PND 10 HI-injured mice
Cupric-silver detects degenerating synaptic terminals, cell bodies, and dendrites at 1–4 days post-injury, whereas axonal degeneration is detected 3–7 days post-injury.48 Silver staining robustly increased throughout the ipsilateral hemisphere at 3 days post-HI, including in the cortex, hippocampus, and thalamus, but with little evidence of delayed diffuse axonal degeneration at 5–7 days post-injury. Patchy dendritic and neuropil cupric-silver positivity persisted 3–7 days across hippocampal subfields, which is indicative of ongoing delayed neuronal death/degeneration. This is consistent with HI in term infants from perinatal asphyxia and contrasts the diffuse non-cystic white matter injury/axonal damage characteristic of encephalopathy of prematurity.49,50,51
Effect of HI on CSP expression in the developing normothermic brain
Homeothermy in newborn mice proceeds in three phases.52 Pups aged PND 0–7 are functionally poikilothermic (Phase I). Pups aged PND 8-14 have limited thermogenesis (Phase II). Pups aged PND 15–17 reach peak (adult) capacity for shivering thermogenesis and can maintain body temperature (Phase III).52 RBM3, CIRBP, and RTN3 are abundant in the mammalian normothermic brain during infancy, then rapidly decrease at an early postnatal age. We examined if HI altered the baseline CSP expression in the brain during phases II–III of the thermogenic transition in neonatal mice. CSPs were measured at PND 11 (early Phase II), PND 14 (late Phase II), and PND 18 (late Phase III). Baseline brain CSP levels in shams were inversely associated with the phase of thermoregulatory development. Whether this relationship is causative or correlative is unclear. Given that hypoxia alone can increase RBM3 and CIRBP levels, surprisingly HI in normothermic pups minimally impacted the age-related decline in brain CSPs.53
Cold-induced RBM3 is a potent neuroprotectant in vivo.10,11 RBM3 KO mice subjected to cold-stress in utero have postnatal brain deformities, suggesting that it safeguards the developing CNS from potentially detrimental antenatal hypothermia.23 Furthermore, in HI-injured RBM3 KOs, neural stem cell proliferation in the subgranular zone (SGZ) of the DG was severely impaired and apoptosis was increased vs. WTs.54 We found that age was associated with a significant decrease in RBM3 levels in the cortex, thalamus, and hippocampus. However, HI did not affect RBM3 levels in the cortex or thalamus. In contrast, the interaction of HI with age was detected in the hippocampus, manifested as increased levels 8 days post-injury. Intriguingly, surrogate markers of insult severity (GFAP, Iba1) did not correlate with RBM3 levels in the hippocampus at 4 and 8 days post-injury. That raises important questions/concepts for additional exploration. Seventy-two hours of TH within 6 h of birth is the standard of care to treat HI in term newborns in high-income countries and decreases long-term disability.30 However, it is unknown if clinically optimized TH slows the age-related loss of RBM3 expression in the developing brain post-insult.55 Similarly, the optimal depth and duration of cooling that maximally increase/prolong brain RBM3 levels in the RV model need to be explored. The lack of a correlation between GFAP/Iba1 and RBM3 levels supports the notion that RBM3-inducing therapies should be studied across insult severity. Characterization here on neurodevelopmental timing of RBM3 changes early after HI, in the RV model, provides insight for future studies.
CIRBP can promote neuroprotection and neurotoxicity.2 Whether CIRBP-mediated effects on neuronal survival are temperature-dependent or modified by developmental age remains to be determined. In adult mice, infarct volume is decreased in normothermic CIRBP KOs after middle cerebral artery occlusion.8 Furthermore, studies in normothermic BV-2 cells suggested that hypoxia induces microglial CIRBP secretion and impacts neuronal injury.8 We found that CIRBP levels in the cortex, thalamus, and hippocampus were associated with neonatal age. In the thalamus and hippocampus, the age-mediated decline was modest. In the cortex, the decrease was steep, mirroring the rapidly fading expression of RBM3 in the cortex and hippocampus by PND 18. Furthermore, an interaction (injury vs. age) was detected in the cortex and hippocampus, manifested as a slight decrease in HI-injured CIRBP levels at 4 days post-injury but a slight increase at 8 days post-injury. Also, surrogate markers of insult severity (GFAP, Iba1) correlated with hippocampal CIRBP levels at 4 days post-injury. Unaddressed questions include—does TH increase CIRBP levels in the RV model, is it neuroprotective/neurotoxic, what depth/duration of TH optimally induces (or avoids) CIRBP induction, and might early/acute serum CIRBP levels serve as a HI severity biomarker? We did not measure serum CIRBP levels to know if the insult-severity-dependent decrease in brain levels was associated with increased serum levels. However, serum CIRBP is increased in patients with brain injury from intracerebral hemorrhage; it may also be detectable post-injury in the clinical NE population and merits investigation.56
RTN3 is a recent addition to the small family of CSPs. Like CIRBP, it may be neuroprotective and/or neurotoxic.2 However, chronically increasing levels at normothermia can produce RTN3-immunoreactive dystrophic neurites.14 RBM3 also regulates RTN3 induction during cold stress.5 We found that age was associated with decreased RTN3 levels (25 and 20 kDa forms) in the cortex and thalamus. RTN3 in the hippocampus was at the limit of detection, precluding analysis; however, hippocampal RTN3 was reported to increase with age from 2 to 18 months in mice.57 In the cortex, injury is independently associated with an increase in RTN3c levels. Its impact on post-HI recovery remains unclear. Finally, we measured the correlation between RBM3/CIRBP (classic CSPs) and RBM3/RTN3 expression for additional insights into their possible co-regulation. In all three brain regions, RBM3/CIRBP were strongly correlated in shams and after HI. RBM3/RTN3 showed weaker correlations in shams. However, after HI, RBM3/RTN3 were no longer correlated. The latter finding raises the possibility that HI injury may modify downstream signaling pathways that link RBM3 with RTN3 induction.5 It remains to be tested if this could translate to a decreased ability of TH to induce RTN3 in neonatal HI.
Our study has limitations, specifically, the exclusion of females and the small sample size. Sex differences are well described in the NE literature; males tend to have larger lesion size vs. females in the RV model.58,59 Also, RBM3 is located on the X chromosome and is among a few genes that escapes X-chromosome inactivation in human female cells.60,61,62 It is unclear if this phenomenon alters the dynamics of RBM3 expression in the normothermic developing female vs. male brain, or if it helps explain observations in terms of why mild TH may offer greater neuroprotection in juvenile female vs male rodents.63 Given the need to improve outcomes in males and females, future studies of the impact of TH on CSPs are warranted in females, and to assess the ability of modulation of these CSPs to provide neuroprotection across temperature and age.64
In summary, we investigated the effect of HI on the neurodevelopmental expression of CSPs at normothermia during a critical time window involving the rapid maturation of thermogenesis in neonatal mice. CSPs rapidly decreased in the neonatal brain as heat-generating mechanisms become operational and the levels were not importantly altered by HI. Given the previously reported neuroprotective effects of RBM3 in adult mice,11 and ongoing efforts to optimize care in perinatal asphyxia,65 our findings support the need to determine if blunting the age-related decline in neuroprotective RBM3 (such as with TH) may promote neuroprotection during critical periods post-NE.
References
Al-Fageeh, M. B. & Smales, C. M. Control and regulation of the cellular responses to cold shock: the responses in yeast and mammalian systems. Biochem. J. 397, 247–259 (2006).
Jackson, T. C. & Kochanek, P. M. A new vision for therapeutic hypothermia in the era of targeted temperature management: a speculative synthesis. Ther. Hypothermia Temp. Manag. 9, 13–47 (2019).
Chappell, S. A., Owens, G. C. & Mauro, V. P. A 5’ leader of Rbm3, a cold stress-induced mRNA, mediates internal initiation of translation with increased efficiency under conditions of mild hypothermia. J. Biol. Chem. 276, 36917–36922 (2001).
Al-Fageeh, M. B. & Smales, C. M. Cold-inducible RNA binding protein (CIRP) expression is modulated by alternative mRNAs. RNA 15, 1164–1176 (2009).
Bastide, A. et al. Rtn3 is a novel cold-induced protein and mediates neuroprotective effects of Rbm3. Curr. Biol. 27, 638–650 (2017).
Lujan, D. A., Ochoa, J. L. & Hartley, R. S. Cold-inducible RNA binding protein in cancer and inflammation. Wiley Interdiscip. Rev. RNA https://doi.org/10.1002/wrna.1462 (2018).
Qiang, X. et al. Cold-inducible RNA-binding protein (CIRP) triggers inflammatory responses in hemorrhagic shock and sepsis. Nat. Med. 19, 1489–1495 (2013).
Zhou, M., Yang, W. L., Ji, Y., Qiang, X. & Wang, P. Cold-inducible RNA-binding protein mediates neuroinflammation in cerebral ischemia. Biochim. Biophys. Acta 1840, 2253–2261 (2014).
Wong, J. J. et al. Rbm3 regulates temperature sensitive Mir-142-5p and Mir-143 (thermomirs), which target immune genes and control fever. Nucleic Acids Res 44, 2888–2897 (2016).
Liu, B. et al. The overexpression of Rbm3 alleviates TBI-induced behaviour impairment and AD-like tauopathy in mice. J. Cell. Mol. Med. 24, 9176–9188 (2020).
Peretti, D. et al. Rbm3 mediates structural plasticity and protective effects of cooling in neurodegeneration. Nature 518, 236–239 (2015).
Su, F. et al. CIRBP ameliorates neuronal amyloid toxicity via antioxidative and antiapoptotic pathways in primary cortical neurons. Oxid. Med. Cell. Longev. 2020, 2786139 (2020).
Sertel, S. M., von Elling-Tammen, M. S. & Rizzoli, S. O. The mRNA-binding protein Rbm3 regulates activity patterns and local synaptic translation in cultured hippocampal neurons. J. Neurosci. 41, 1157–1173 (2021).
Hu, X. et al. Transgenic mice overexpressing reticulon 3 develop neuritic abnormalities. EMBO J. 26, 2755–2767 (2007).
Kim, D. Y., Kim, K. M., Kim, E. J. & Jang, W. G. Hypothermia-induced RNA-binding motif protein 3 (Rbm3) stimulates osteoblast differentiation via the Erk signaling pathway. Biochem. Biophys. Res. Commun. 498, 459–465 (2018).
Cooper, S. T. et al. Effects of hibernation on bone marrow transcriptome in thirteen-lined ground squirrels. Physiol. Genomics 48, 513–525 (2016).
Dresios, J. et al. Cold stress-induced protein Rbm3 binds 60s ribosomal subunits, alters microrna levels, and enhances global protein synthesis. Proc. Natl Acad. Sci. USA 102, 1865–1870 (2005).
Elabbassi, E. B. et al. Head insulation and heat loss in naked and clothed newborns using a thermal mannequin. Med. Phys. 29, 1090–1096 (2002).
Baum, J. D. Keeping babies warm. Bull. Am. Coll. Nurse Midwives 16, 39–46 (1971).
Totapally, A. et al. Epidemiology and outcomes of children with accidental hypothermia: a propensity-matched study. J. Trauma Acute Care Surg. 82, 362–367 (2017).
Rosenthal, M., Poliquin, V. & Yu, A. Maternal hypothermia from environmental exposure in the third trimester. Int. J. Circumpolar Health 79, 1710894 (2020).
Pilotte, J., Cunningham, B. A., Edelman, G. M. & Vanderklish, P. W. Developmentally regulated expression of the cold-inducible RNA-binding motif protein 3 in euthermic rat brain. Brain Res. 1258, 12–24 (2009).
Xia, W., Su, L. & Jiao, J. Cold-induced protein Rbm3 orchestrates neurogenesis via modulating Yap mRNA stability in cold stress. J. Cell Biol. 217, 3464–3479 (2018).
Chip, S. et al. The RNA-binding protein Rbm3 is involved in hypothermia induced neuroprotection. Neurobiol. Dis. 43, 388–396 (2011).
Jackson, T. C., Kotermanski, S. E. & Kochanek, P. M. Infants uniquely express high levels of Rbm3 and other cold-adaptive neuroprotectant proteins in the human brain. Dev. Neurosci. 40, 325–336 (2018).
Jackson, T. C., Janesko-Feldman, K., Carlson, S. W., Kotermanski, S. E. & Kochanek, P. M. Robust Rbm3 and beta-Klotho expression in developing neurons in the human brain. J. Cereb. Blood Flow Metab. 39, 2355–2367 (2019).
Di Scala, F. et al. Tissue specificity and regulation of the N-terminal diversity of reticulon 3. Biochem. J. 385, 125–134 (2005).
Chakkarapani, A. A. et al. Therapies for neonatal encephalopathy: targeting the latent, secondary and tertiary phases of evolving brain injury. Semin. Fetal Neonatal Med. 26, 101256 (2021).
Marlow, N. et al. Neurological and developmental outcomes following neonatal encephalopathy treated with therapeutic hypothermia. Semin. Fetal Neonatal Med. 26, 101274 (2021).
Jacobs, S. E. et al. Cooling for newborns with hypoxic ischaemic encephalopathy. Cochrane Database Syst. Rev. CD003311 (2013).
Jacobs, S. E. et al. Whole-body hypothermia for term and near-term newborns with hypoxic-ischemic encephalopathy: a randomized controlled trial. Arch. Pediatr. Adolesc. Med. 165, 692–700 (2011).
El-Dib, M. et al. Should therapeutic hypothermia be offered to babies with mild neonatal encephalopathy in the first 6 h after birth? Pediatr. Res. 85, 442–448 (2019).
Hess, S. E. et al. Home improvement: C57bl/6j mice given more naturalistic nesting materials build better nests. J. Am. Assoc. Lab. Anim. Sci. 47, 25–31 (2008).
Gaskill, B. N. et al. Heat or insulation: behavioral titration of mouse preference for warmth or access to a nest. PLoS ONE 7, e32799 (2012).
Uchihara, T. Silver diagnosis in neuropathology: principles, practice and revised interpretation. Acta Neuropathol. 113, 483–499 (2007).
Semple, B. D., Blomgren, K., Gimlin, K., Ferriero, D. M. & Noble-Haeusslein, L. J. Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 106-107, 1–16 (2013).
Reinboth, B. S. et al. Endogenous hypothermic response to hypoxia reduces brain injury: implications for modeling hypoxic-ischemic encephalopathy and therapeutic hypothermia in neonatal mice. Exp. Neurol. 283, 264–275 (2016).
Nakajima, W. et al. Apoptosis has a prolonged role in the neurodegeneration after hypoxic ischemia in the newborn rat. J. Neurosci. 20, 7994–8004 (2000).
Santagostino, S. F., Spinazzi, M. & Radaelli, E. Restricted sensitivity of Fj-C staining to assess neuronal degeneration and death in preclinical mouse studies. Vet. Pathol. 58, 643–649 (2021).
Meier, S. et al. The P75 neurotrophin receptor is required for the survival of neuronal progenitors and normal formation of the basal forebrain, striatum, thalamus and neocortex. Development 146, dev181933 (2019).
Sun, M. Y. et al. Bax inhibiting peptide reduces apoptosis in neonatal rat hypoxic-ischemic brain damage. Int. J. Clin. Exp. Pathol. 8, 14701–14708 (2015).
Annink, K. V. et al. The long-term effect of perinatal asphyxia on hippocampal volumes. Pediatr. Res. 85, 43–49 (2019).
Chalak, L. F. et al. Biomarkers for severity of neonatal hypoxic-ischemic encephalopathy and outcomes in newborns receiving hypothermia therapy. J. Pediatr. 164, 468.e1–474.e1 (2014).
Li, Y. et al. Osteopontin is a blood biomarker for microglial activation and brain injury in experimental hypoxic-ischemic encephalopathy. eNeuro 4, ENEURO.0253-16.2016 (2017).
Umekawa, T., Osman, A. M., Han, W., Ikeda, T. & Blomgren, K. Resident microglia, rather than blood-derived macrophages, contribute to the earlier and more pronounced inflammatory reaction in the immature compared with the adult hippocampus after hypoxia-ischemia. Glia 63, 2220–2230 (2015).
Shankaran, S. et al. Neonatal magnetic resonance imaging pattern of brain injury as a biomarker of childhood outcomes following a trial of hypothermia for neonatal hypoxic-ischemic encephalopathy. J. Pediatr. 167, 987.e3–993.e3 (2015).
Northington, F. J., Ferriero, D. M., Graham, E. M., Traystman, R. J. & Martin, L. J. Early neurodegeneration after hypoxia-ischemia in neonatal rat is necrosis while delayed neuronal death is apoptosis. Neurobiol. Dis. 8, 207–219 (2001).
Switzer, R. C. 3rd Application of silver degeneration stains for neurotoxicity testing. Toxicol. Pathol. 28, 70–83 (2000).
Gopagondanahalli, K. R. et al. Preterm hypoxic-ischemic encephalopathy. Front. Pediatr. 4, 114 (2016).
Volpe, J. J. Brain injury in premature infants: a complex amalgam of destructive and developmental disturbances. Lancet Neurol. 8, 110–124 (2009).
Agut, T. et al. Preterm white matter injury: ultrasound diagnosis and classification. Pediatr. Res. 87, 37–49 (2020).
Lagerspetz, K. Y. H. Postnatal development of thermoregulation in laboratory mice. Helgoländer wissenschaftliche Meeresuntersuchungen 14, 559–571 (1966).
Wellmann, S. et al. Oxygen-regulated expression of the RNA-binding proteins RBM3 and CIRP by a HIF-1-independent mechanism. J. Cell Sci. 117, 1785–1794 (2004).
Zhu, X. et al. RBM3 promotes neurogenesis in a niche-dependent manner via IMP2-IGF2 signaling pathway after hypoxic-ischemic brain injury. Nat. Commun. 10, 3983 (2019).
Shankaran, S. et al. Effect of depth and duration of cooling on death or disability at age 18 months among neonates with hypoxic-ischemic encephalopathy: a randomized clinical trial. JAMA 318, 57–67 (2017).
Zhou, K. et al. Cold-inducible RNA-binding protein contributes to intracerebral hemorrhage-induced brain injury via TLR4 signaling. Brain Behav. 10, e01618 (2020).
Shi, Q., Hu, X., Prior, M. & Yan, R. The occurrence of aging-dependent reticulon 3 immunoreactive dystrophic neurites decreases cognitive function. J. Neurosci. 29, 5108–5115 (2009).
Hill, C. A. & Fitch, R. H. Sex differences in mechanisms and outcome of neonatal hypoxia-ischemia in rodent models: implications for sex-specific neuroprotection in clinical neonatal practice. Neurol. Res. Int. 2012, 867531 (2012).
Mirza, M. A., Ritzel, R., Xu, Y., McCullough, L. D. & Liu, F. Sexually dimorphic outcomes and inflammatory responses in hypoxic-ischemic encephalopathy. J. Neuroinflammation 12, 32 (2015).
Danno, S. et al. Increased transcript level of RBM3, a member of the glycine-rich rna-binding protein family, in human cells in response to cold stress. Biochem. Biophys. Res. Commun. 236, 804–807 (1997).
Zhang, Y. et al. Genes that escape X-inactivation in humans have high intraspecific variability in expression, are associated with mental impairment but are not slow evolving. Mol. Biol. Evol. 33, 302 (2016).
Wainer Katsir, K. & Linial, M. Human genes escaping X-inactivation revealed by single cell expression data. BMC Genomics 20, 201 (2019).
Dietz, R. M. et al. Therapeutic hypothermia protects against ischemia-induced impairment of synaptic plasticity following juvenile cardiac arrest in sex-dependent manner. Neuroscience 325, 132–141 (2016).
Back, S. A. White matter injury in the preterm infant: pathology and mechanisms. Acta Neuropathol. 134, 331–349 (2017).
Benninger, K. L. et al. Perspectives from the Society for Pediatric Research. Neonatal encephalopathy clinical trials: developing the future. Pediatr. Res. 89, 74–84 (2021).
Funding
This work was supported by NIH/NINDS grants R01NS105721 to T.C.J., by the University of South Florida Morsani College of Medicine start-up funds to T.C.J., by a Lloyd Reback Family Gift and T32 (2T32HD040686) to J.R.H., and by the Ake N. Grenvik Chair in Critical Care Medicine to P.M.K.
Author information
Authors and Affiliations
Contributions
T.C.J. conceived the study. T.C.J. and P.M.K. contributed to the study design. T.C.J. and J.R.H. drafted the manuscript. R.H.G., R.D.K., V.A.V., K.G., K.J.-F., and J.S. contributed to experiments and data acquisition. T.C.J., P.M.K., R.H.G., and J.R.H. contributed to data analysis. R.H.G., R.D.K., V.A.V., K.G., K.J.-F., and J.S. edited the draft and contributed to the final submitted version.
Corresponding author
Ethics declarations
Competing interests
T.C.J. and P.M.K. are co-inventors on a USPTO Application (No. 15/573,006) titled: “Method to Improve Neurologic Outcomes in Temperature Managed Patients.”
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
About this article

Cite this article
Jackson, T.C., Herrmann, J.R., Garman, R.H. et al. Hypoxia–ischemia-mediated effects on neurodevelopmentally regulated cold-shock proteins in neonatal mice under strict temperature control. Pediatr Res (2022). https://doi.org/10.1038/s41390-022-01990-4
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41390-022-01990-4
This article is cited by
-
RBM3 Promotes Anti-inflammatory Responses in Microglia and Serves as a Neuroprotective Target of Ischemic Stroke
Molecular Neurobiology (2024)
-
Hypothermia increases cold-inducible protein expression and improves cerebellar-dependent learning after hypoxia ischemia in the neonatal rat
Pediatric Research (2023)
-
FGF21 modulates hippocampal cold-shock proteins and CA2-subregion proteins in neonatal mice with hypoxia–ischemia
Pediatric Research (2023)
